
Method Article
In Vivo Modeling of the Morbid Human Genome using Danio rerio
In This Article
Summary
Here, we present a systematic approach for developing physiologically relevant, sensitive and specific in vivo assays for interpreting variation in human pathology. Transient genetic manipulation via microinjection of WT and mutant human mRNA and morpholino (MO) antisense oligonucleotides harness the tractability of the developing zebrafish embryo to rapidly assay pathogenic mutations, especially, but not exclusively, in the context of human developmental disorders.
Abstract
Here, we present methods for the development of assays to query potentially clinically significant nonsynonymous changes using in vivo complementation in zebrafish. Zebrafish (Danio rerio) are a useful animal system due to their experimental tractability; embryos are transparent to enable facile viewing, undergo rapid development ex vivo, and can be genetically manipulated.1 These aspects have allowed for significant advances in the analysis of embryogenesis, molecular processes, and morphogenetic signaling. Taken together, the advantages of this vertebrate model make zebrafish highly amenable to modeling the developmental defects in pediatric disease, and in some cases, adult-onset disorders. Because the zebrafish genome is highly conserved with that of humans (~70% orthologous), it is possible to recapitulate human disease states in zebrafish. This is accomplished either through the injection of mutant human mRNA to induce dominant negative or gain of function alleles, or utilization of morpholino (MO) antisense oligonucleotides to suppress genes to mimic loss of function variants. Through complementation of MO-induced phenotypes with capped human mRNA, our approach enables the interpretation of the deleterious effect of mutations on human protein sequence based on the ability of mutant mRNA to rescue a measurable, physiologically relevant phenotype. Modeling of the human disease alleles occurs through microinjection of zebrafish embryos with MO and/or human mRNA at the 1-4 cell stage, and phenotyping up to seven days post fertilization (dpf). This general strategy can be extended to a wide range of disease phenotypes, as demonstrated in the following protocol. We present our established models for morphogenetic signaling, craniofacial, cardiac, vascular integrity, renal function, and skeletal muscle disorder phenotypes, as well as others.
Introduction
The functional interpretation of genetic information and assignment of the predictive clinical value to a genotype represents a major problem in medical genetics and is becoming increasingly poignant with the accelerating technical and economic feasibility of genome-wide sequencing. Therefore, it is necessary to develop and implement new paradigms to test the pathogenicity of variants of unknown significance (VUS) detected in patients. These assays must then be accurate, time- and cost-efficient, and harbor the potential to catalyze a transition to clinical utility.
While the mouse has been traditionally the tool of choice in the field of human disease modeling, zebrafish are emerging as a scientifically and economically favorable surrogate. Unlike the mouse, zebrafish biology allows easy and timely access to all developmental stages, aided by optical clarity of embryos which allows for real-time imaging of developing pathologies.1 The relatively recent generation of mutant zebrafish lines has provided additional testing and modeling options, employed by many in functional studies, but this technology continues to be limited (reviewed in 1,38 ). Not only are genetic mutants with knock-ins of specific mutations laborious to attain, they are also not amenable to medium- or high-throughput analysis for the testing of a range of mutations within a single gene. Importantly, a single suite of tests can provide information not only for the pathogenic potential of alleles, but also for the direction of effect at the cellular level (e.g. loss of function vs. gain of function), which is critical for informing mode of inheritance in families, especially when small human pedigrees harbor limited information on the mode of genetic transmission. For further comparison of the uses of available mouse and zebrafish models, see Table 1.
We also note that there are inherent limitations to the zebrafish model system. Although D. rerio have rapid initial development of organ systems, sexual maturity requires approximately three months. Because of this, prenatal and pediatric-onset disorders are the most amenable to this transient expression model. While ideal for conducting large chemical compound screens, the use of genetic mutants is not feasible for the systematic testing of thousands of nonsynonymous variants that contribute to, and continue to be detected in pediatric disorders.
The complementation tests described here take advantage of this experimental tractability, high degree of homology, and preservation of function between human and zebrafish proteins, particularly so for molecules necessary for conserved developmental processes. Figure 1 outlines the testing and identification strategy for various allele effects. Both loss of function (LOF) and dominant assays can be performed. For LOF, the experiment begins with the suppression of the gene of interest with a morpholino knockdown, and assaying for phenotypes that might be relevant to the clinical phenotype under investigation. Suppression can be achieved either by blocking translation by targeting a MO at or near the translational start site of the zebrafish locus (translation blocker morpholino; tbMO) or by interfering with splicing by placing a MO on a splice junction, typically inducing either inclusion of an intron or aberrant exon skipping (splice blocking morpholino; sbMO).
Subsequently, capped mRNA from the orthologous human transcript is introduced and quantifiable rescue of the phenotype is measured. Once the assay is established, candidate mutations in the human message can be introduced and assayed for their ability to rescue the MO-induced phenotype at the same efficiency as WT human mRNA. Conversely, for candidate dominant alleles, human mRNA (but not MO) is introduced with an expectation that WT human mRNA will not grossly affect zebrafish anatomy and physiology, whereas introduction of test mutations that have a dominant effect will induce phenotypes analogous to those observed in the human clinical condition. This experiment can be fine-grained further to dissect whether the dominant effect occurs by a gain of function (GOF) or a dominant-negative mechanism by blending WT and mutant human mRNA; for GOF events, addition of WT human mRNA is expected to be irrelevant, whereas for dominant-negative alleles, blending of WT and mutant mRNA should alter the severity of the phenotype induced by mutant message. In all cases, we recommend that all combinations of injections (MO with WT human mRNA vs. morpholino with mutant human mRNA etc. be performed, preferably within the same clutch of embryos (see Figure 1). Interpretation is as follows:
For LOF tests:
- If the knockdown produces a phenotype which can be rescued equivalently by mutant and WT mRNA, the allele is likely benign.
- If the mutant rescue of the knockdown phenotype is indistinguishable from the knockdown phenotype itself, the allele is a likely functional null. The experiment cannot discriminate between true nulls (no functional protein) and ultralow protein activity levels that have no rescue capacity.
- If the mutant rescue of the knockdown phenotype is statistically better than the MO, but worse than the WT, the allele is likely a hypomorph as this result demonstrates partial loss of function.
For dominant tests:
- If there is no knockdown phenotype, but injection of WT mRNA produces a phenotype, a contingency plan must be used if the experiment is to proceed (see below).
- If there is no knockdown phenotype and injection of WT mRNA produces no phenotype, the experiment proceeds as usual.
- If injection of mutant mRNA is equivalent to that of wild-type mRNA, the allele may be either benign or loss of function, or the assay may have failed. This requires further experimentation to discriminate between these options.
- If injection of mutant mRNA is indistinguishable from MO knockdown, the function of the gene product is likely altered in some way. To discern the change in function, a titration of mutant mRNA with wild-type mRNA should be carried out.
- If the result of this titration is indistinguishable to wild-type mRNA alone, it has been shown that the mutant protein product uses the wild-type protein as a substrate, as its effect is varied with the amount of titration. This indicates a dominant negative phenotype.
- If the result of this titration is indistinguishable to mutant mRNA alone, it has been shown that the mutant protein product no longer has the same function as the wild-type, and thus is not affected by the amount of wild-type protein product present. This demonstrates that the allele is likely gain of function.
Contingency plan:
- If no phenotype presents from MO knockdown, but does present with WT mRNA, further experimentation can occur, although we emphasize that this situation is not ideal. WT human mRNA should be titrated to minimize the phenotype, and can also be used as a new set point. Further coinjection of WT and mutant human mRNA can be evaluated based on the rescuing ability of the mutant.
Protocol
1. Bioinformatics Analysis
- Determine if the human gene of interest has a zebrafish ortholog, and if so, how many copies. We recommend reciprocal BLAST (http://blast.ncbi.nlm.nih.gov/) of the human protein against the zebrafish genome, and subsequent BLAST of the best zebrafish hit against the human genome. True orthologs will be the best hit in each instance.
- Determine the size of the open reading frame (ORF) of the human gene. If longer than 6 kb, this model is intractable at present because of limitations of high quality in vitro transcription of long templates.
- Obtain or generate a construct containing the human ORF in the pCS2+ vector backbone (or equivalent vector with a 5' SP6 transcription site and 3' polyA signal).
- Design a MO to block splicing or translation of the targeted zebrafish gene. If multiple copies exist in the zebrafish genome, there are two options: a) design of additional MOs; or b) identification of a splice site fully conserved between copies against which a single MO can be effective. Some published MO sequences are available at www.zfin.org.
2. Expression Analysis in the Developing Zebrafish Embryo
- Determine if the zebrafish ortholog is expressed in a spatiotemporal context relevant to the phenotypic readout. If no expression data are available for the gene of interest, conduct reverse transcription (RT)-PCR using cDNA from whole zebrafish embryos or in situ hybridization. (See 2,3).
3. Site-Directed Mutagenesis
- Design mutagenesis primers 25-45 bases in length, with the desired mutation in the center. The primer melting temperature should be greater than or equal to 78 °C. Design a forward and reverse mutagenesis primer to anneal to opposite strands of the plasmid.
- Obtain primers for sequence confirmation of the ORF post-mutagenesis; these should tile across ~300 base-pair sections to cover the entire ORF.
- Assemble the mutagenesis reaction with a high-fidelity polymerase and cycle as follows (1: 95 °C 30 sec, 2: 95 °C 30 sec , 3: 55 °C 1 min, 4: 68 °C 6 min, 5: Go to 2 18x, 6: 4 °C forever, 7: end).
- Add 1 μl DpnI restriction endonuclease per reaction to remove dam methylated template; incubate at 37 °C for 2 hr.
- Transform 2 μl of mutagenesis reaction into 20 μl competent cells according to standard protocols.
- Pick 3-4 colonies and inoculate 5 ml of LB media containing appropriate antibiotic. Shake at 37 °C overnight at 225 rpm.
- Miniprep DNA and determine concentration.
- Sequence the mutation site to confirm the presence of the mutation of interest.
- Sequence entire ORF to confirm sequence integrity.
4. In vitro mRNA Transcription
- Using a linearized pCS2+ template, generate capped mRNA using the mMessage mMachine SP6 kit (Ambion). We recommend using half of the reaction component quantities.
- Purify the RNA sample with LiCl precipitation or phenol chloroform as described in the kit instructions.
- Determine the concentration of mRNA using absorbance, ensure the integrity of the mRNA using gel electrophoresis, and store the sample in three or more aliquots at -80 °C until ready for use. We do not recommend multiple freeze-thaw cycles of mRNA aliquots.
5. In vivo Assay of Variant Loss of Function
- Obtain zebrafish embryos from natural zebrafish matings, and maintain them at 28 °C in embryo water in 6 or 10 cm dishes.
- Conduct a morpholino dose response curve to evaluate phenotype specificity, MO efficiency and MO toxicity. Inject a dosage curve of at least three different concentrations between 1-10 ng MO into 50-100 (1-4 cell stage) zebrafish embryos/batch. Efficient MOs should give rise to dose-dependent increases of the proportion of affected embryos in a batch.
- Phenotype embryos at appropriate developmental stage based on expression of targeted zebrafish gene of interest and stage at which a relevant phenotype will be observed. This may either be quantitative (such as a measurement between anatomical structures) or qualitative (based on standardized phenotypic criteria). For all injections scored >24 hpf: embryos should be treated with PTU (0.003% 1-phenyl-2-thiourea in embryo media) at 24 hpf to maximum reduction of melanocyte formation.
- For splice-blocking MOs, test MO efficiency by extracting total RNA from whole embryo lysates at the time point of phenotypic scoring, generate cDNA and conduct RT-PCR of the target gene using primers flanking the MO target site.
- To verify the suppression efficiency of a tb-MO, harvest whole embryo protein lysates and conduct immunoblotting to compare levels of the targeted protein versus control. However, this approach is not amenable to all target genes because there is limited cross-reactivity for many commercial antibodies to zebrafish proteins. Two indirect methods of showing MO specificity, include: a) demonstrating that there is a dose-dependent effect on phenotype; and b) showing that co-injection of wildtype mRNA with tb-MO rescues the phenotype efficiently. For these reasons, we recommend a splice blocker when possible because efficiency can be monitored directly.
- If a phenotype is observed proceed to step 5.7, if no phenotype is observed proceed to step 6.1.
- For qualitative phenotypes, select a MO dose in which 50-75% of embryos are affected; for quantitative phenotypes, select a MO dose in which the phenotypic measure is significantly different from wild-type (p < 0.001). Inject new batches of zebrafish embryos (1-4 cell stage; n = 50-100/batch) with a cocktail containing the "assay" dose of MO and a dosage curve of human WT mRNA (ranging from 10-200 pg mRNA; these doses ensure substantial overexpression above the baseline of any single transcript, representing 0.25-0.5% of total polyA mRNA in a zebrafish embryo).4
- Conduct masked scoring of injection batches; choose the WT mRNA dose with the most significant rescue in comparison to MO alone; this is the "assay" dose of mRNA.
- Inject new batches (1-4 cell stage; n = 50-100/batch) with assay dose of MO and assay dose of mutant human mRNA. Phenotype embryos at the appropriate stage and compare results to WT human mRNA rescue using an appropriate statistical test (t-test or chi-squared). See Figure 1 for outcomes and proceed to step 7. Inject the assay doses of WT and mutant mRNA alone to control for mRNA toxicity effects.
6. In vivo Assay of Variant Dominant Negative or Gain of Function Effects
- If no loss of function phenotype is observed (Step 5.5) or if mutant mRNA gives rise to phenotypes not significantly different from MO alone (Step 5.7), inject a dosage curve ranging from 10-200 pg WT human mRNA (we recommend 25, 50, and 100 pg as an initial test) into embryo batches (1-4 cell stage; 50-100 embryos/batch).
- At the appropriate stage (see 5.3 above), conduct phenotypic scoring, and determine the highest dose at which there is not a statistically significant number of dead and/or affected embryos in comparison to uninjected controls. This is the "assay" dose.
- Inject the assay dose of mutant human mRNA (1-4 cell stage; 50-100 embryos/batch). Phenotype embryos and compare results to scoring from the assay dose of WT human mRNA injection or the assay MO concentration. See Figure 1 for outcomes.
- If results are indistinguishable from MO, titrate human mutant mRNA with WT mRNA and compare to human WT and mutant mRNA injections. Improvement of phenotypes with mutant plus WT mRNA-injected batches indicates a dominant negative. No improvement indicates a gain of function.
7. Reproduce in vivo Testing Results
- Repeat in vivo assays a minimum of three times.
8. Integrate Zebrafish in vivo Pathogenicity Data with Other Lines of Evidence
- Compare pathogenicity data obtained from zebrafish experiments to: genetic data within a pedigree (if applicable), control population frequency data, in vitro studies (cell-based assays of protein stability, cellular localization, signaling output, or enzymatic activity).
Results
Recessive and Pseudorecessive Disorders
Primary cilia are near-ubiquitous structures on the vertebrate body plan that play cellular signaling roles in multiple cell fates, including proliferation, polarity, differentiation, and tissue maintenance.5 Dysfunction of these organelles leads to a broad range of human genetic disorders referred collectively as ciliopathies.6,7 One such clinical entity is Bardet-Biedl syndrome (BBS), a multisystemic pediatric disorder characterized by retinal degeneration, obesity, hypogonadism, polydactyly, and renal dysfunction.7 The development of in vivo assays of allele pathogenicity was necessary for BBS because a) it is a genetically heterogeneous disorder caused by primarily private nonsynonymous changes occurring in at least 17 genes7-10; and b) oligogenic inheritance in >25% of BBS families, wherein the presence of heterozygous changes in a second BBS gene (in addition to recessive primary causal mutations) can modulate clinical penetrance and expressivity. Typically, such third alleles are nonsynonymous heterozygous changes of, from a genetic standpoint, unclear pathogenic potential, thus necessitating accurate interpretation of their biological effect on protein function.11-13
To investigate the pathogenic potential of mutations contributing to mutational load in BBS, we initially tested all missense changes identified in BBS1-BBS14. Both we and others have shown that loss of basal body proteins gives rise to dysregulation of planar cell polarity (PCP; non-canonical Wnt signaling) manifesting as convergent extension defects in mid-somitic zebrafish embryos.14 Using this physiologically relevant phenotypic readout, we have found that the suppression of BBS genes resulted in shortened body axes, broader and thinner somites, and wide, kinked notochords.15 (Figure 2) MO-induced suppression produced gastrulation defects, and co-injection with human mRNA rescued significantly (and reproducibly) these phenotypes as scored by three different in vivo methods. First, embryos were scored live according to qualitative phenotypic criteria (Normal, Class I and Class II, for detailed definitions of phenotypic classes see 15). Next, we quantitated cell migration during epiboly (an early developmental stage characterized by the thinning and spreading of cell layers over the yolk cell16) by employing a fluorescein tracer to visualize migrating cells. Finally, we measured somite trunk length in nine-somite embryos in situ hybridized with a cocktail of krox20, pax2 and myoD riboprobes, which were flat mounted for morphological analysis.
This methodology has been used to test in excess of 500 alleles in the ciliary mutational space. In one study alone, in vivo testing of >130 alleles produced a range of phenotypic scores; as indicated by our protocol (Figure 1) complete rescues were classified as benign (not significantly different from WT rescue), partial rescues were classified as hypomorphs (significantly improved from MO, but more severe than WT rescue), failure to rescue were classified as functional null (not significantly different from MO), and phenotypes induced by mutant mRNA alone compared to MO were classified as dominant negatives.
We have also evaluated sensitivity and specificity of the in vivo complementation assay in zebrafish. Specificity was confirmed by co-injecting common SNPs (>5% minor allele frequency in healthy control populations); these were found to give benign phenotypes in 14/17 tested (>82%), and sensitivity was shown to be 98% as indicated by concordance between in vivo data and genetic arguments sufficient to attribute an allele as causal in a BBS pedigree.17 In addition, phenotypic effects observed using the three in vivo measures (live scoring, epiboly tracking, and ISH morphometrics) were validated in vitro using immunoblotting and cellular localization studies. While the interpretation of these results required prior knowledge at least one mechanism of disease pathogenesis, this example provides evidence to substantiate the utility and robustness of our protocol. We have since corroborated our in vivo scoring with multiple other lines of experimental evidence in an unbiased mutational screening and functional analysis study of TTC21B, a retrograde intraflagellar transport protein.18
Dominant Disorders
Limb girdle muscular dystrophies (LGMD) are an autosomal class of muscular dystrophy, causing slow progressing muscle weakness in the hips and shoulders. This genetically and mechanistically heterogeneous group of disorders is caused by both dominant and recessive mutations across multiple sarcolemmal, sarcomeric, cytoplasmic, and nuclear proteins. Based on the presentation of clinical phenotypes and evidence of muscle involvement from magnetic resonance imaging, we investigated the cause of a dominant LGMD found in Finnish, US, and Italian families19. Sequencing of positional candidates within the mapped locus revealed mutations in DNAJB6, a gene encoding a co-chaperone of HSP70 family expressed as at least two splice isoforms (nuclear and cytoplasmic) in humans. To gain further insights into DNAJB6 function and its relevance to LGMD, we examined its role in muscle integrity in zebrafish. RT-PCR of the zebrafish ortholog (dnajb6b) detected expression as early as the five-somite stage, which was followed by injection of embryos with a splice-blocking morpholino. At 48 hr post fertilization, masked scoring showed detachment of slow fibers from their insertion points. The specificity of this phenotype was then tested with a second non-overlapping MO and rescued subsequently with WT human DNAJB6 mRNA.
To query how the loss of DNAJB6 function leads to defects in muscular integrity, we introduced missense mutations found in patients into human transcripts of both isoforms and injected them into zebrafish embryos. While injection of WT human mRNA produced no appreciable phenotype, these changes phenocopied the loss of function effects of the MO when engineered in the cytoplasmic, but not nuclear isoform. This was followed by coinjection of equimolar amounts of mutant and WT mRNA, which showed enhanced severity of the muscular phenotype, suggesting a dominant effect. To test this notion, further injections were carried out with altering molar ratios of mutant and WT mRNA. Consistent with the prediction, an excess of mutant mRNA compared with WT induced lethality in embryos, while an excess of WT produced a progressively increased rescue. This was followed by in vitro experiments to determine oligimerization properties and possible protein interactions. These showed that the mutations impair the antiaggregation activity of cytoplasmic DNAJB6 and interfere with turnover of both mutant and WT, as well as interact with another molecule, BAG3, which is also relevant to the pathomechanism of LGMD, since LOF BAG3 causes a pediatric form of muscular dystrophy20. We then asked whether BAG3 could modulate the phenotype induced by mutant DNAJB6b in zebrafish. While injection of WT BAG3 alone produced no phenotype, coinjection with DNAJB6 mutants significantly increased phenotypic severity, suggesting that BAG3 plays a role in mediating the pathogenicity of such mutants19.
| Transient Zebrafish Model | Mutant Zebrafish Line | Transgenic Mouse Line | |
| Age of onset of human phenotype under investigation |
|
|
|
| Feasible Phenotypes |
|
|
|
| Time until use (from birth) | 1-7 days | >3 months | >6 months |
| Throughput | medium-high | low | low |
| Advantages |
|
|
|
Table 1. Comparison of in vivo models.

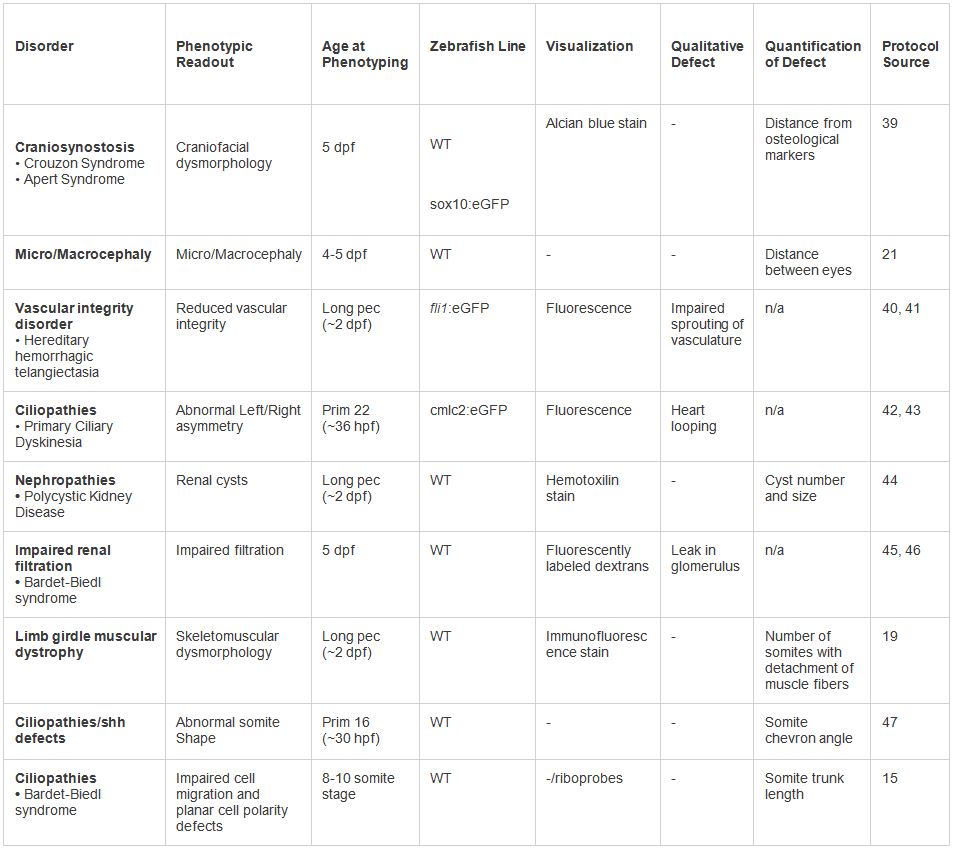
Table 2. Examples of in vivo modeling of human dysmorphologies. Various phenotypes tested under the presented protocol. A range of phenotypic readouts and visualization techniques may be employed based on the type of disorder. Click here to view larger table.
{kind=link}

Figure 1. In vivo functional testing of nonsynonymous variants. A systematic approach to functional testing of alleles of unknown or hypothesized significance. Gene knockdown via morpholino microinjection is followed by a series of (co)injections of both WT and mutant human mRNA. Statistical analyses of phenotypic outcomes inform the allele pathogenicity and molecular function. Briefly, for loss of function tests: If the knockdown produces a phenotype which can be rescued equivalently by mutant and WT mRNA, the allele is likely benign (green box). If the mutant rescue of the knockdown phenotype is indistinguishable from the knockdown phenotype, the allele is a likely functional null (yellow box). If the mutant rescue of the knockdown phenotype is statistically better than the MO, but worse than the WT, the allele is likely a hypomorph (green box). For dominant tests: If injection of mutant mRNA is equivalent to that of wild-type mRNA, the allele may be either benign or loss of function, or the assay may have failed (green box). If injection of mutant mRNA is indistinguishable from MO knockdown, the function of the gene product is likely altered in some way. To discern the change in function, titrate mutant mRNA with wild-type mRNA. If the result of this titration is indistinguishable to wild-type mRNA alone, the mutant protein product uses the wild-type protein as a substrate, thereby indicating a dominant negative phenotype (blue box). If the result of this titration is indistinguishable to mutant mRNA alone, the mutant protein product no longer has the same function as the wild-type, and thus is likely a gain of function (blue box). If no phenotype presents from MO knockdown, but does present with WT mRNA, further experimentation can occur, WT human mRNA should be titrated to minimize the phenotype, and can also be used as a new set point. Further coinjection of WT and mutant human mRNA can be evaluated based on the rescuing ability of the mutant (pink box). Click here to view larger figure.
{kind=link}

Figure 2. Quantitative and qualitative evaluation of MKS1 mutations detected in humans. Developmental defects in mks1 morphant embryos. Based on severity, phenotypes were classified into three groups. Examples of each class are shown (a), and their prevalence within their embryo cohort (n = 100-160 embryos) was tabulated (not shown). MO injected embryos with Class I phenotypes had grossly normal morphology but were shorter, with excessive embryonic tissue on the yolk compared to control injected embryos at the same somitic age (8-9 somites). Class II morphants were thinned, short and had poorly developed head and tail structures, and additionally lacked somitic definition and symmetry. Class III embryos were severely delayed with poorly developed and misshapen somites, undulated notochords, and typically did not survive past the 10-somite stage. Co-injection of human MKS1 mRNA rescued each of these defects, demonstrating specificity of the phenotypes to mks1 suppression. In situ hybridization of embryos at the 11-somite stage (±1 somite) stained with krox20, pax2 and myoD riboprobes (b, c). Phenotypes were quantified by measurements from the first to the last appreciable somite of each embryo (arrows), quantified in c. Figure adapted with permission from15.

Figure 3. Examples of in vivo modeling of human dysmorphology. (a) Craniofacial dysmorphology. Control mRNA injected embryo (left) and mutant injected embryo (right) stained with Alcian blue at 5 dpf. Mutants mRNA-injected embryos display notably small and misshapen heads with a general disorganization of the cartilaginous craniofacial skeleton including splayed branchial arches and missing or malformed structures. (b) Micro/macrocephaly. Control uninjected embryo (left) and kctd13 MO-injected embryo (right) at 5 dpf. Morphants display widening of the head, as seen by the space between the eyes.21 (c) Reduced vascular integrity. Control uninjected embryo (top) and eng MO-injected embryo (bottom) imaged using fluorescence microscopy at 2 dpf in a fli1:eGFP transgenic reporter line. Morphants display impaired sprouting of intersegmental vessels and other vascular structures.41 (d) Altered heart looping. In situ hybridization of uninjected wild-type embryos (left) show spaw expression in the left lateral plate mesoderm, while ccdc39 morphant embryos showed bilateral (right) or, in most cases, undetectable spaw expression (not shown).43 (e) Kidney cysts. Uninjected WT embryo (top) and ift80 morphant (bottom). Morphants displayed large kidney cysts (arrow), pericardial edema (arrowhead) and a curled tail.44 (f) Reduced glomerular filtration. Fluorescent visualization of a control injected embryo (top) and ift80 morphant (bottom) 24 hr after injection of rhodamine dextran into the heart. Fluorescence disperses throughout the vascular system and is almost completely evacuated by the kidney as seen the complete absence of fluorescence in the control. The morphant displays persistent fluorescent dextran, suggesting reduced glomerular filtration.46 (g) Muscular dystrophy. WT DNAJB6 mRNA injected embryos (top) show normal slow myofibers spanning the somites normally between adjacent myosepta as determined by immunostaining using anti-slow myosin antibody. Mutant DNAJBb (bottom) showed partial to complete detachment of myofibers from myosepta in one or multiple somites.19 (h) Somite angle. Magnified live lateral views of control uninjected (top) or kif7 morphants (bottom) imaged at 30 hpf. Morphants display abnormally shaped somites, attributable to ectopic Hedgehog signaling in the zebrafish myotome.47
All figures adapted with permission.
Discussion
The methods described here represent a general protocol applicable to the assay of nonsynonymous changes associated with a variety of human genetic disease phenotypes (Table 2, Figure 3). Our approaches have proven useful to evaluate the potential impact of variation on disease phenotypes, and to help dissect disease mechanisms (such as the contribution of dominant negative mutations to Bardet-Biedl Syndrome, a primarily autosomal recessive disorder 17). To date, through the development of the presented decision tree, we have modeled at reasonable cost and time in excess of 200 genes causally associated with genetic disorders, to an excess of 1,000 alleles.
Although not discussed in detail here, we have also shown that these methods are appropriate to model other types of genetic lesions, such as copy number variants (CNVs) as well as and genetic interactions. Analyses of such events are beyond the scope of the present method description, although they fundamentally rely on the same principle of systematic testing of candidate genes (including pairs of genes injected concurrently) to determine induction or exacerbation of clinically-appropriate phenotypes. For example, to elucidate which of the 29 genes in the 16p11.2 CNV might be relevant to the observed microcephaly observed in patients with duplications of a 660 kb genomic segment, mRNAs corresponding to each of the 29 genes within the segment were injected and head size measurements were conducted at 2 dpf and 5 dpf, revealing a major contribution of a single transcript, KCTD13.21 Additionally, we have used this model to assay genetic interactions of genomic lesions in patients with both Bardet-Biedl syndrome and Hirschsprung disease.22 By comparison of MO suppression of the causal genes of the two clinical identities separately and simultaneously, we were able to identify the resulting phenotype as being a synergistic interaction rather than merely additive severity.
Despite having established high sensitivity (98%) and specificity (>82%) for variants contributing to ciliopathies17, we do not yet have sufficient data to determine if these are generalizable to all phenotypic readouts in zebrafish models. To do so, a large number of alleles, predicted genetically to be either benign or pathogenic, must be tested within each phenotypic category. This will be particularly important for the implementation of such assays in the clinical setting, wherein functional interpretation of VUSs can inform diagnosis and management only if a robust understanding of false positives and false negatives can accompany the delivery of such results to physicians and patients. Nonetheless, these methods can contribute significantly toward a better understanding of the landscape of human genetic disease. We anticipate that these models will not only serve as a foundation for improved interpretation of clinical genetic information, but will also be employed as useful models to conduct therapeutic screens. In vivo data may also be compared to in silico computational predictions from sources such as PolyPhen23, SIFT24, SNPs&GO25, or MutPred26 to show concordance. Note that in a previous study of prediction databases SNPs&GO and MutPred were found to be the most accurate, with accuracies reaching only 0.82 and 0.81, respectively.27
Although we have outlined the robustness of these methods for a subset of pediatric anatomical defects (Table 2, Figure 3), certain phenotypes are less tractable by these methods. Some exceptions notwithstanding, there are three main classes of disorders not amenable to our protocol. Adult-onset disorders (such as Parkinson's disease) represent a challenge to model in an embryonic system. Slow progression degenerative phenotypes (such as frontotemporal dementia) may require more time than the seven dpf window of MO activity to produce a phenotype. Other gene knockdown technologies such as RNAi and siRNA are available to interfere with or degrade the gene target, but it has been shown that none are as specific, stable, non-toxic, or long-lasting as MOs28, thus also limiting the timeframe for phenotyping. Third, some vertebrate structures, such as the mammalian lung, do not have a sufficiently orthologous structure in the zebrafish embryo. We have also provided a suggested contingency plan for investigation of those cases in which WT human mRNA injection leads to a phenotype, though we caution this is an unusual and undesirable situation.
Certain disease phenotypes may then require a greater degree of abstraction and surrogacy. It is possible that gene function has diverged sufficiently to weaken the phenotypic similarity between model and true phenotype, or that zebrafish physiology inherently complicates the effects of the induced disease. In such cases, we suggest further dissection of the produced phenotype prior to dismissal. We have produced some successful examples in which problematic phenotypes for this assay have been modeled in zebrafish embryos. For example, mutations in TCF8, a gene associated with Fuchs corneal dystrophy (FCD), were assayed using our protocol by using gastrulation defects as a surrogate phenotypic readout based on the known roles of this transcript in early development.29 In other instances, such as adult-onset muscular dystrophy caused by mutations in DNAJB6, we were able to generate myofiber phenotypes in 5dpf embryos despite the fact that humans are bereft of appreciable muscle pathology in their first three to four decades of life.19
In addition to the transient mutant models presented here, others have also taken advantage of this transient system to model human disease in a variety of body systems. In one example, retinitis pigmentosa was modeled in zebrafish by the knockdown of the gene RP2, resulting in retinal cell death and decreased retinal lamination. Rescue with human wild-type mRNA resulted in development of all three layers of retinal lamination, while four out of five mutant mRNAs failed to rescue.30 Although this model of a human sensory disorder is based on a morphological phenotype, it is also possible to assay response to stimuli such as acoustic startle or prepulse inhibition.47
Recently, a zebrafish model was used to investigate Alzheimer disease pathogenesis through amyloid precursor protein.31 The authors showed that gene knockdown caused impaired axonal outgrowth of motor neurons, which could be rescued with human mRNA. This model has proven to be especially informative, as mouse models display only subtle phenotypes (single knockdown) or postnatal lethality (double knockdown). The ability to evaluate the zebrafish embryos in vivo throughout development helped to discern the pathogenic effect of reduced amyloid precursor protein, as well as provided direct evidence that the protein requires both an extracellular and intracellular domain for proper function. Other notable models include that of additional muscular dystrophies32, Diamond Blackfan anemia33, Axenfeld-Reiger syndrome (ocular and craniofacial development)34, inflammatory bowel disease35 (antibacterial activity), Parkinson disease36 (neuron and locomotion loss) , and seizure37(hydrocephalus and hyperactivity).
More common are mutant zebrafish lines that have been shown also to recapitulate a human disease phenotype. Reviewed in 1,38, models include leukemia, melanoma, dilated cardiomyopathies, Duchenne muscular dystrophy, and many others.
Disclosures
Unsolicited by the authors, Open Access fees were contributed by Gene-Tools LLC.
Acknowledgements
We acknowledge support from a Duke University Dean's Summer Research Fellowship (A.N.), American Heart Association (AHA) fellowship 11POST7160006 (C.G.), National Institutes of Health (NIH) grants R01-EY021872 from the National Eye Institute (E.E.D.), R01HD04260 from the National Institute of Child Health and Development (N.K.), R01DK072301 and R01DK075972 from the National Institute of Diabetes Digestive and Kidney Disorders (N.K.), and the European Union (Funded by EU 7th FP under GA nr. 241955, project SYSCILIA; E.E.D., N.K.) NK is a Distinguished Jean and George W. Brumley Professor.
Materials
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
References
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M., Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209 (2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914 (2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596 (2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved