
Method Article
In Vivo Modellierung des Morbid Human Genome mit Danio rerio
In diesem Artikel
Zusammenfassung
Hier präsentieren wir einen systematischen Ansatz für die Entwicklung physiologisch relevant, sensitive und spezifische In vivo Assays für die Interpretation Variation in der menschlichen Pathologie. Transient Genmanipulation via Mikroinjektion von WT und mutiertes menschliches mRNA und Morpholino (MO) Antisense-Oligonukleotide nutzen die Lenkbarkeit der Entwicklung Zebrafisch schnell Assay pathogenen Mutationen, insbesondere, aber nicht ausschließlich, im Zusammenhang mit menschlichen Entwicklungsstörungen.
Zusammenfassung
Hier präsentieren wir Methoden für die Entwicklung von Assays, um potenziell klinisch signifikante nichtsynonymen Änderungen mit in vivo Komplementation in Zebrafisch abzufragen. Zebrafisch (Danio rerio) sind ein nützliches Tier System aufgrund ihrer experimentellen Handhabbarkeit; Embryonen sind transparent für einfache Betrachtung zu ermöglichen, durchlaufen eine schnelle Entwicklung ex vivo und können genetisch manipuliert werden 1 Diese Aspekte für bedeutende Fortschritte in der Analyse der Embryogenese erlaubt haben,. molekulare Prozesse und morphogenetic Signalisierung. Zusammengenommen machen die Vorteile dieser Wirbeltiere Modell Zebrafisch sehr gut für die Modellierung der Entwicklungsstörungen in pädiatrische Erkrankung, und in einigen Fällen, Altersdiabetes Störungen. Da der Zebrafisch ist eng mit dem des Menschen (~ 70% orthologous) konserviert sind, ist es möglich, menschlichen Krankheitszuständen im Zebrafisch zusammenzufassen. Dies wird entweder durch die Injektion von mutiertes menschliches m erreichtRNA oder dominant negative gain of function Allele oder Verwendung Morpholino (MO) Antisense-Oligonukleotide, um Gene zu unterdrücken Funktionsverlust Varianten imitieren induzieren. Durch Ergänzung der MO-induzierte Phänotypen mit capped menschlichen mRNA, ermöglicht unser Ansatz die Interpretation der schädlichen Auswirkungen von Mutationen auf die menschliche Protein-Sequenz auf die Fähigkeit der mutierten mRNA, um einen messbaren, physiologisch relevanten Phänotyp retten basiert. Modellierung des menschlichen Krankheit Allele erfolgt durch Mikroinjektion von Zebrafisch Embryonen mit MO und / oder menschlichen mRNA im 1-4 Zell-Stadium, und Phänotypisierung bis zu sieben Tage nach der Befruchtung (DPF). Diese allgemeine Strategie kann auf eine Vielzahl von Krankheitsphänotypen erweitert werden, wie in dem folgenden Protokoll gezeigt. Wir präsentieren unsere etablierte Modelle für morphogenetic Signalisierung, kraniofaziale, Herz-, Gefäß-Integrität, Nierenfunktion und Skelettmuskel Störung Phänotypen sowie andere.
Einleitung
Die funktionale Interpretation der genetischen Information und Zuordnung der prädiktiven klinischen Wert zu einem Genotyp stellt ein großes Problem in der medizinischen Genetik und wird immer ergreifend mit der Beschleunigung technischen und wirtschaftlichen Durchführbarkeit der genomweite Sequenzierung. Daher ist es notwendig, die Entwicklung und Umsetzung neuer Paradigmen, die Pathogenität von Varianten unbekannter Signifikanz (MUS) in Patienten nachgewiesen testen. Diese Tests müssen dann genau sein, zeit-und kosten-effizient und Hafen das Potenzial, einen Übergang zu klinischen Nutzen zu katalysieren.
Während sich die Maus ist traditionell das Werkzeug der Wahl auf dem Gebiet der Modellierung menschlichen Krankheit gewesen sind Zebrafisch als wissenschaftlich und wirtschaftlich günstige Surrogat Schwellenländern. Im Gegensatz zur Maus, Zebrafisch erlaubt Biologie einfach und zeitnah Zugang zu allen Entwicklungsstadien, durch optische Klarheit der Embryonen, die für Echtzeit-Bildgebung zu entwickeln Pathologien ermöglicht unterstützt. 1 Die relativ neue Generation von Mutanten Zebrafischlinien hat zusätzliche Tests und Modellierung Optionen, die von vielen in funktionelle Studien verwendet werden, vorausgesetzt, diese Technologie weiter zu beschränken (Bewertung in 1,38) werden. Nicht nur genetische Mutanten mit Knock-ins spezifischer Mutationen mühsam zu erreichen, sind sie auch nicht geeignet, um Medium-oder High-Throughput-Analyse für die Prüfung von einer Reihe von Mutationen in einem Gen. Wichtig ist, dass eine einzige Reihe von Tests nicht nur Informationen für die pathogene Potential der Allele zu schaffen, sondern auch für die Richtung der Wirkung auf zellulärer Ebene (zB Verlust der Funktion vs gain of function), die entscheidend für die Unterrichtung Erbgang ist in Familien, vor allem, wenn kleine menschliche Stammbäume beherbergen begrenzte Informationen über die Art der genetischen Übertragung. Zum weiteren Vergleich der Verwendungen der verfügbaren Maus und Zebrafisch Modelle, siehe Tabelle 1.
Wir stellen ferner fest, dass diere sind inhärente Beschränkungen der Zebrafisch Modellsystem. Obwohl D. rerio haben schnelle anfängliche Entwicklung von Organsystemen, erfordert Geschlechtsreife etwa drei Monate. Aus diesem Grund sind pränatale und pädiatrische-onset Erkrankungen die am stärksten für eine transiente Expression dieses Modell. Während ideal für die Durchführung von großen chemischen Verbindung Bildschirme, ist die Verwendung von genetischen Mutanten nicht zu einer systematischen Prüfung von Tausenden von synonymen Varianten denkbar, die dazu beitragen, und weiterhin in der pädiatrischen Erkrankungen nachgewiesen werden.
Die Komplementierung hier beschriebenen Tests nutzen diese experimentellen Lenkbarkeit hohen Grad an Homologie, und die Erhaltung der Funktion zwischen Mensch und Zebrafisch-Proteine, insbesondere so für Moleküle, die für konservierte Entwicklungsprozesse. Abbildung 1 skizziert die Prüfung und Kennzeichnung Strategie für verschiedene Allel-Effekte. Beide Verlust der Funktion (LOF) und dominant Assays durchgeführt werden kann. Für LOF, beginnt das Experiment mit der Unterdrückung des Gens von Interesse mit einem Morpholino Knockdown und Testen auf Phänotypen, die relevant für die klinische Phänotyp der Untersuchung könnte. Oder durch Eingriffe Spleißen indem ein MO auf Spleißstelle, typischerweise induziert entweder die Aufnahme eines, Suppression kann entweder durch Blockieren Übersetzung von Aktionen in einem MO an oder nahe der translationalen Startstelle des Zebrafisch-Locus (tbMO Übersetzung Blocker Morpholino) erreicht werden, Intron oder Exon-Skipping aberrante (splice Blockierung Morpholino; SBMO).
Anschließend wird capped mRNA aus dem menschlichen orthologe Transkript eingeführt und quantifizierbare Rettung der Phänotyp gemessen. Sobald der Test aufgebaut ist, kann Kandidat Mutationen im humanen Botschaft vorgestellt und untersucht für ihre Fähigkeit, die MO-induzierte Phänotyp auf der gleichen Effizienz wie WT menschlichen mRNA retten. Umgekehrt ist für Bewerber dominante Allele, menschliche mRNA (aber nicht MO) ist Einführunged mit der Erwartung, dass WT menschlichen mRNA nicht grob beeinflussen Zebrafisch Anatomie und Physiologie, wohingegen Einführung von Test-Mutationen, die eine dominante Wirkung Phänotypen analog zu denen in der menschlichen klinischen Zustand beobachtet induzieren. Dieses Experiment kann feinkörnigen weiter zu sezieren, ob der dominierende Effekt durch eine gain of function (GOF) oder einer dominant-negativen Mechanismus durch Mischen WT und mutierten humanen mRNA auftritt; GOF für Ereignisse, die Zugabe von WT menschlichen mRNA wird erwartet, dass irrelevant, sollte während für dominant-negative Allele, Mischen von WT und Mutant mRNA verändern die Schwere der Phänotyp durch Mutante Nachricht induziert. In allen Fällen empfehlen wir, dass alle Kombinationen von Injektionen (MO mit WT menschlichen mRNA vs morpholino mit mutierten humanen mRNA usw. durchgeführt, vorzugsweise in der gleichen Kupplung von Embryonen (siehe Abbildung 1) Interpretation ist wie folgt.:
Für LOF Tests:
- Wenn der Zuschlagerzeugt einen Phänotyp auf, die entsprechend durch Mutanten und WT mRNA gerettet werden kann, ist das Allel wahrscheinlich gutartig.
- Wenn das mutierte Rettung der Knockdown Phänotyp ist nicht von der Knockdown Phänotyp selbst, ist das Allel eine wahrscheinliche funktionelle null. Das Experiment kann nicht zwischen wahr Nullen (kein funktionelles Protein) und ultralow Protein Aktivität, die keine Rettung Kapazität haben diskriminieren.
- Wenn das mutierte Rettung der Knockdown Phänotyp ist statistisch besser als die MO, aber schlechter als der WT, ist das Allel wahrscheinlich ein hypomorph da dies Ergebnis zeigt teilweisen Verlust der Funktion.
Für dominant Tests:
- Wenn es keinen Zuschlag Phänotyp, aber Injektion von WT mRNA produziert einen Phänotyp, muss ein Notfallplan verwendet werden, wenn das Experiment, um fortzufahren (siehe unten).
- Wenn es keine Knockdown Phänotyp und Injektion von WT mRNA produziert keine Phänotyp, geht der Versuch wie üblich.
- Wenn Injektionvon mutierten mRNA ist äquivalent zu der Wildtyp-mRNA kann das Allel entweder gutartig oder Verlust der Funktion, oder kann der Test versagt haben. Dies erfordert weitere Experimente, um zwischen diesen Optionen zu unterscheiden.
- Wenn Injektion von mutierten mRNA ist nicht von MO Knockdown, wird die Funktion des Genprodukts wahrscheinlich in irgendeiner Weise verändert. Um die Änderung in der Funktion zu erkennen, sollte eine Titration der mutierten mRNA mit Wildtyp-mRNA vorgenommen werden.
- Wenn das Ergebnis dieser Titration ununterscheidbar zu Wildtyp-mRNA allein hat es sich gezeigt, dass das mutierte Protein Produkt das Wildtyp-Protein verwendet als Substrat für die Wirkung mit der Menge der Titration verändert wird. Dies deutet auf eine dominant negative Phänotyp.
- Wenn das Ergebnis dieser Titration nicht zu unterscheiden mutierten mRNA allein hat es sich gezeigt, dass das mutierte Protein Produkt nicht mehr die gleiche Funktion wie der Wildtyp, und ist somit nicht durch die Menge an Wildtyp-Protein-Produkt vor betroffengesendet. Dies zeigt, dass das Allel wahrscheinlich gain of function ist.
Notfallplan:
- Wenn kein Phänotyp präsentiert von MO Knockdown, aber tut Gegenwart mit WT mRNA können weitere Experimente auftreten, obwohl wir betonen, dass diese Situation nicht ideal ist. WT menschlichen mRNA titriert, um den Phänotyp zu minimieren, und kann auch als neuer Sollwert verwendet werden. Weitere Co-Injektion von WT und mutiertes menschliches mRNA kann basierend auf der Rettung Fähigkeit des mutierten ausgewertet werden.
Protokoll
1. Bioinformatik-Analyse
- Bestimmen Sie, ob das menschliche Gen von Interesse hat ein Zebrafisch ortholog, und wenn ja, wie viele Kopien. Wir empfehlen gegenseitige BLAST ( http://blast.ncbi.nlm.nih.gov/ ) des humanen Proteins gegen Zebrafisch und anschließende BLAST der besten Zebrafisch gegen das menschliche Genom zu treffen. Wahre Orthologe wird die beste Hit in jedem Fall sein.
- Bestimmen der Größe des offenen Leserahmens (ORF) des menschlichen Gens. Wenn mehr als 6 kb, dieses Modell ist derzeit aufgrund von Einschränkungen von hoher Qualität in vitro-Transkription von langer Templates unlösbar.
- Erhalten oder erzeugen ein Konstrukt, das den Menschen in der ORF pCS2 + Vektor-Rückgrat (oder gleichwertig Vektor mit einem 5 'SP6 Transkription Website und 3'-Poly-A-Signal).
- Entwerfen Sie ein MO bis Spleißen oder Übersetzung der gezielten Zebrafisch-Gen blockieren. Wenn mehrere Kopien existieren im Zebrafisch Genom there es zwei Möglichkeiten: a) Gestaltung der zusätzlichen MOs, oder b) die Feststellung einer Spleißstelle vollständig zwischen den Kopien erhalten, gegen die ein einzelner MO effektiv sein kann. Einige veröffentlichte MO-Sequenzen sind bei www.zfin.org .
2. Expression Analysis in der Entwicklung von Zebrafisch Embryo
- Bestimmen Sie, ob der Zebrafisch ortholog in einem räumlich-zeitlichen Kontext relevant für die phänotypische Auslesen exprimiert wird. Wenn keine Daten vorhanden Ausdruck für das Gen von Interesse sind, führen reversen Transkription (RT)-PCR unter Verwendung von ganzen Zebrafischembryonen oder in-situ-Hybridisierung cDNA. (Siehe 2,3).
3. Ortsspezifische Mutagenese
- Design Mutageneseprimer 25-45 Basen Länge, mit der gewünschten Mutation in der Mitte. Der Primer Schmelztemperatur sollte größer als oder gleich 78 ° C. Entwerfen Sie ein Forward-und Reverse Mutageneseprimers auf gegenüberliegenden GlühenStränge des Plasmids.
- Erhalten Primer für Sequenz Bestätigung des ORF post-Mutagenese, sollten diese Fliese über ~ 300 Basenpaar-Abschnitte, um den gesamten ORF zu decken.
- Montieren Sie die Mutagenese Reaktion mit einer High-Fidelity-Polymerase und Zyklus wie folgt (1: 95 ° C 30 sec, 2: 95 ° C 30 sec, 3: 55 ° C 1 min, 4: 68 ° C 6 min, 5: Gehen 18x 2, 6: 4 ° C immer, 7: Ende).
- 1 ul DpnI Restriktionsendonuklease pro Reaktion auf dam methylierte Vorlage zu entfernen; bei 37 ° C für 2 Stunden.
- Transform 2 ul Mutagenesereaktion in 20 ul kompetente Zellen nach Standard-Protokollen.
- Wählen Sie 3-4 Kolonien und impfen 5 ml LB-Medium mit entsprechenden Antibiotika. Schütteln bei 37 ° C über Nacht bei 225 Umdrehungen pro Minute.
- Miniprep DNA bestimmen und Konzentration.
- Sequenz die Mutation Ort zur Bestätigung der Anwesenheit der Mutation von Interesse.
- Sequence gesamten ORF zu sequenzieren Integrität bestätigen.
4. In vitro mRNA Transkription
- Mit einem linearisierten pCS2 + Vorlage erzeugen capped mRNA mit der mMessage mMachine SP6 Kit (Ambion). Wir empfehlen die Verwendung der Hälfte der Reaktion Komponente Mengen.
- Reinigen Sie die RNA-Probe mit LiCl Fällung oder Phenol Chloroform als in den Kit Anleitung beschrieben.
- Bestimmen Sie die Konzentration der mRNA mit Absorption, um die Integrität der mRNA mittels Gelelektrophorese, und speichern Sie die Probe in drei oder mehr Portionen bei -80 ° C bis zur Verwendung. Wir empfehlen nicht mehrere Gefrier-Auftau-Zyklen von mRNA Aliquots.
5. In vivo Assay von Variant Verlust der Funktion
- Erhalten Zebrafischembryonen aus natürlichen Zebrafisch Paarungen, und halten sie bei 28 ° C in Wasser Embryo in 6 oder 10 cm Gerichte.
- Führen Sie eine Morpholino Dosisreaktionskurve zu Phänotyp Spezifität, MO MO Effizienz und Toxizität zu bewerten. Injizieren einer Dosis-Kurve vonmindestens drei verschiedenen Konzentrationen zwischen 1-10 ng MO in 50-100 (1-4 Zell-Stadium) Zebrafischembryonen / Charge. Effiziente MOs sollte Vorrücken in dosisabhängige Erhöhung des Anteils der betroffenen Embryonen in einer Charge zu ergeben.
- Phänotyp Embryonen im Entwicklungsstadium angemessen auf die Expression von gezielten Zebrafisch Gen von Interesse und in welchem Stadium ein relevant Phänotyp beobachtet werden basierend. Dies kann entweder quantitative (zB Messung zwischen anatomischen Strukturen) oder qualitativ (auf Basis standardisierter phänotypischen Kriterien). Für alle Injektionen> 24 erzielt HPF: Embryonen mit PTU (0,003% 1-Phenyl-2-thioharnstoff in Medien Embryo) bei 24 HPF, um eine maximale Reduzierung der Melanozyten Bildung behandelt.
- Für Spleiß-blocking MOs Test MO Effizienz durch Extrahieren von Gesamt-RNA aus ganzen Embryo Lysate zum Zeitpunkt der phänotypischen Scoring, Herstellung von cDNA und führen RT-PCR des Zielgens unter Verwendung der Primer flankieren MO Zielstelle.
- Um die Unterdrückung effic überprüfeniency einer tb-MO, Ernte ganze Embryo Proteinlysate und führen Immunoblot auf Ebenen der gezielten Protein im Vergleich zur Kontrollgruppe zu vergleichen. Allerdings ist dieser Ansatz nicht geeignet für alle Zielgene weil es nur begrenzte Kreuzreaktivität für viele kommerzielle Antikörper Zebrafisch Proteinen. Zwei indirekte Methoden zu zeigen, Mo Spezifität, gehören: a) Nachweis, dass es eine Dosis-abhängige Wirkung auf den Phänotyp, und b) zeigen, dass Co-Injektion von Wildtyp-mRNA mit tb-MO den Phänotyp rettet effizient. Aus diesen Gründen empfehlen wir einen Spleiß-Blocker, wenn möglich, weil die Effizienz direkt überwacht werden kann.
- Wenn ein Phänotyp beobachtet gehen 5.7 Schritt, wenn kein Phänotyp beobachtet gehen 6.1 fort.
- Für qualitative Phänotypen, wählen Sie eine MO Dosis, in denen 50-75% der Embryonen betroffen sind; zur quantitativen Phänotypen, wählen Sie eine MO Dosis, in der die phänotypische Maßnahme unterscheidet sich deutlich von Wildtyp (p <0,001). Spritzen Sie neue Chargen von Zebrafisch embRYOS (1-4 Zell-Stadium, n = 50-100/batch) mit einem Cocktail mit dem "Test" Dosis von MO und eine Dosis-Kurve des menschlichen WT mRNA (im Bereich von 10 bis 200 pg mRNA; diese Dosen für wesentliche Überexpression oben die Grundlinie eines einzelnen Transkript, was 0,25-0,5% der gesamten polyA mRNA in einer Zebrafischembryo). 4
- Führen maskiert Scoring der Einspritzung Chargen; wählen Sie die WT mRNA Dosis mit dem bedeutendsten Rettung im Vergleich zu MO allein, das ist der "Test"-Dosis von mRNA.
- Spritzen Sie neue Chargen (1-4 Zell-Stadium, n = 50-100/batch) mit Assay-Dosis von MO und Assay-Dosis von mutierten humanen mRNA. Phänotyp Embryonen im geeigneten Stadium und vergleichen Sie die Ergebnisse auf WT menschlichen mRNA Rettung mit Hilfe eines geeigneten statistischen Test (t-Test oder Chi-Quadrat). Siehe Abbildung 1 für die Ergebnisse und fahren Sie mit Schritt 7 fort. Injizieren Sie den Test Dosen von WT und mutierten mRNA allein für mRNA Toxizität Effekte zu steuern.
6. In vivo Assay von Variant Dominant Negative oder gain of function Effects
- Wenn kein Funktionsverlust-Phänotyp beobachtet wird (Schritt 5.5) oder mutierten mRNA entsteht Phänotypen nicht signifikant von MO allein (Schritt 5.7), eine Dosis zu injizieren Kurve im Bereich von 10 bis 200 pg WT menschlichen mRNA (Empfehlung 25, 50 und 100 pg ein erster Test) in Embryo Chargen (1-4 Zell-Stadium; 50-100 Embryonen / Charge).
- Zum geeigneten Zeitpunkt (siehe 5.3 oben), führen phänotypische Scoring, und bestimmen Sie die höchste Dosis, bei der es nicht eine statistisch signifikante Anzahl von Toten und / oder betroffene Embryonen im Vergleich zu nicht injizierten Kontrollen. Dies ist der "Test"-Dosis.
- Injizieren Sie die Assay-Dosis von mutierten humanen mRNA (1-4 Zell-Stadium; 50-100 Embryonen / Charge). Phänotyp Embryonen und vergleichen Sie die Ergebnisse auf Scoring aus dem Assay Dosis von WT menschlichen mRNA Injektion oder dem Test MO Konzentration. Siehe Abbildung 1 für die Ergebnisse.
- Wenn die Ergebnisse nicht von MO sind, titrieren menschlichen mutierten mRNA mit WT mRNAund die menschliche WT und mutierten mRNA Injektionen zu vergleichen. Verbesserung der Phänotypen mit mutierten zzgl. WT mRNA-injizierten Chargen zeigt eine dominant negative. Keine Verbesserung gibt eine gain of function.
7. Reproduzieren in vivo-Testergebnisse
- Wiederholen Sie in vivo-Tests mindestens dreimal.
8. Integrieren Zebrafisch in vivo Pathogenität Daten mit anderen Lines of Evidence
- Vergleichen Pathogenität Daten von Zebrafisch Experimenten zu erhalten: genetische Daten in einem Stammbaum (falls zutreffend), Kontrolle der Bevölkerung Frequenz Daten, in-vitro-Studien (Zell-basierte Assays von Protein Stabilität, zelluläre Lokalisation, Signal-Ausgang oder enzymatische Aktivität).
Ergebnisse
Recessive und Pseudorecessive Disorders
Primäre Zilien sind nahezu allgegenwärtigen Strukturen auf der Wirbeltiere Körper Plan, zelluläre Signalwege Rollen in mehreren Zellschicksale, einschließlich Proliferation, Polarität, Differenzierung und Wartung Gewebe spielen. 5 Dysfunction dieser Organellen führt zu einem breiten Spektrum von menschlichen genetischen Erkrankungen bezeichnet zusammenfassend als ciliopathies. 6,7 Eine solche klinische Entität ist Bardet-Biedl-Syndrom (BBS), eine Erkrankung, die durch multisystemic pädiatrische die Degeneration der Netzhaut, Adipositas, Hypogonadismus, Polydaktylie und Nierenfunktionsstörungen gekennzeichnet. 7 Die Entwicklung der in-vivo-Untersuchungen von Allel Pathogenität war notwendig für BBS, weil a) es ist eine genetisch heterogene Erkrankung, die durch vor allem private nichtsynonymen Veränderungen in mindestens 17 Genen verursacht 7-10; und b) oligogenic Erbschaft in> 25% der BBS Familien, wobei die Anwesenheit von heterozygoten Veränderungen in einem zweitenBBS-Gen (zusätzlich zu rezessive primäre ursächlicher Mutationen) modulieren können klinische Penetranz und Expressivität. Typischerweise sind solche dritte Allele nichtsynonymen heterozygoten Veränderung der von einer genetischen Sicht unklar pathogenes Potential, so erfordern genaue Auslegung ihrer biologischen Wirkung auf Proteinfunktion. 11-13
Um das pathogene Potential von Mutationen Beitrag zur Mutationsgrad in BBS untersuchen, haben wir zunächst alle getestet missense Änderungen in BBS1 identifiziert - BBS14. Sowohl wir als auch andere haben gezeigt, dass der Verlust der basalen Körper Proteine Anlass zu Fehlregulation der planaren Zellpolarität (PCP; nicht-kanonischen Wnt-Signalweg). Manifestiert als konvergente Extension Mängel in Mitte somitischen Zebrafischembryonen 14 Mit dieser physiologisch relevanten phänotypische Auslesen wir festgestellt, dass die Unterdrückung der BBS Gene verkürzt Körperachsen, breiter und dünner Urwirbel und breit, geknickt notochords geführt. 15 </ Sup> (Abbildung 2) MO-induzierte Unterdrückung erzeugt Gastrulation Defekte und Co-Injektion mit menschlichen mRNA gerettet signifikant (und reproduzierbar) diese Phänotypen als Tor von drei verschiedenen in vivo-Methoden. Zunächst wurden die Embryonen Live nach qualitativen Kriterien phänotypische (Normal, Klasse I und Klasse II, für detaillierte Definitionen der phänotypischen Klassen siehe 15.) erzielte. Weiter, quantifiziert wir Zellwanderung während epiboly (einem frühen Entwicklungsstadium gekennzeichnet durch die Ausdünnung und Ausbreitung von Zellschichten über den Dotter Zelle 16) durch den Einsatz einer Fluorescein Tracer wandernden Zellen sichtbar zu machen. Schließlich messen wir somite Oberkörperlänge in neun somite Embryonen in situ mit einem Cocktail aus Krox20, PAX2 und myoD Ribosonden, die flach für morphologische Analyse wurden montiert hybridisiert.
Diese Methodik wurde verwendet, um mehr als 500 Allele in der Zilien Mutations Raum testen. In einerstudieren allein, in-vivo-Tests von> 130 Allele produziert eine Reihe von phänotypische Partituren; von unseren Protokoll angezeigt (Abbildung 1) vollständige rettet wurden als gutartig eingestuft (nicht signifikant verschieden von WT Rettung), rettet teilweise wurden als hypomorphs klassifiziert (deutlich verbessert von MO, aber schwerer als Rettung WT) wurden fehlende Rettung als funktionelle null (nicht signifikant verschieden von MO) und Phänotypen von Mutanten mRNA allein im Vergleich zu MO wurden als dominant eingestuft Negative induziert eingestuft.
Wir haben auch die Empfindlichkeit und Spezifität der in-vivo-Assay in Komplementierung Zebrafisch ausgewertet. Die Spezifität wurde bestätigt durch Coinjektion gemeinsamen SNPs (> 5% kleineren Allel-Häufigkeit in gesunden Bevölkerung), diese wurden gefunden, um gutartige Phänotypen in 14/17 geprüft (> 82%) zu ergeben, und Sensitivität ist 98% wie angegeben durch Übereinstimmung zwischen in vivo-Daten und genetische Argumente ausreichend to zuschreiben ein Allel als kausale in einer BBS Stammbaum. 17. Darüber hinaus beobachtet phänotypische Effekte mit den drei in vivo Maßnahmen (live scoring, epiboly Verfolgung und ISH Morphometrie) wurden in vitro validiert mit Immunoblot und zelluläre Lokalisation Studien. Während die Interpretation dieser Ergebnisse erforderlich Vorkenntnisse mindestens einen Mechanismus der Krankheitsentstehung stellt dieses Beispiel Beweise für den Nutzen und die Robustheit unserer Protokoll zu untermauern. Wir haben seit unserer in vivo bestätigt Scoring mit mehreren anderen Linien der experimentelle Nachweis in einem unvoreingenommenen Mutations-Screening und funktionelle Analyse der Studie TTC21B, eine retrograde intraflagellar Transportprotein. 18
Dominant Disorders
Gliedmaßen Muskeldystrophien (LGMD) sind eine Klasse von autosomal-Muskeldystrophie, was langsam fortschreitende Muskelschwäche in den Hüften und Schultern. Diese genetisch und michchanistically heterogene Gruppe von Erkrankungen wird von den beiden dominanten und rezessiven Mutationen in mehreren sarkolemmalen, sarcomeric, zytoplasmatische und nukleäre Proteine verursacht. Basierend auf die Darstellung der klinischen Phänotypen und der Nachweis der Beteiligung von Muskel Kernspintomographie, untersuchten wir die Ursache einer beherrschenden LGMD in Finnisch, USA und italienischen Familien 19 gefunden. Die Sequenzierung von Positionsdaten Kandidaten innerhalb der zugeordneten Locus zeigte Mutationen in DNAJB6 äußerte ein Gen, das eine Co-Chaperon von HSP70 Familie mindestens zwei Spleiß-Isoformen (Kern-und Zytoplasma) beim Menschen. Um weitere Einblicke in DNAJB6 Funktion und ihre Bedeutung für LGMD gewinnen, untersuchten wir seine Rolle im Muskel Integrität im Zebrafisch. RT-PCR des Zebrafisch-Ortholog (dnajb6b) detektiert Ausdruck bereits in der fünf-somite Stufe, die durch Injektion von Embryonen mit einer Spleiß-blocking Morpholino folgte. Bei 48 Stunden nach der Befruchtung, zeigte maskierte Scoring Ablösung des langsamenFasern von ihren Ansatzstellen. Die Spezifität dieser Phänotyp wurde dann mit einer zweiten nicht-überlappenden MO getestet und rettete anschließend mit WT menschlichen DNAJB6 mRNA.
Um abzufragen, wie der Verlust von DNAJB6 Funktion zu Defekten der muskulären Integrität führt, haben wir Missense-Mutationen bei Patienten in menschliche Transkripte beider Isoformen gefunden und injiziert sie in Zebrafisch Embryonen. Während Injektion von WT menschlichen mRNA erzeugt keine nennenswerte Phänotyp phenocopied diese Änderungen den Funktionsverlust Auswirkungen der MO wenn in der zytoplasmatischen entwickelt, aber nicht nuklearen Isoform. Dies wurde durch Co-Injektion von äquimolaren Mengen von Mutanten und WT-mRNA, die eine erhöhte Härte des Muskel Phänotyp zeigte gefolgt, was auf eine dominante Wirkung. Um diese Idee zu testen, wurden weitere Injektionen mit Änderung molaren Verhältnissen von Mutante und WT mRNA durchgeführt. In Übereinstimmung mit der Vorhersage, ein Überschuss von mutierten mRNA mit WT induzierte Letalität in Embryonen gegenüber, während einn über WT produziert eine schrittweise erhöht Rettung. Dies wurde durch in-vitro-Experimente folgen zu oligimerization Eigenschaften und möglichen Protein-Wechselwirkungen zu bestimmen. Dies zeigte, dass die Mutationen die antiaggregation Aktivität von cytoplasmatischen DNAJB6 beeinträchtigen und stören Umsatz sowohl Mutante und WT als auch mit einem anderen Molekül, BAG3, der auch die für die Pathomechanismus LGMD interagieren, da LOF BAG3 bewirkt eine pädiatrische Form Muskeldystrophie 20. Wir haben dann gefragt, ob BAG3 könnte modulieren den Phänotyp von Mutanten DNAJB6b im Zebrafisch induziert. Während Injektion von WT BAG3 allein produziert keine Phänotyp, Koinjektion mit DNAJB6 Mutanten deutlich erhöhte phänotypische Schwere, was darauf hindeutet, dass BAG3 spielt eine Rolle bei der Vermittlung der Pathogenität von solchen Mutanten 19.
| Transient Zebrafischmodell | Mutant Zebrafisch-Linie | Transgene Mauslinie | |
| Alter bei Beginn der menschlichen Phänotyp untersucht |
|
|
|
| Machbar Phenotypes |
|
|
|
| Zeit bis zur Verwendung (von der Geburt) | 1-7 Tage | > 3 Monate | > 6 Monate |
| Durchsatz | mittel-hoch | niedrig | niedrig |
| Vorteile |
|
|
|
Tabelle 1. Vergleich der in-vivo-Modellen.

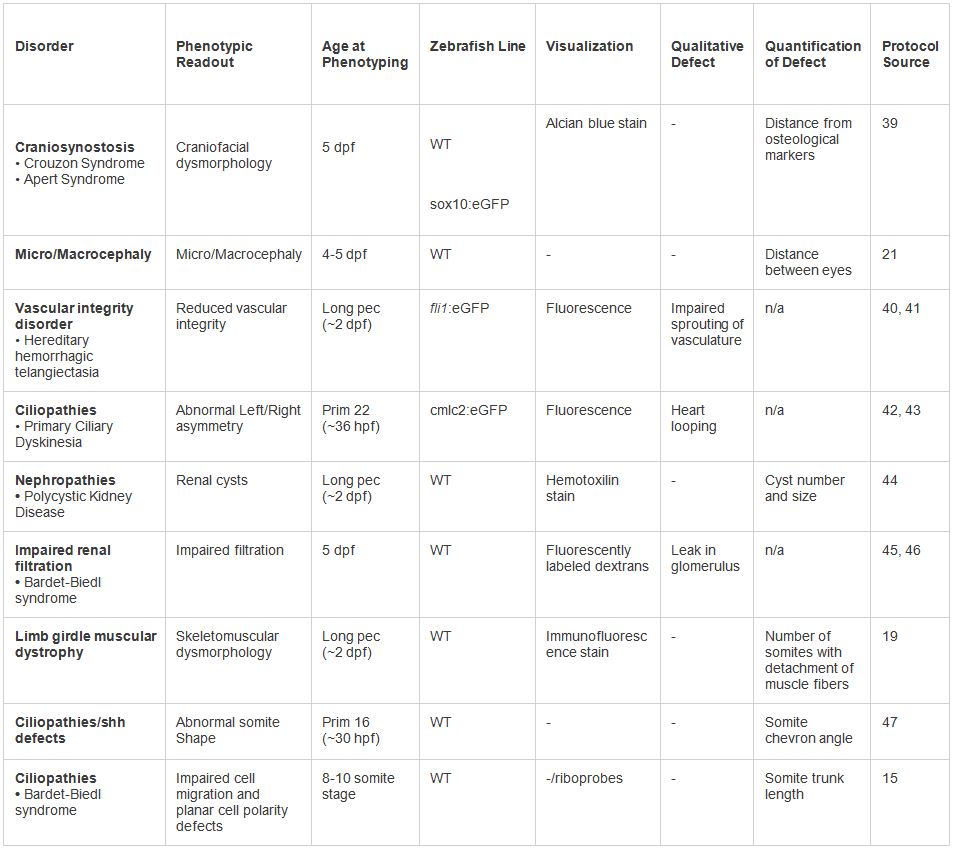
Tabelle 2. Beispiele für in vivo Modellierung menschlicher dysmorphologies. Verschiedene Phänotypen unter der dargebotenen Protokoll getestet. Eine Reihe von Anzeigen und phänotypische Visualisierungstechniken kann basierend auf der Art der Erkrankung eingesetzt werden. Klicken Sie hier, um eine größere Tabelle anzuzeigen.
{kind=link}

Abbildung 1. In vivo Funktionstests nichtsynonymen Varianten. Ein systematischer Ansatz zur Funktionsprüfung allereles unbekannter oder vermutet Bedeutung. Gene Knockdown über Morpholino Mikroinjektion wird durch eine Reihe von (Co) Bolusinjektionen von WT und mutiertes menschliches mRNA gefolgt. Statistische Analysen der phänotypischen Ergebnisse informieren das Allel Pathogenität und molekulare Funktion. Kurz gesagt, für den Verlust von Funktionstests: Wenn der Knockdown erzeugt einen Phänotyp, die entsprechend durch Mutante und WT mRNA gerettet werden kann, ist das Allel wahrscheinlich gutartig (green box). Wenn das mutierte Rettung der Knockdown Phänotyp ist nicht von der Knockdown Phänotyp ist das Allel eine wahrscheinliche funktionelle null (gelber Kasten). Wenn das mutierte Rettung der Knockdown Phänotyp ist statistisch besser als die MO, aber schlechter als der WT, ist das Allel wahrscheinlich ein hypomorph (green box) Für dominant Tests:. Falls Injektion von mutierten mRNA ist äquivalent zu der Wildtyp-mRNA das Allel kann entweder gutartig oder Verlust der Funktion oder der Test kann nicht (grünes Feld) haben. Wenn Injektion von mutierten mRNA ist unver unterscheidbar von MO Knockdown, wird die Funktion des Genprodukts wahrscheinlich in irgendeiner Weise verändert. Um die Änderung in der Funktion zu erkennen, titrieren mutierte mRNA mit Wildtyp-mRNA. Wenn das Ergebnis dieser Titration ist ununterscheidbar zu Wildtyp-mRNA allein, verwendet das mutierte Protein Produkt das Wildtyp-Protein als Substrat, wodurch ein dominant-negativen Phänotyp (blauer Kasten). Wenn das Ergebnis dieser Titration ist nicht zu unterscheiden, um mutierte mRNA allein, das mutierte Protein Produkt hat nicht mehr die gleiche Funktion wie der Wildtyp, und damit ist wahrscheinlich ein gain of function (blauer Kasten). Wenn kein Phänotyp zeigt von MO Knockdown, aber tut vorliegenden WT mit mRNA, weitere Experimente ausgeschlossen ist, WT menschlichen mRNA titriert, um den Phänotyp zu minimieren, und kann auch als eine neue Sollwert verwendet werden. Weitere Co-Injektion von WT und mutiertes menschliches mRNA kann basierend auf der Rettung Fähigkeit der Mutante (pink box) ausgewertet werden.et = "_blank"> Klicken Sie hier für eine größere Abbildung zu sehen.

Abbildung 2. Quantitative und qualitative Auswertung der MKS1 Mutationen beim Menschen nachgewiesen. Developmental Mängel in MKS1 morphanten Embryonen. Basierend auf Schwere wurden Phänotypen in drei Gruppen eingeteilt. Beispiele für jede Klasse (a) gezeigt, und ihre Verbreitung in ihrem Embryo Kohorte (n = 100-160 Embryonen) wurden aufgezeichnet (nicht gezeigt). MO injizierten Embryonen mit Klasse-I-Phänotypen hatte grob normale Morphologie waren aber kürzer, mit übermäßiger embryonalem Gewebe auf dem Eigelb im Vergleich zu injizierten Embryonen gleichzeitig somitischen Alter (8-9 Somiten) zu steuern. Klasse II morphants wurden ausgedünnt, kurz und hatte schlecht Kopf und Schwanz Strukturen entwickelt und zusätzlich fehlten somitischen Definitionund Symmetrie. Klasse III Embryonen wurden schwer mit schlecht entwickelt und unförmig Somiten verzögert, gewellte notochords, und in der Regel nicht über die 10-Somiten Stadium überleben. Co-Injektion von menschlichen MKS1 mRNA gerettet jede dieser Mängel zeigt Spezifität der Phänotypen MKS1 Suppression. In situ Hybridisierung von Embryonen im 11-somite Stufe (± 1 somite) mit Krox20, PAX2 und myoD Riboproben gefärbt (b, c ). Die Phänotypen wurden durch Messungen von der ersten bis zur letzten nennenswerten somite jedes Embryos (Pfeile) in c quantifiziert quantifiziert. Abbildung mit freundlicher Genehmigung von 15 angepasst.

Abbildung 3. Beispiele für in vivo Modellierung menschlicher Dysmorphologie. (A) Craniofacial Dysmorphologie. Control-mRNA injiziert Embryo (links) und mutierten Embryo injiziert (rechts) mit Alcianblau bei 5 dpf gefärbt. Mutanten mRNA-injizierten Embryonen weisen bemerkenswert klein und unförmig Köpfe mit einer allgemeinen Desorganisation des knorpeligen kraniofazialen Skeletts einschließlich gespreizt Kiemenbögen und fehlenden oder fehlerhaften Strukturen. (B) Micro / Makrozephalie. Kontrolle injizierten Embryo (links) und kctd13 MO-injizierten Embryonen (rechts) bei 5 dpf. Morphants anzuzeigen Verbreiterung des Kopfes, wie durch den Raum zwischen den Augen gesehen. 21 (c) Reduzierte vaskulären Integrität. Kontrolle injizierten Embryos (oben) und eng MO-injizierten Embryonen (unten) abgebildet mittels Fluoreszenzmikroskopie bei 2 dpf in einem FLI1: eGFP transgenen Reporter Linie. Morphants Anzeige beeinträchtigt sprießen Zwischengewinneliminie Schiffe und andere vaskuläre Strukturen. 41 (d) Altered Herz Looping. In-situ-Hybridisierung von uninjected Wildtyp-Embryonen (links) zeigen spaw Ausdruck in der linken seitlichen Mesoderm, während ccdc39 morphanten Embryonen zeigten bilaterale (rechts) oder, in den meisten Fällen nicht nachweisbar spaw Ausdruck (nicht gezeigt). 43 (e) Nieren Zysten. Injizierten WT Embryo (oben) und ift80 morphanten (unten). Morphants angezeigt großen Zysten (Pfeil), Herzbeutel Ödeme (Pfeilspitze) und eine geringelte Rute. 44 (f) Reduzierte glomeruläre Filtration. Fluoreszierende Visualisierung Kontrolle injiziert Embryo (oben) und ift80 morphanten (unten) 24 Stunden nach der Injektion von Rhodamin Dextran in das Herz. Fluoreszenz verteilt sich im ganzen Gefäßsystem und ist fast vollständig von der Niere evakuiert gesehen das völlige Fehlen von Fluoreszenz in der Kontrollgruppe. Die morphanten zeigt persistent fluoreszierenden Dextran, was darauf hindeutet, reduzierten glomerulären Filtrationsrate. 46 (g) Muskeldystrophie. WT DNAJB6 mRNA injizierten Embryonen (oben) zeigen normale langsame Muskelfasern überspannt den Somiten normalerweise zwischen benachbarten myosepta als durch Immunfärbung mit Anti-Myosin-Antikörper bestimmt langsam. Mutant DNAJBb (unten) zeigte teilweise zur Ablösung der Muskelfasern von myosepta vervollständigen in einer oder mehreren Somiten. 19 (h) Somiten Winkel. Vergrößerte Live Seitenansichten Kontrolle injizierten (oben) oder kif7 morphants (unten) bei 30 hpf abgebildet. Morphants anzuzeigen abnorm geformte Somiten, zuzurechnen ectopic Hedgehog-Signalgebung in der Zebrafisch Myotom. 47
Alle Angaben mit Genehmigung angepasst.
Diskussion
Die hier beschriebenen Verfahren stellen eine allgemeine Vorschrift für die Bestimmung von nicht-synonymen Änderungen mit einer Vielzahl von menschlichen genetischen Krankheitsphänotypen (Tabelle 2, Abbildung 3) zugeordnet ist. Unsere Ansätze haben sich bewährt, um die möglichen Auswirkungen der Variation Krankheitsphänotypen bewerten und zu helfen, sezieren Krankheitsmechanismen (wie der Beitrag von dominant negative Mutationen Bardet-Biedl-Syndrom, eine autosomal rezessiv vererbte Erkrankung in erster Linie 17). Bis heute, durch die Entwicklung der vorgestellten Entscheidungsbaum, haben wir zu vernünftigen Kosten und Zeit in mehr als 200 Genen ursächlich mit genetischen Störungen assoziiert modelliert, zu einem Überschuss von 1.000 Allele.
Obwohl hier nicht im Detail diskutiert, haben wir auch gezeigt, dass diese Verfahren gegebenenfalls auf andere Typen von genetischen Läsionen, wie Kopienzahl Varianten (CNV) sowie genetische und Interaktionen zu modellieren sind. Analysen solcher Ereignisse sind über den Umfang derdas vorliegende Verfahren Beschreibung, obwohl sie im Grunde verlassen sich auf dem gleichen Prinzip der systematischen Prüfung von Kandidaten-Gene (einschließlich Genpaare injiziert gleichzeitig) auf die Induktion oder Verschlechterung eines klinisch entsprechenden Phänotypen zu bestimmen. Zum Beispiel könnte, welche der 29 Gene in der 16p11.2 CNV aufzuklären relevant sein zu der beobachteten Mikrozephalie bei Patienten mit Vervielfältigungen eines 660 kb genomische Segment beobachtet, mRNAs, die jedem der 29 Gene innerhalb des Segments injiziert wurden und Kopf Größe Messungen bei 2 und 5 dpf dpf durchgeführt wurden, enthüllt einen wichtigen Beitrag eines einzelnen Transkript KCTD13. 21. Darüber hinaus haben wir dieses Modell auf genetische Interaktionen Assay von genomischer Läsionen bei Patienten sowohl mit Bardet-Biedl-Syndrom und Morbus Hirschsprung. 22 Durch Vergleich der MO Unterdrückung der ursächlichen Gene der beiden klinischen Identitäten getrennt und gleichzeitig konnten wir die resultierende Phänotyp als bei Identifizierungng eine synergistische Interaktion anstatt lediglich additive Schwere.
Trotz etablierten hohe Empfindlichkeit (98%) und Spezifität (> 82%) für die Varianten Beitrag zur ciliopathies 17, wissen wir noch nicht genügend Daten, um festzustellen, ob diese verallgemeinerbar auf alle phänotypische Auslesen im Zebrafisch Modelle. Dazu vorhergesagt eine große Anzahl von Allelen, genetisch entweder gutartig oder pathogenen muss innerhalb jedes phänotypische Kategorie getestet werden. Dies ist besonders wichtig für die Durchführung eines solchen Tests in der klinischen Einstellung kann bei funktionellen Auslegung VUSS informieren Diagnose und Behandlung nur dann, wenn eine robuste Verständnis von False Positives und False Negatives kann die Lieferung solcher Ergebnisse für Ärzte und Patienten zu begleiten. Dennoch können diese Methoden einen wichtigen Beitrag zu einem besseren Verständnis der Landschaft der menschlichen Erbkrankheit. Wir erwarten, dass diese Modelle nicht nur als Stiftungen dienenstein für eine verbesserte Interpretation der klinischen genetische Information, sondern auch als nützliche Modelle eingesetzt werden, um therapeutische Bildschirme leiten. In-vivo-Daten können auch in silico rechnerischen Vorhersagen aus Quellen wie PolyPhen 23 verglichen werden, SIFT 24. MutPred SNPs & GO 25 oder 26 Konkordanz zu zeigen. Beachten Sie, dass in einer früheren Studie der Vorhersage Datenbanken SNPs & GO und MutPred wurden gefunden, um die genaueste sein, mit Genauigkeiten erreicht nur 0,82 und 0,81, beziehungsweise. 27
Obwohl wir die Robustheit dieser Methoden für eine Teilmenge der pädiatrischen anatomische Defekte (Tabelle 2, Abbildung 3) skizziert haben, sind bestimmte Phänotypen weniger gefügig mit diesen Methoden. Einige Ausnahmen ungeachtet gibt es drei Hauptklassen von Erkrankungen nicht zugänglich unser Protokoll. Altersdiabetes Störungen (wie Parkinson-Krankheit) stellen auf Modell in einem embryonalen System. Langsam progression degenerative Phänotypen (wie Pickatrophie) erfordern mehr Zeit als die sieben dpf Fenster von MO Aktivität einen Phänotyp zu erzeugen. Andere Gen Knockdown Technologien wie RNAi und siRNA zur Verfügung zu stören oder verschlechtern die Gen Ziel, aber es hat sich gezeigt, dass keiner so genau, stabil, ungiftig, oder langlebig wie MOs 28 sind, also auch die Begrenzung der Zeitrahmen für Phänotypisierung. Drittens haben einige Wirbeltier Strukturen wie der Säuger Lunge, keine ausreichend orthologous Struktur im Zebrafisch. Wir haben auch eine vorgeschlagene Notfallplan für die Untersuchung von Fällen, in denen die menschliche WT mRNA Injektion führt zu einem Phänotyp vorgesehen, obwohl wir vorsichtig Dies ist eine ungewöhnliche und unerwünschte Situation.
Bestimmte Krankheitsphänotypen kann dann verlangen, einen höheren Grad an Abstraktion und Leihmutterschaft. Es ist möglich, dass Genfunktion abgewichen ist ausreichend, um die phänotypische Ähnlichkeit zwischen Modell und tr schwächenue Phänotyp oder dass Zebrafisch Physiologie inhärent kompliziert die Wirkungen des induzierten Krankheit. In solchen Fällen empfehlen wir weitere Präparation des erzeugten Phänotyp vor Entlassung. Wir haben einige erfolgreiche Beispiele, in denen problematische Phänotypen für diesen Test in Zebrafischembryonen modelliert wurden produziert. Zum Beispiel können Mutationen in TCF8 wurden ein Gen mit Fuchs Hornhautdystrophien (FCD) verbunden sind, untersucht unter Verwendung unseres Protokolls unter Verwendung gastrulation Defekte als Surrogat phänotypische Anzeige auf den bekannten Rollen dieses Transkript in der frühen Entwicklung. 29. In anderen Fällen, wie als Altersdiabetes Muskeldystrophie durch Mutationen in DNAJB6 verursacht, konnten wir myofiber Phänotypen in 5dpf Embryonen erzeugen trotz der Tatsache, dass die Menschen beraubt nennenswerte Muskelpathologie sind in den ersten drei bis vier Jahrzehnten des Lebens. 19
Zusätzlich zu den transienten Mutante vorgestellten Modelle, andere haben auch vorteilhaft getroffenge dieser vergänglichen System beim Menschen eine Krankheit in einer Vielzahl von Körper-Systeme zu modellieren. In einem Beispiel wurde Retinitis pigmentosa in Zebrafisch von dem Zuschlag des Gens RP2 modelliert, was in retinalen Zelltod und verringert retinalen Laminierung. Rettungs mit menschlichem Wildtyp-mRNA führte zu der Entwicklung aller drei Schichten von retinalen Laminierung, während vier von fünf mutierten mRNAs nicht zu retten. 30 Obwohl dieses Modell eines menschlichen sensorischen Störung auf einer morphologischen Phänotyps beruht, ist es auch möglich, Assay Reaktion auf Reize wie akustische Schreckreaktion oder Präpulsinhibition. 47
Kürzlich wurde ein Zebrafisch-Modell wurde verwendet, um Alzheimer Krankheitsentstehung durch Amyloid-Vorläufer-Protein zu untersuchen. 31 Die Autoren zeigten, dass Gen-Knockdown verursacht beeinträchtigt axonalen Auswachsen von Motoneuronen, die mit der menschlichen mRNA gerettet werden konnte. Dieses Modell hat sich als besonders informativ, wie Maus-Modellen nur subtile phenot anzuzeigenypes (single Knockdown) oder postnatale Letalität (Doppel-Knockdown). Die Fähigkeit, die Zebrafischembryonen in vivo während der Entwicklung bewerten geholfen, die pathogene Wirkung von reduziertem Amyloid-Vorläufer-Protein, sowie versehen direkten Beweis dafür, dass das Protein sowohl eine extrazelluläre und intrazelluläre Domäne für die ordnungsgemäße Funktion erfordert erkennen. Andere bemerkenswerte Modelle beinhalten, dass der zusätzliche Muskeldystrophien 32, Diamant Blackfan Anämie 33, Axenfeld-Reiger-Syndrom (Augen-und kraniofaziale Entwicklung) 34, entzündliche Darmerkrankung 35 (antibakterielle Wirkung), Parkinson-Krankheit 36 (Neuron und Fortbewegung Verlust), und Beschlagnahme 37 (Hydrocephalus und Hyperaktivität).
Häufiger sind mutierten Zebrafisch Linien, die gezeigt haben auch eine menschliche Krankheit Phänotyp rekapitulieren. Bewertet in 1,38, gehören Modelle Leukämie, Melanom, dilatative Kardiomyopathien, Duchenne Muskeldystrophie,und viele andere.
Offenlegungen
Die Autoren erklären, dass sie keine finanziellen Interessen haben.
Danksagungen
Wir danken für die Unterstützung von einem Duke University Deans Summer Research Fellowship (AN), American Heart Association (AHA) Gemeinschaft 11POST7160006 (CG), National Institutes of Health (NIH) Zuschüsse R01-EY021872 vom National Eye Institute (EED), R01HD04260 aus dem National Institute of Child Health and Development (NK), R01DK072301 und R01DK075972 vom National Institute of Diabetes verdauungsfördernden und Niere Disorders (NK) und der Europäischen Union (EU Gefördert durch 7. FP unter GA nr 241955, Projekt SYSCILIA;. EED, NK) NK ist ein Distinguished Jean und George W. Brumley Professor.
Materialien
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
Referenzen
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M., Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209 (2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914 (2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596 (2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten