
Method Article
Anreicherung von Ureinium Lipoprotein-Partikeln mit Mikro-RNA und anschließender Bestimmung ihrer Absolute/Relative MicroRNA-Inhalte und ihrer Celluläre Transferquote
In diesem Artikel
Zusammenfassung
Hier wird ein quantitatives Echtzeit-Polymerase-Kettenreaktionsprotokoll zur Bestimmung des nativen Mikro-RNA-Gehalts (absolute/relativ) von Lipoprotein-Partikeln vorgestellt. Darüber hinaus wird eine Methode zur Erhöhung des Mikro-RNA-Niveaus sowie eine Methode zur Bestimmung der zellulären Aufnahmevorrate von Lipoprotein-Partikeln demonstriert.
Zusammenfassung
Lipoprotein-Partikel sind überwiegend Transporter von Lipiden und Cholesterin im Blutkreislauf. Darüber hinaus enthalten sie kleine Mengen von Strängen der nicht-kodierenden microRNA (miRNA). In der Regel verändert miRNA das Protein-Expressionsprofil durch Interaktionen mit messenger-RNA (mRNA). Daher ist das Wissen um den relativen und absoluten miRNA-Gehalt von Lipoprotein-Partikeln unerlässlich, um die biologische Wirkung der Zellteilchenaufnahme abzuschätzen. Hier wird ein quantitatives Echtzeit-Polymerase-Kettenreaktion (qPCR) vorgestellt, um den absoluten miRNA-Gehalt von Lipoprotein-Teilchen zu bestimmen — für native und miRNA-angereicherte Lipoprotein-Teilchen exemplarisch dargestellt. Der relative miRNA-Inhalt wird mit multiwell mikrofluidischen Array-Karten quantifiziert. Darüber hinaus ermöglicht dieses Protokoll Wissenschaftlern, die zelluläre miRNA und damit die Lipoprotein-Partikelaufnahme zu schätzen. Eine signifikante Erhöhung des zellulären MiRNA-Niveaus ist bei der Verwendung von hochdichten Lipoprotein-Partikeln (HDL) zu beobachten, die künstlich mit miRNA beladen sind, während die Inkubation mit nativen HDL-Partikeln aufgrund ihres eher niedrigen MiRNA-Gehalts keine signifikante Wirkung hat. Im Gegensatz dazu — die zelluläre Aufnahme von Lipoprotein-Partikeln mit geringer Dichte (LDL) weder mit der einheimischen MiRNA noch künstlich beladen — den zellulären miRNA-Wert nicht verändert hat.
Einleitung
Lipoprotein-Partikel bestehen aus einem Monolayer aus Amphiphilen und Cholesterinschalen, die einen Kern aus Cholesterystestern und Triglyceridfetten umschließen. Stabilisiert wird das gesamte Teilchen durch Membran-eingebettete Apolipoproteine, die die biologische Funktionalität des Teilchens definieren. Lipoprotein-Partikel können je nach ihrer jeweiligen zunehmenden Dichte und damit abnehmender Größe unterschieden werden, nämlich als sehr wenig Dichte Lipoprotein (VLDL), mitteldichte Lipoprotein (IDL), LDL und HDL-Partikel. Trotz des Transports von wasserunlöslichen Komponenten im Blutkreislauf hat sich gezeigt, dass HDL-Partikel nicht-kodierende Stränge von miRNA1,2tragen. Mikro-RNAs sind eine Klasse von kurzen (in der Regel zwei Dutzend Nukleotiden) RNA-Strängen, die intrazellulär ergänzende mRNA-Stränge abbauen und dadurch das Expressionsprofil bestimmter Proteine 3, 4,5verändern, 6. Platz Darüber hinaus wurden Veränderungen des miRNA-Profils bei einer Vielzahl von Krankheiten festgestellt und somit als Biomarker für Diagnose und Prognose eingesetzt. Der extrazelluläre Transport von miRNAs zwischen Zellen über Lipoprotein-Partikel kann als zusätzlicher Mechanismus für die interzelluläre mRNA-Niveaumodulation dienen. Um den biologischen Effekt quantitativ zu schätzen, ist das Wissen über den absoluten und relativen miRNA-Gehalt von Lipoprotein-Partikeln erforderlich.
Quantitative Echtzeit-PCR ist eine geeignete und relativ schnelle Methode, um diese Informationen zu erhalten. So kann der relative Quantifizierungswert (RQ) berechnet werden, und es sind relative Unterschiede zwischen verschiedenen Proben und Lipoprotein-Braktionen abschätzbar. Multiwell mikrofluidische Array-Karten sind eine schnelle und einfach zu bedienende Methode, um die relative Präsenz (entspricht dem RQ-Wert) von miRNAs in einer Probe zu bestimmen. Multiwell mikrofluidische Array-Karten bestehen aus 96 oder 384 einzelnen Reaktionskammern für einzelne QPCR-Reaktionen, die in ein mikrofluidisches Gerät eingebettet sind. Jede Kammer enthält die erforderliche Hydrolyse-Sonde und spezielle QPCR-Primer für eine einzelne miRNA. Die Vorteile sind eine kurze Bearbeitungszeit durch Standardisierung, ein einfacher Arbeitsablauf und eine reduzierte Anzahl von Pipettierschritten. Zudem wird das benötigte Probenvolumen minimiert. Im Gegensatz zur relativen Quantifizierung erfordert der absolute miRNA-Gehalt einen Vergleich von QPCR-Probenergebnissen mit Standard-Kurven bekannter absoluter MiRNA-Stränge. Es sollte darauf hingewiesen werden, dass aufgrund ihres relativ niedrigen MiRNA-Gehalts, Standard-und darüber hinaus auch einmolekül-empfindliche bildgebende Verfahren nicht machbar sind — die künstliche Anreicherung von Lipoprotein-Partikeln mit miRNA ist unvermeidlich, um zelluläre Studie zu studieren Lipoprotein-Partikelinteraktion und miRNA-Übertragung. In diesem Zusammenhang ermöglicht die Delikatesse des HDL-Teilchens, das mit anschließender Verlässlichkeit7 einhergeht, die Einbindung und damit die Anreicherung mit miRNA-Strängen. Eine ähnliche Anreicherung von LDL-Partikeln mit miRNA ist aufgrund der Hydrophobizität des ApoB-100-Proteins, das der Hauptbestandteil des LDL-Teilchens ist, nicht möglich. Durch die Zugabe des polaren Lösemittels Dimethylsulfoxid (DMSO), das in der Lage ist, in Lipidmembranen einzudringen, können LDL-Partikel aber auch künstlich mit MiRNA-Strängen beladen werden.
Die Hochgeschwindigkeits-Atomkraftmikroskopie (HS-AFM) ist ein leistungsfähiges Werkzeug zur Charakterisierung biologischer Proben, die subnanometer räumliche und subsekundenlange zeitliche Auflösung8bieten. Daher ist es eine gut geeignete Technik für die Qualitätskontrolle von modifizierten Lipoprotein-Partikeln, da native/reconstitetetete Lipoprotein-Partikel unter einer nahezu physiologischen Umgebung abgebildet werden können.
Hier wird Schritt für Schritt ein QPCR-basiertes Protokoll vorgestellt, um den absoluten/relativen miRNA-Gehalt von Lipoprotein-Partikeln und Zellproben zu bestimmen, was eine Abschätzung der zellulären Lipoprotein-Partikelaufnahme ermöglicht. Darüber hinaus wird eine Methode zur Anreicherung von Lipoprotein-Partikeln mit miRNA demonstriert. Diese Methode kann für die allgemeine Manipulation des Lipoprotein-Gehalts angepasst werden und demonstriert damit die Anwendbarkeit von Lipoprotein-Partikeln als Ziel für die Arzneimittelzufuhr.
Protokoll
Blutspenden wurden von der Ethikkommission der Medizinischen Universität Wien genehmigt (EK-Nr. 511/2007, EK-Nr. 1414/2016). Die Nomenklatur ist nach den Mindestangaben für die Veröffentlichung von Quantitativen Real-Time-PCR-Experimenten (MIQE) 9 Richtlinien.
1. Lipoprotein-Partikelisolierung von menschlichem Blut
- Precool eine Ultrakentrifuge auf 4 ° C. Ziehen Sie Blut von gesunden Freiwilligen nach dem Fastenzeit.
Hinweis: Typischerweise werden drei Spender benötigt, die jeweils 80 ml spenden, und Blutentnahrohre, die Ethylenediaminetetraacetikinsäure (EDTA) als Gerinnungsmittel enthalten. - Zentrifuge bei 2.000 x g für 20 min bei 4 ° C und Ernteplasma (obere Phase); Scherkräfte vermeiden. Bestimmen Sie das Gesamtvolumen V und passen Sie bei Bedarf mit phosphatgepufferter Saline (PBS) ein Vielfaches des Zentrifugationsrohrvolumens an. Messen Sie die Masse von 1 mL 3x und berechnen Sie die durchschnittliche Dichte- .
- Berechnen Sie die erforderliche Menge an Kaliumbromid (KBr) für die Dichteverstellung mit der folgenden Gleichung10 (für die gewünschte Dichte in grams/Milliliter, verwenden Sie A = 1.019, und für das spezifische Volumen von KBr, verwenden
 = 0,364 mL/g). KBr zum Plasma hinzufügen und sanft umrühren, um Scherkräfte zu vermeiden, bis KBr sich vollständig aufgelöst hat.
= 0,364 mL/g). KBr zum Plasma hinzufügen und sanft umrühren, um Scherkräfte zu vermeiden, bis KBr sich vollständig aufgelöst hat.
- Messen Sie die Dichte, wie sie in Schritt 1,2 beschrieben ist, und passen Sie gegebenenfalls wieder an, indem Sie weitere KBr. Füll-und Dichtungsrifugenröhren hinzufügen, die für die Ultrakentrifugation mit Plasma geeignet sind. Vermeiden Sie Luftblasen; Sonst könnte die Röhre zusammenbrechen. Legen Sie die Rohre nach Herstellerangaben und Zentrifuge bei 214.000 x g für 20 h bei 4 ° C in den Rotor.
- Öffnen Sie die Rohre nach den Anweisungen des Herstellers und werfen Sie die obere Phase mit VLDL und IDL ab. Bestimmen Sie das Gesamtvolumen V und passen Sie sich bei Bedarf an ein Vielfaches des Zentrifugationsrohrvolumens mit PBS an.
- Bestimmen Sie die Dichte der unteren Fraktion. Berechnen Sie die erforderliche KBr zur Dichteverstellung (Verwendung A = 1.063). Rühren Sie sanft, um Scherkräfte zu vermeiden, bis KBr aufgelöst ist. Wiederholungsschritt 1.4.
- Entfernen und sammeln Sie die obere Phase, die LDL-Partikel enthält. Die LDL-Partikellösung unter einer Inertgasatmosphäre bei 4 ° C lagern. Bestimmen Sie das Gesamtvolumen V und passen Sie sich bei Bedarf an ein Vielfaches des Zentrifugationsrohrvolumens mit PBS an. Bestimmen Sie die Dichte der unteren Fraktion , wie in Schritt 1.2 beschrieben.
- Berechnen Sie die erforderliche Menge an KBr für die Dichteverstellung (verwenden Sie A = 1.220) und fügen Sie sie hinzu. Rühren Sie sanft, um Scherkräfte zu vermeiden, bis KBr aufgelöst ist. Wiederholungsschritt 1.4.
- Entfernen und sammeln Sie die obere Phase, die HDL-Partikel enthält. Bestimmen Sie ihr Gesamtvolumen V und passen Sie sich bei Bedarf an ein Vielfaches des Zentrifugationsrohrvolumens mit PBS an. Bestimmen Sie die Dichte, die sich in der Dichte der Dichte, die Dichte, die Dichte, die Dichte, Ein zweiter Zentrifugationsschritt der oberen Phase bei 214.000 x g für 20 h bei 4 ° C wird empfohlen, um Albumin zu entfernen. Bei Bedarf wiederholen Sie den Schritt 1.8. Entfernen und sammeln Sie die obere Phase, die HDL-Partikel enthält.
- Bereiten Sie mindestens 20 L Dialysepuffer (0,9% NaCl, 0,1% EDTA [pH 7,4]) auf 4 ° C vor. Prewet Dialysebröhren (molekulares Gewichtsabteil: 12 – 14 kDa) und die LDL und HDL-Partikellösung nach Herstellerangaben hinzufügen. Dialysieren Sie gegen 5 L Dialysepuffer bei 4 ° C und ändern Sie den Puffer nach 1, 2 und 4 Stunden.
- Nach 24 Stunden die Lipoprotein-Partikellösungen aus den Dialysegröhren zurückholen und die Proteinkonzentration mit dem Bradford-Test11 oder einem anderen geeigneten bestimmen. Speichern Sie die HDL und LDL-Partikellösungen unter Inertgas-Atmosphäre bei 4 ° C.
2. Synthetische miRNA-Aliquots
NOTE: Beim Umgang mit RNA-Oligonukleotiden, Arbeit RNase-free. Arbeiten Sie nur mit frischen, einwegfähigen Plastikverbrauchsmaterialien und tragen Sie immer Handschuhe, die häufig gewechselt werden sollten. Verwenden Sie nur nukleasfreie Lösungen. Arbeiten Sie immer auf Eis.
- Drehen Sie die vom Hersteller erworbene Ampullitin mit maximaler Kraft zu einem Pellet der lyophilisierten synthetischen MiRNA. Fügen Sie für eine Endkonzentration von 10 μM (Bestandskonzentration) miRNA ein entsprechendes Volumen von 10 mM Tris (Hydroxymeyl) Aminomethan (TRIS) Puffer, pH 7,5, hinzu.
- Sanft pipette ein paar Mal auf und ab, um eine Wiedereinlage zu erhalten. In sterilen Rohren Aliquots von je 100 μL zubereiten. Speichern Sie sie bei-20 ° C, wenn sie nicht sofort verwendet werden. Vermeiden Sie wiederholtes Auftauen und Einfrieren.
3. Rückstellung von HDL-Teilchen
-
Die Lieferung
- Bereiten Sie Puffer A (150 mM NaCl, 0,01% EDTA, 10 mM Tris/HCl [pH 8.0]). Die Zentrifuge auf-10 ° C. Precool 100 mL einer Mischung aus Ethanol: Diethylether (3:2) bei-20 ° C.
CAUTION: Tragen Sie eine entsprechende persönliche Schutzausrüstung und arbeiten Sie in einer Rauchhaube, während Sie mit Diethyläther umgehen, da er extrem entflammbar und schädlich für die Haut ist. - Mischen Sie ein Volumen, das 5 mg HDL-Partikeln entspricht, mit 50 ml des vorgekühlten Ethanols: Diethylether (3:2) Gemisch und Inkubat für 2 h bei-20 ° C. Zentrifuge bei 2.500 x g für 10 min bei-10 ° C.
- Das Supernatant abwerfen, das Pellet in 50 ml vorgekühltes Ethanol zurückgeben: Diethyleäther-Gemisch durch Wirbel, und ein zweites Mal für 2 h bei-20 ° C inkubieren. Zentrifuge wieder bei 2.500 x g für 10 min bei-10 ° C.
NOTE: Auf Wunsch Lyophilisierung des Supernatants für eine Analyse des MiRNA-Gehalts in der Fettfraktion der HDL-Partikel. - Das Pellet unter N2 Gasstrom trocknen und in 250 μL Puffer A wiederbeleben (siehe Schritt 3.1.1). Bestimmen Sie die Proteinkonzentration mit dem oder anderen geeigneten Bradford-Protein-Test und verdünnen Sie eine Endkonzentration von 1 mg protein/250 μL Puffer A.
Hinweis: Das Protokoll kann hier angehalten werden. Bewahren Sie die Lösung über Nacht bei 4 ° C unter Inertgasatmosphäre auf.
- Bereiten Sie Puffer A (150 mM NaCl, 0,01% EDTA, 10 mM Tris/HCl [pH 8.0]). Die Zentrifuge auf-10 ° C. Precool 100 mL einer Mischung aus Ethanol: Diethylether (3:2) bei-20 ° C.
-
Wiederherstellung
- Bereiten Sie eine Phosphatidylcholin (PC)-Lösung mit einem Gemisch von Chloroform vor: Methanol (2:1) bei einer Konzentration von 5,6 mg PC/mL. In ähnlicher Weise bereiten Sie Bestandslösungen für Cholesterin-oleat (5 mg CO/mL) und Cholesterin (5 mg C/mL) vor. Alle Lösungen bei-20 ° C.
- In einem sauberen Glasrohr, mischen 500 μL PC, 100 μL CO und 13,5 μL von C. Diese Volumina entsprechen einem ungefähren molaren Verhältnis von 100 PC:22 CO: 4,8 C. Trocknen Sie das Gemisch unter N 2-Gasstrom, während Sie das Rohr drehen, um eine homogene Oberflächenschicht zu erzeugen.
Hinweis: Das Protokoll kann hier angehalten werden. Die Glasviale (auf Wunsch Vorratshaltung möglich) unter Inertgas-Atmosphäre bei-20 ° C lagern. - Bereiten Sie eine frische 30-MM-Spermin-Lösung in Puffer A. Mischen Sie einen Aliquot (100 μL, 10 μM) von synthetischem MiRNA (siehe Schritt 2.2) mit 100 μL Sperminenlösung und Inkubat für 30 min bei 30 ° C.
NOTE: Für negative Kontrollexperimente, ersetzen Sie die miRNA and/oder Spermin-Lösung mit dem gleichen Volumen von Puffer A. - Lösen Sie einen PC/CO/C-Master-Aliquot in der Mischung aus Schritt 3.2.3 auf.
- Bereiten Sie eine Lösung von 30 mg/mL Natriumdeoxycholat im Puffer A vor und fügen Sie 50 μL von Schritt 3.2.4 in die Lösung ein. Rühren Sie bei 4 ° C für 2 Stunden.
- Fügen Sie 250 μL der delipidated HDL-Lösung von Schritt 3.1.4 hinzu. Dieser Volumen entspricht einem ungefähren molaren Verhältnis von 100 PC:22 CO:4 C:1 Delipidated HDL-Proteinen. Über 4 ° C umrühren.
-
Dialyse
- Precool mindestens 15 L PBS bei 4 ° C. 50 g adsorbierende Perlen zu 800 ml doppeltem destilliertem Wasser (ddH2 O)dazugeben und 1 min umrühren. 15 min abwarten, den Supernatant abstellen und den Vorgang mit PBS wiederholen.
- Prewet Dialyse-Kassetten (molekulares Gewichtsabteil: 20 kDa) oder entsprechende Dialysebuben und fügen Sie die Lösung aus Schritt 3.2.6 mit einer Spritze nach Herstellerangaben hinzu.
- Die adsorbierenden Perlen von Schritt 3.3.1 auf 3 L PBS hinzufügen und bei 4 ° C dialysieren. Ändern Sie den Puffer und die Perlen nach 1 h und 2 h.
- Nach 24 Stunden die wiederhergestellte HDL-Partikellösung (rHDL) zurückholen und die Proteinkonzentration mit dem Bradford-Test bestimmen. Die rHDL-Partikellösung unter Inertgas-Atmosphäre bei 4 ° C lagern.
4. Kennzeichnung von LDL-Teilchen
- Bereiten Sie 10x LDL-Puffer (1,5 M NaCl, 3 mM EDTA, 1 mM Ethylen-Glykol-bis (β-Aminethylethy-Äther)-N, N, N ', N '-Tetraacetikinsäure [EGTA, pH 7.4]) vor und speichern Sie sie bei Raumtemperatur.
- Bereiten Sie eine frische 30-MM-Spermin-Lösung in RNase-freien Wasser. Einen Aliquot (100 μL, 10 μM) synthetischer MiRNA (siehe Schritt 2.2) mit 100 μL Sperminenlösung und Inkubat für 30 min bei 30 ° C vermischen.
NOTE: Für negative Kontrollexperimente, ersetzen Sie die miRNA and/oder die Sperminen-Lösung mit dem gleichen Volumen von 1x LDL-Puffer. - Fügen Sie 100 μL von DMSO der miRNA/Spermine-Lösung ab Schritt 4.2 hinzu und verdünnen Sie sie mit 1,2 ml von 1x LDL-Puffer weiter.
- Die LDL-Partikellösung auf eine Endkonzentration von ca. 4 mg/mL mit PBS verdünnen und 450 μL mit 50 μL 10x-LDL-Puffer mischen. Inkubieren Sie es für 10 Minuten auf Eis.
- Kombinieren Sie die LDL-Partikellösung aus dem vorherigen Schritt mit der miRNA/spermine/DMSO-Lösung (ab Schritt 4.3) und inkubieren Sie sie für 2 h bei 40 ° C.
- Führen Sie Dialyse ähnlich wie in Abschnitt 3.3 beschrieben durch und speichern Sie die beschriftete LDL-Partikellösung entsprechend.
5. Qualitätskontrolle von rekonstituierten/beschrifteten Lipoprotein-Teilchen
Hinweis: Zur Qualitätskontrolle können Durchmesser und allgemeine Form von Lipoprotein-Teilchen beispielsweise mit AFM oder Elektronenmikroskopie (EM) bestimmt werden. Hier wird HS-AFM verwendet, um die Größenverteilung von native/rekonstituierten Lipoprotein-Partikeln zu messen.
- Die HDL/LDL-Partikellösung in PBS (1:100 – 1:1000) verdünnen und auf frisch getakteten Glimmer 5 min inkubieren. Zum Rischen des Glimmerbandes 12, drücken Sie Klebeband gegen das Substrat und entfernen Sie die oberen Glimmerschichten, indem Sie das Klebeband abziehen.
Hinweis: Je nach wichtigem AFM-Instrument, Beobachtungsbereich und Anfangskonzentration der Partikel muss der Verdünnungsfaktor an die Beobachtung einzelner Partikel angepasst werden. - Nach der Inkubation die Probe mit PBS spülen und HS-AFM-Bildgebung in PBS und im Abtastmodus mit Freischwellen mit Federkonstanten von kcant < 0,2 N/m durchführen. Es wird empfohlen, Scangrößen und lt;1 μm 2 zu verwenden und die Bildkräfte so gering wie möglich zu halten.
- Bestimmen Sie die Höhe der abgebildet Partikel in Bezug auf die Glimmeroberfläche mit geeigneter Software.
- Laden Sie die Daten in Gwyddion (Freeware), erkennen Sie die Partikel mittelsKornanalyse (Mark-Körner nach Schwelle) und setzen Sie die Schwelle über dem Substrathintergrund. Das Bild verflachen(polynomiale Hintergründeentfernen), das die Option " Maskte Region ausschließen" wählt.
- Exportieren Sie die Höhenwerte der entdeckten Partikel (Verteilung verschiedener Korneigenschaften) und wiederholen Sie diese Schritte für alle aufgenommenen Bilder. Führen Sie statistische Analysen durch, indem Sie entweder Histogramme erstellen oder die Wahrscheinlichkeitsdichte der gewonnenen Partikelhöhen berechnen.
- Wiederholen Sie die Schritte 5.1 – 5.3 mit nativen und rekonstituierten/beschrifteten Lipoprotein-Partikeln und vergleichen Sie die erhaltenen Ergebnisse, um die Partikelqualität zu überprüfen. Wenn Sie Trümmer and/oder Konglomerate beobachten, werfen Sie die Probe ab.
6. Zellkultur
- Wachende Zellen nach einem etablierten Protokoll anbauen (z.B. ldlA7-SRBI13) bis zur Konfluenz.
NOTE: Je nach Anzahl der Experimente (empfohlen werden zwei Kammern pro Versuchseinstellung) und Negativkontrollversuche (empfohlen werden zwei Kammern ohne Zusatz von Lipoprotein-Partikeln) benötigt werden. Zusätzlich sind zwei Kammern für die Bestimmung der Zellnummer erforderlich. - Waschen Sie die Zellen 3x mit der vorgewärmten Salzlösung Hank, um Zellschutt zu entfernen und die Zellschicht mit einem entsprechenden Volumen an serumfreiem Wachstumsmedium zu bedecken. Fügen Sie eine entsprechende Lautstärke von Lipoprotein-Partikellösung hinzu, um eine endgültige Konzentration von 50 μg/mL lipoprotein Partikel zu erreichen. Inkubat bei 37 ° C und 5% CO2 für 16 h.
Hinweis: Je nach experimentellem Design und Zelllinie muss die Inkubationszeit angepasst werden, um eine ausreichende (d.h. messbare) Erhöhung des zellulären miRNA-Niveaus zu erreichen. - Waschen Sie die Zellen 3x mit vorgewärmtem (37 ° C) HBSS, um Zell-Trübris/lipoprotein-Partikel zu entfernen und die Zellschicht mit einem entsprechenden Volumen an serumfreiem Medium zu bedecken.
- Bestimmen Sie die Zelldichte mit einer geeigneten Methode (z.B. Hämozytometer, automatisierter Zellzähler) in mindestens zwei unabhängigen Kammern, um die Anzahl der Zellen im Probenvolumen ab Schritt 6.3 zu berechnen. Diese Zahl wird zur Normalisierung verwendet.
7. miRNA-Extraktion aus Zell-und Lipoprotein-Partikelproben
Hinweis: Die Extraktion von miRNA aus Zellen erfolgt mit dem miRNA-Extraktionskit mit den folgenden Modifikationen.
-
Zellproben
- Die Zentrifuge auf 4 ° C vorstocken. Fügen Sie jeweils 350 μL Lyse-Reagenz zu zwei Kammern mit Zellen hinzu. Als negatives Kontrollexperiment eine Kammer ohne Zellen verwenden.
CAUTION: Tragen Sie eine entsprechende persönliche Schutzausrüstung und arbeiten Sie in einer Rauchhaube, während Sie mit Lyse-Reagenz umgehen, da es Phenol und Thiocyanat enthält. - Warten Sie auf 3 – 5 min (je nach Zelllinie) auf die Zellablösung. Bei Bedarf auf die Zellablösung mit der Brightfield-Mikroskopie achten. Die beiden Kammern in einem 1,5 mL-Rohr einlegen.
- Mit einer 20-G-Nadel und einer 5 ml Spritze stören homogenize/die Probe 5x–10x durch Aspiration und Inkubat für 5 min. 140 μL Chloroform (CHCl 3) hinzufügen, 15 s kräftig schütteln und 3 min inkubieren.
- Zentrifuge bei 12.000 x g für 15 min bei 4 ° C. Danach hören Sie auf, die Zentrifuge abzukühlen.
- Übertragen Sie die obere wässrige Phase auf ein neues 1,5 ml Rohr; Vermeiden Sie Phasenmischungen/Verunreinigungen, da die Zwischenphase DNA enthält und die untere Phase Proteine enthält. Das Volumen von 100% Ethanol hinzufügen und durch Pipettieren gründlich mischen.
- Legen Sie eine Spinnsäule in ein 2 mL Sammelrohr und fügen Sie 700 μL des Gemisches aus Schritt 7.1.5 hinzu. Zentrifuge bei 8.000 x g für 15 s bei Raumtemperatur. Den Durchflussdurchgang ablegen und diesen Schritt mit dem Restprobenvolumen wiederholen.
- Den Durchfluss abwerfen und 700 μL des ersten Waschpuffers in die Spin-Spalte einfügen. Zentrifuge es bei 8.000 x g für 15 s bei Raumtemperatur.
- Verwerfen Sie den Durchfluss und fügen Sie 500 μL des zweiten Waschpuffers in die Spin-Spalte. Zentrifuge es bei 8.000 x g für 15 s bei Raumtemperatur. Wiederholen Sie diesen ganzen Schritt ein zweites Mal mit 2 Minuten Zentrifugationszeit.
- Die Spinnsäule in ein neues 2 mL Sammelrohr legen und mit voller Geschwindigkeit für 1 Minute zentrifugen, um die Membran zu trocknen.
- Legen Sie die Spinnsäule in ein 1,5 mL Sammelrohr. 30 μL RNase-freies Wasser in der Mitte der Membran für die Ellung und Zentrifuge mit 8.000 x g für 1 min hinzufügen. Der umgekehrte Transkriptionsschritt erfolgt unmittelbar nach der Extraktion; Ansonsten lagern Sie die extrahierten miRNA-Proben bei-20 ° C.
- Die Zentrifuge auf 4 ° C vorstocken. Fügen Sie jeweils 350 μL Lyse-Reagenz zu zwei Kammern mit Zellen hinzu. Als negatives Kontrollexperiment eine Kammer ohne Zellen verwenden.
-
Lipoprotein-Teilchen
- Stellen Sie das Volumen der Probe mit der niedrigsten Proteinkonzentration auf 100 μL (= maximales Probenvolumen) ein. Berechnen Sie die Probenvolumina der anderen Proben nach dieser Normalisierung, umgekehrt, auf ihre Konzentration. Für negative Kontrollversuche, 100 μL RNase-freies Wasser verwenden.
Hinweis: Eine Normalisierung ist nicht erforderlich, sondern vereinfacht den direkten Ergebnisvergleich während des qPCR-Schrittes und der Analyse. - Die Zentrifuge auf 4 ° C vorstocken. Fügen Sie 700 μL Lyse-Reagenz in das Probenvolumen 7.2.1.
- Die MiRNA-Extraktion nach Schritten 7.1.3 – 7.1.10 durchführen.
- Stellen Sie das Volumen der Probe mit der niedrigsten Proteinkonzentration auf 100 μL (= maximales Probenvolumen) ein. Berechnen Sie die Probenvolumina der anderen Proben nach dieser Normalisierung, umgekehrt, auf ihre Konzentration. Für negative Kontrollversuche, 100 μL RNase-freies Wasser verwenden.
8. Umkehr-Transkription
Hinweis: Die umgekehrte Transkription von miRNA wird mit einem umgekehrten Transkriptionssatz mit den folgenden Änderungen durchgeführt.
- Tauschen Sie die Bausatz-Reagenzien und die umgekehrte Transkription Primeln auf Eis. Bereiten Sie den folgenden Master-Mix in einem Reaktionsrohr auf Eis vor: 45,7 μL RNase-freier H2 O,16,5 μL 10x Reverzept-Puffer, 11 μL Reverse Transkript-Enzym, 2,1 μL RNase-Inhibitor und 1,7 μL dNTP-Mix. Mischen Sie sanft, nicht wirbeln.
Hinweis: Die Skala hängt von der Probenmenge ab; Hier wird für 10 Reaktionen gerechnet. - Etikett 0,2 mL röhren entsprechend und mischen 7 μL des Master-Mix aus Schritt 8.1 mit 5 μL der extrahierten Probe aus Schritt 7.1.10 und 3 μL der umgekehrten Transkriptionsprimer. Die Mischung vorsichtig verrühren, zentrifugen und auf Eis aufbewahren.
- Um für jede Zelllinie die gleiche Zellzahl zu verwenden, reduzieren Sie das Probenvolumen der Zelllinie mit einer höheren Gesamtzahl der Zellen; Verwenden Sie dies als Restvolumen, um das gesamte Probenvolumen von 5 μL RNase-freies Wasser zu erreichen.
Hinweis: Die Lipoprotein-Partikelproben werden bereits in Schritt 7.2.1 normalisiert. Für die Standardkurvenvorbereitung wird ein Aliquot von miRNA nacheinander in rNase-freiem Wasser verdünnt und die Anzahl der Stränge pro Probenvolumen berechnet. Erforderlich sind mindestens fünf Datenpunkte im Bereich des daraus resultierenden Probenzifizierungszyklus (cq). In der Regel sind Gesamtverdünnungsfaktoren von 102 bis 106 geeignet . - Legen Sie die Rohre in die Thermozykler-Maschine und starten Sie folgendes Programm: 30 min bei 16 ° C (Glühschritt), 30 min bei 42 ° C (Rückschrift), 5 min bei 85 ° C (Schmelzschritt) und Pause bei 4 ° C (Lagerung). Führen Sie den qPCR-Schritt unmittelbar nach der umgekehrten Transkription durch; Speichern Sie ansonsten die komplementäre DNA (cDNA), die aus den miRNA-Proben synthetisiert wird, bei-20 ° C.
9. qPCR
- Führen Sie eine qPCR der cDNA (umgekehrte Transkription von miRNA) mit Hilfe eines kommerziell erhältlichen Tests (siehe Materialtabelle) mit den folgenden Änderungen durch.
- Alle Reagenzien (Supermix, RNase-freie H2 O), cDNA-Proben (ab Schritt 8.4) und die Primeln auf Eis aufwerfen. Bereiten Sie den folgenden Master-Mix in einem Reaktionsrohr auf Eis vor: 75 μL Supermix, 47,5 μL RNase-freies H2 O,7,56 μL Grundierung. Mischen Sie sanft, nicht wirbeln.
Hinweis: Die Skala hängt von der Probenmenge ab; Hier wird für 10 Reaktionen gerechnet. - Etikettieren Sie die Rohre von 0,2 mL entsprechend und fügen Sie 2 μL der cDNA-Probe auf 13 μL des Master-Mixes und mischen Sie sanft. In der Regel messen Sie jedes Beispiel 2x.
Hinweis: Zusätzlich werden mindestens zwei negative Kontrollproben — Verwendung von RNase-freiem Wasser als Probe benötigt. Wird die Kalibrierkurve nicht im gleichen Lauf wie die Probe ermittelt, ist auch eine Probe aus der Kalibrierkurve erforderlich. Es wird verwendet, um jeden einzelnen Lauf auf die gleiche Reaktionseffizienz zu kalibrieren. - Legen Sie die Rohre in die PCR-Maschine und starten Sie folgendes Programm: 2 min bei 50 ° C, 10 min bei 95 ° C, 15 s bei 95 ° C und 60 s bei 60 ° C. Wiederholen Sie die letzten beiden Schritte des Programms bis zu 50x.
- Für eine Analyse der c q-Werte mit dem Softwarepaket der PCR-Maschine aktivieren Sie die DynamicTube-Normalisierung (für die Kompensation verschiedener Hintergrundebenen mit dem zweiten Derivat jeder Probenspur) und Noise Slope Korrektur (Normalisierung des Geräuschpegels).
Hinweis: Der cq-Wert der Negativkontrolle sollte mehrere Zyklen höher sein als der höchste Probenwert cq .- Für eine Kalibrierkurvenanalyse bestimmen Sie die Schwelle für die Berechnung des cq für jede miRNA aus den Standardkurven jeder miRNA einzeln, mit der Auto-Fingerschwellenfunktion des Softwarepakets, und halten Sie sie konstant Für jede bestimmte miRNA.
- Bei der Probenanalyse können bei Bedarf unterschiedliche Reaktionseffizienzen des Probenlaufs mit dem Datenpunkt aus der Kalibrierkurvenprobe ausgeglichen werden. Die Software berechnet die cq-Werte der Proben aus der Schwelle der jeweiligen Kalibrierkurvenmessung.
10. Berechnung des miRNA-Inhalts
-
Kalibrierkurve
- Berechnen Sie aus der Anfangszahl der miRNA-Stränge pro Aliquot (100 μL von 10 μM miRNA, molekulares Gewicht aus Datenblatt) und den anschließenden seriellen Verdünnungsschritten die Anzahl der miRNA-Stränge im Probenvolumen (5 μL Probenvolumen ab Schritt 8.3).
Hinweis: Wenn man eine Reverse Transkription von 1 ansieht, entspricht diese Zahl der Anzahl der cDNA-Stränge. - Berechnen Sie die Anzahl der cDNA-Stränge im Probenvolumen von 2 μL ab Schritt 9.3. Betrachten Sie dabei den zusätzlichen Verdünnungsfaktor von 7,5 (das 2 μL-Probenvolumen aus Schritt 9.4 aus dem 15 μL-Probenvolumen ab Schritt 8.4).
- Zeichnen Sie den cq-Wert aus Schritt 9.5.1 gegen die Anzahl der Stränge n Strängen, die in Schritt 10.1.2 in einem Basis-10-halnogradmischen Diagramm berechnet werden, und passen Sie die Datenpunkte mit der folgenden Regressionskurve (M = die Neigung der linearen Regressionskurve, B = Offset).

Bestätigen Sie, dass der Korrelationskoeffizient (R 2) für die Linieistund gt;0.99.
- Berechnen Sie aus der Anfangszahl der miRNA-Stränge pro Aliquot (100 μL von 10 μM miRNA, molekulares Gewicht aus Datenblatt) und den anschließenden seriellen Verdünnungsschritten die Anzahl der miRNA-Stränge im Probenvolumen (5 μL Probenvolumen ab Schritt 8.3).
-
Lipoprotein-Teilchen
- Berechnen Sie die Anzahl der miRNA-Stränge pro Probenvolumen mit dem gemessenen Proben-Q-Wert (Schritt 9.5.2) und der folgenden Gleichung (M und B sind die Kalibrierungskurven-Parameter der spezifischen miRNA).

- Berechnen Sie die entsprechende Anzahl von Lipoprotein-Partikeln im Probenvolumen, beginnend mit dem Volumen im Schritt 7.2.1 (100 μL), seiner bekannten Konzentration und den anschließenden Verdünnungsschritten (100 μL-> 30 μL Probenvolumen im Schritt 7.1.10 5 μL [Verdünnung 1:6] Probe im 15 μL-Gesamtvolumen in Schritt 8.4-> 2 μL [Verdünnung 1:7.5] in Schritt 9.4). Nehmen Sie ein molekulares Gewicht von 250 kDa für HDL-Partikel und 3 MDa für LDL-Partikel und eine 100% ige Rückgewinnung von miRNA während des miRNA-Absaugschrittweises an (das Ignorieren eines Lipidbeitrags zum molekularen Gewicht ergibt eine leichte Überschätzung der Anzahl der miRNA-Stränge pro Lipoprotein-Teilchen).
- Teilen Sie die Anzahl der miRNA-Stränge von Schritt 10.2.1 durch die Anzahl der Partikel, die im vorherigen Schritt berechnet wurden, um die Anzahl der miRNA-Stränge pro Lipoprotein-Teilchen zu erzeugen.
- Berechnen Sie die Anzahl der miRNA-Stränge pro Probenvolumen mit dem gemessenen Proben-Q-Wert (Schritt 9.5.2) und der folgenden Gleichung (M und B sind die Kalibrierungskurven-Parameter der spezifischen miRNA).
-
Zellen
- Berechnen Sie die Anzahl der miRNA-Stränge pro Probenvolumen nach Schritt 10.2.1.
- Berechnen Sie die entsprechende Anzahl von Zellen im Probenvolumen nach Schritt 10.2.2, beginnend mit der Anfangszellenzahl, die in Schritt 6.4 gemessen wird.
- Teilen Sie die Anzahl der miRNA-Stränge von 10.3.1 durch die Anzahl der Zellen, die im vorherigen Schritt berechnet wurden, um die Anzahl der miRNA-Stränge pro Zelle zu erzeugen.
- Berechnen Sie die Aufnahmevorrate von Lipoprotein-Partikeln, indem Sie die Gesamtmenge der miRNA-Stränge aus dem vorherigen Schritt nach der Korrektur für den Hintergrund-miRNA-Wert der Zellen, die aus einem negativen Kontrollexperiment durch das miRNA/Partikelverhältnis gewonnen wurden, teilen (Schritt 10.2.3 ) und die Inkubationszeit (16 Uhr, siehe Schritt 6.2).
11. Multiwell mikrofluidische Arrays
-
MiRNA-Extraktion
- Führen Sie die miRNA-Extraktion, wie sie in Schritt 7 beschrieben ist.
-
Umgekehrte Transkription
- Tauschen Sie die umgekehrten Transkriptionsgrundierungen, die umgekehrten Transkriptionskomponenten und MgCl 2 (25 mM) auf Eis. Für acht Proben, mischen 8 μL umgekehrte Transkriptionsgrundierung (10x), 2,25 μL dNTPs mit dTTP (100 mM), 16,88 μL von Reverse Transkriptase (50 U/μL), 9,00 μL von 10x Reverse-Transkript-Puffer, 10,12 μL MgCl2, 1.12 μL von RNase inhibitor (20 U/μL) , und 1 μL natomfreies Wasser.
- Kurz mit Zentrifuge verrühren. 4,3 μL umgekehrter Transkriptionsreaktionsmix auf 3,5 μL extrahierte MiRNA in einem Rohr hinzufügen und mischen, abdrehen und 5 min auf Eis inkubieren. Die Röhre in eine Thermozykler-Maschine legen und das folgende Programm starten: 16 ° C für 2 min, 42 ° C für 1 min , und 50 ° C für 1 s wiederholte 40x insgesamt, und dann, als Stopp-Reaktion, 85 ° C für 5 min und halten bei 4 ° C, bis gestoppt.
-
Vorverstärker
- Die Primeln auf Eis auftürmen und kurz umkehren und zentrifugen. Mischen Sie den Vorverstärker-Master-Mix (2x), indem Sie die Flasche schwirren. Bereiten Sie den Vorverstärkungs-Reaktionsmix nach folgender Anweisung für acht Proben vor: 112,5 μL Vorverstärker-Master-Mix (2x), 22,5 μL Vorverstärkungsgrundierung und 67,5 μL Nuklease-freies Wasser. Invertiert und zentrifuge kurz.
- Vermischen Sie 2,5 μL des umgekehrten Transkriptionsreaktionsprodukts aus Schritt 11.2.2 mit 22,5 μL Vorverstärkungsreaktionsmix aus dem vorherigen Schritt und Invert und Zentrifuge kurz. Die Proben auf Eis 5 min einwirken.
- Legen Sie die Rohre in eine Thermozykler-Maschine und inkubieren Sie sie an folgenden Einstellungen: Enzymaktivierung bei 95 ° C für 10 min, Glühen bei 55 ° C für 2 min, Verlängerung bei 72 ° C für 2 min, wiederholte 12x: Verdichtung bei 95 ° C für 15 s und bei 60 ° C für 4 min , Enzym-Inaktivierung bei 99,9 ° C für 10 min und 4 ° C auf Halt.
- Drehen Sie sich, fügen Sie 7,5 μL von 1x TE (pH 8,0) und 67,5 μL von nukleasfreiem Wasser, Invert und Zentrifuge kurz hinzu. Die Proben können bis zu einer Woche bei-20 ° C gelagert werden.
-
Qpcr
- Den Master-Mix vermischen, indem man die Flasche wirbelt. Bereiten Sie den PCR-Reaktionsmix für eine Karte vor: 450 μL Master-Mix, 441 μL kernfreies Wasser und 9 μL der Vorverstärkungsprobe ab Schritt 11.3.4. Invertiert und zentrifuge kurz.
- Laden Sie jedes Füllreservoir der multiwell mikrofluidischen Array-Karte mit 100 μL vorbereitetem PCR-Reaktionsmix nach Herstelleranleitung und Zentrifuge 2x für 1 min bei 3.000 x g. Die Beschleunigung während der beiden aufeinanderfolgenden Zentrifugationsschritte ist wichtig, um die Karte richtig zu füllen. Die Karte nach Herstelleranweisung versiegelt.
- Verwenden Sie ein PCR-System mit folgendem Programm: Enzymaktivierung bei 95 ° C für 10 min und dann, wiederholt 40x: Denaturierung bei 95 ° C für 15 s und annealing/bei 60 ° C für 1 min.
- Importieren Sie die Ergebnisdatei aus dem PCR-System und berechnen Sie die RQ-Werte mit dem Softwarepaket des Systems mit den folgenden Analyseeinstellungen: Ein maximal zulässiger c q-Wert von 40,0, maximale cq-Werte in den Berechnungen inklusive und Ausreißer Unter Replikaten ausgeschlossen. Aktivieren Sie die Fehlentdeckungsrate von Benjamini – Hochberg als Option für die p-Wertanpassung (Korrektur des Auftretens falscher Positivwerte 14) und die globale Normalisierung als Normalisierungsmethode (Verwendung eines Median-Schwellenwertes für alle Proben). 15).
Ergebnisse
Ein allgemeines Schema der Lipoprotein-Partikelisolierung
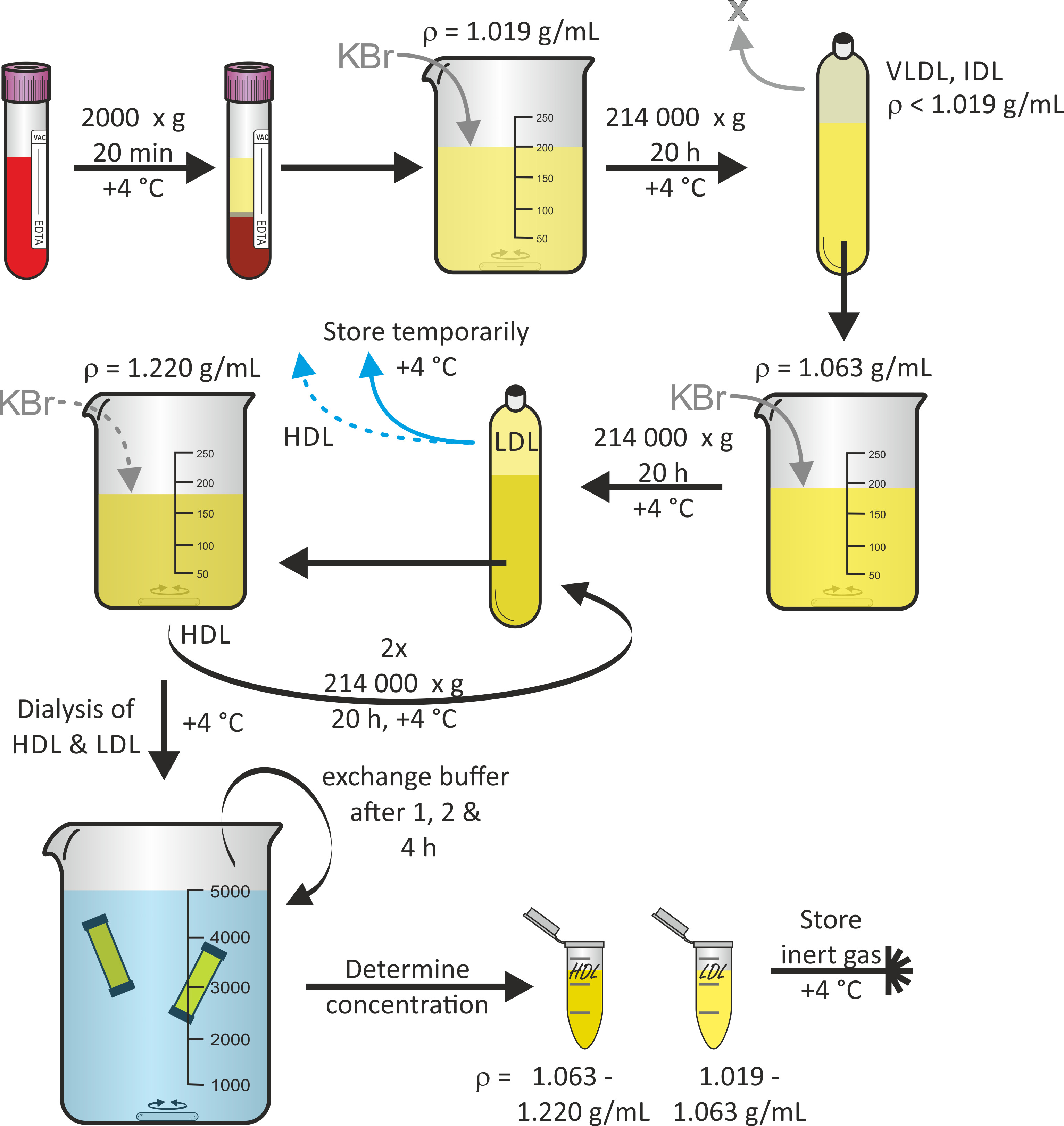
Abbildung 1 zeigt das allgemeine Schema der Lipoprotein-Partikelisolierung ab Vollblut, wobei sequenzielleFlotationUltraentrifugation 16. Auf Wunsch können während dieses Protokolls auch andere Lipoprotein-Teilchenfraktionen wie VLDL und IDL-Partikel geerntet werden. Der fixierte Titan-Rotor in Kombination mit Polypropylen-Schnelldichtrohren ist geeignet, den Zentrifugationskräften standzuhalten. Um einen Rohrkollaps zu vermeiden, ist es wichtig, Luftblasen im Rohr zu vermeiden. Die Zentrifugation wird bei 4 ° C durchgeführt, um den Proteinabbau zu minimieren. In der Regel ist mit einem Ertrag von LDL und HDL-Partikellösungsvolumen von jeweils 3 mL mit Konzentrationen im Bereich von 1-3 mg/mL zu rechnen. Die ganze Prozedur, beginnend mit der Blutspende, dauerte rund 7 Tage.

Bild 1: Flowchart der Lipoprotein-Isolierung. Zentrifugenblut von gesunden Freiwilligen in Vakuumbehälterröhren und sammeln Plasma (obere Phase). Nach der Anpassung der Dichte auf , die mit KBr auf die Ebene der Zentrifuge mit der Zentrifuge die Lösung bei 214.000 x g für 20 h bei 4 ° C. Nach der Anpassung der Dichte der unteren Fraktion auf ■ = 1.063 g/mL mit KBr, Zentrifuge die Lösung wieder bei 214.000 x g für 20 h bei 4 ° C. Speichern Sie die obere Fraktion, die LDL-Partikel vorläufig bei 4 ° C enthält. Nach der Anpassung der Dichte der unteren Fraktion auf ■ = 1.220 g/mL mit KBr, Zentrifuge die Lösung zweimal bei 214.000 x g für 20 h bei 4 ° C. Sammeln Sie die obere Fraktion, die HDL-Partikel enthält, dialysieren Sie sowohl die HDL-Partikellösungen als auch die LDL-Partikellösungen und tauschen Sie den Puffer nach 1, 2 und 4 Stunden aus. Nach 24 Stunden die Proteinkonzentration bestimmen und die Proben unter Inertgas bei 4 ° C lagern. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Rückstellung von HDL-Teilchen
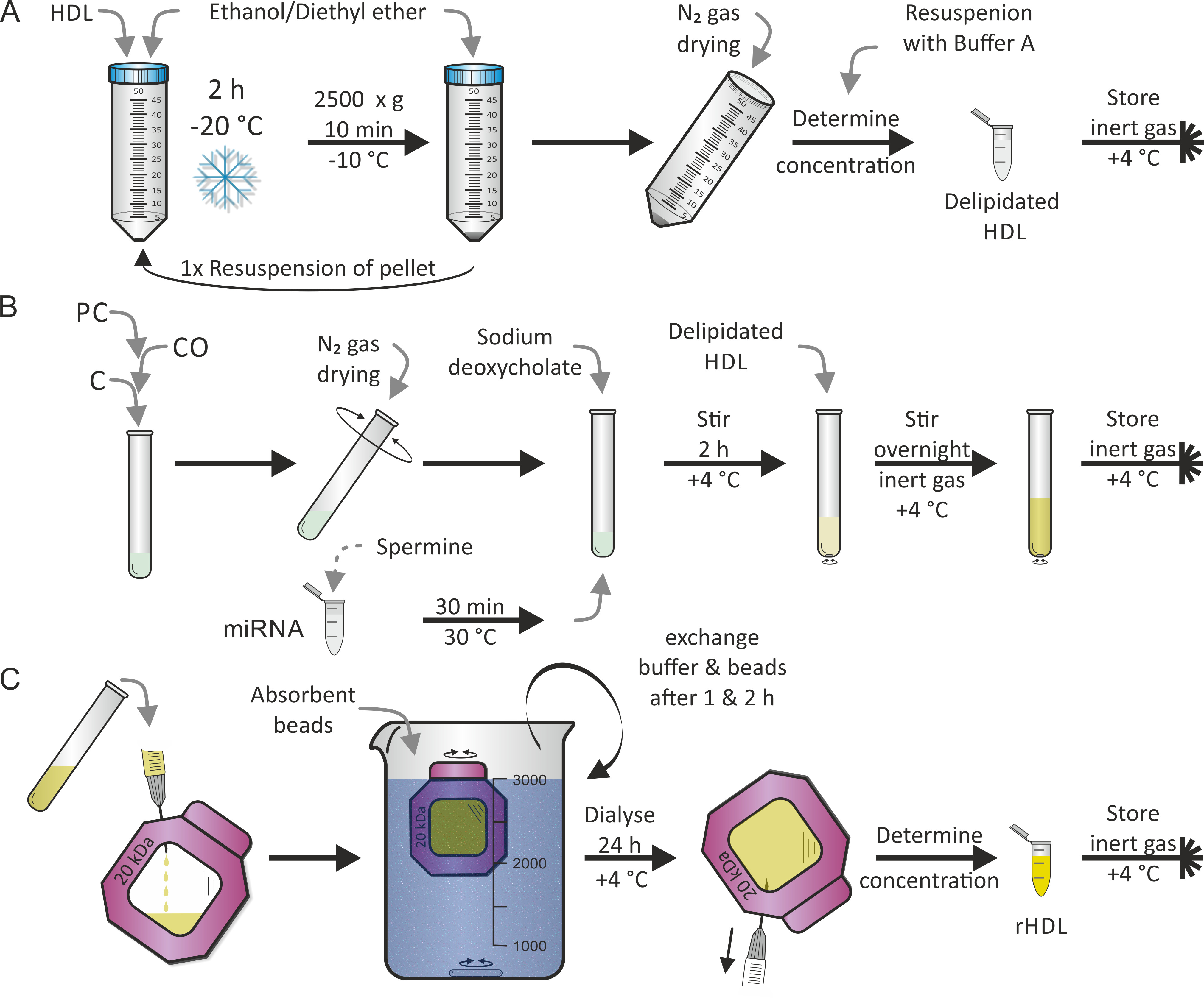
Die Rekrekonstitution von HDL-Partikeln erfolgte nach einem zuvor von Jonas7 veröffentlichtenProtokoll. Der erste Schritt war die Verflüssigung von HDL-Partikeln, wie sie in Abbildung 2A gezeigt wurde, gefolgt von dem zweiten Schritt der Verlässlichkeit (d.h. der Wiederherstellung ), wie sie in Abbildung 2B gezeigt wurde, mit LipidPC, CO und C zusätzlich zu einer Mischung aus MiRNA undSperma. Wir haben uns für die menschlichen reifen MiR-223 und MiR-155 entschieden, weil miR-223 eine hohe Fülle aufweist und miR-155 bei Lipoprotein-Teilchen 17 selten ist. In der Regel werden beide Schritte an zwei aufeinander folgenden Tagen durchgeführt. Bei der Rekonstruktion konnten nach Belieben weitere lipophile und/oder amphiphile Komponenten hinzugefügt werden. Die vollständige Verdunstung von Ethanol/Diethyläther und Methanol/Chloroform Lösungsmittel von PC, CO und C war entscheidend. Der letzte Schritt —, wie in Abbildung 2Cgezeigt — war das Dialyseverfahren, um wiederhergestellte HDL-Partikel (rHDL) von freien Lipids/miRNA/Waschmittel zu trennen. Dies dauerte zusätzlich 1-2 Tage. Durch die Zugabe von absorbierenden Perlen in die Dialyselösung wurde der Dichtegradienten entlang der Dialysembran konstant gehalten. Es ist mit einem Ertrag von 50% der rHDL-Partikel zu rechnen.

Bild 2: Flowchart der HDL-Teilchenkonstitution. (A) Lieferung: Mischen Sie die HDL-Partikellösung mit vorgekühltem Ethanol/Diethyläther und Inkubat bei-20 ° C für 2 Stunden. Nach dem Ablegen des Supernatans, das Pellet wieder ausrichten und das Verfahren wiederholen. Das Pellet mit N2-Gas trocknen und in Puffer A wieder auffüllen. Nach der Bestimmung der Konzentration die entzückende HDL unter inerter Gasatmosphäre bei 4 ° C lagern. (B) Konterstellung: Nach dem Mischen von Phosphatidyl-Cholin (PC), Cholesteryl (CO) und Cholesterin (C) verdampft das Lösungsmittel mit N2-Gas beim Drehen des Rohres. Den MiRNA-Aliquot mit Sperminallösung für 30 min bei 30 ° C unterwischen, Natriumdeoxycholat hinzufügen und den getrockneten Lipidfilm wiederbeleben. Die Probe 2 Stunden bei 4 ° C umrühren, die delipidierte HDL-Lösung dazugeben und die Probe wieder umrühren, diesmal über Nacht bei 4 ° C unter inerter Gasatmosphäre. (C) Dialyse: Übertragung der Lösung von der Platte B mit rekonstruierten HDL-Partikeln (rHDL) auf eine Dialysemher-Kammer (molekularschweres Abschneiden: 20 kDa) und Dialyse gegen PBS und saugfähige Perlen bei 4 ° C. Tauschen Sie den Puffer und die Perlen nach 1 h und 2 h aus. Nach 24 Stunden die rHDL-Partikellösung zurückholen, die Konzentration bestimmen und die Probe unter Inertgasatmosphäre bei 4 ° C speichern. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Kennzeichnung von LDL-Teilchen
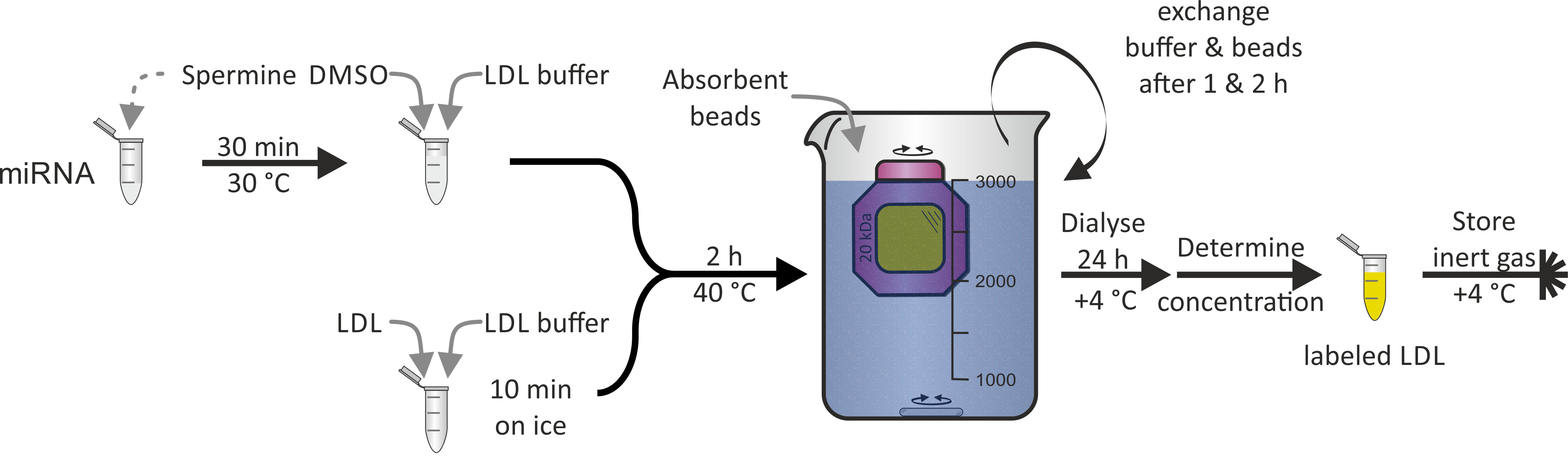
Die Kennzeichnung von LDL-Partikeln mit miRNA(Abbildung 3), wie sie bei HDL-Partikeln nachgewiesen wurde, war aufgrund der Hydrophobizität des ApoB-100-Proteins, das der Hauptbestandteil des LDL-Teilchens ist, nicht möglich. DMSO wurde für die Durchdringung des Lipidmonolayers des LDL-Teilchens verwendet und vermittelte damit die miRNA-Assoziation. Die gesamte Prozedur dauerte 1-2 Tage mit einer Rendite von fast 100%.

Bild 3: Fließdiagramm der LDL-Partikelkennzeichnung. Inkubieren Sie den miRNA-Aliquot mit einer Sperminallösung für 30 min bei 30 ° C und fügen Sie DMSO und LDL-Puffer hinzu. Inkubieren Sie die LDL-Probe mit dem LDL-Puffer für 10 Minuten auf Eis und fügen Sie miRNA/spermine/DMSO-Mischung hinzu. Nach der Inkubation bei 40 ° C für 2 Stunden die Lösung in eine Dialysemher-Kammer (molekulares Gewichtsabteil: 20 kDa) und Dialyse gegen PBS und saugfähige Perlen bei + 4 ° C übertragen. Tauschen Sie Puffer und Perlen nach 1 & 2 h. Recovertieren Sie die beschriftete LDL-Partikellösung nach 24 Stunden, bestimmen Sie die Konzentration und speichern Sie unter Inertgas-Atmosphäre bei + 4 ° C. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Qualitätskontrolle von Lipoprotein-Teilchen
HS-AFM kann verwendet werden, um die Größe und Form von einheimischen und rekonstituierten/beschrifteten Lipoprotein-Partikeln auf Glimmer zu untersuchen. Kurz vor dem Gebrauch muss Glimmer frisch gespalten werden (mit Klebeband die obere Schicht entfernen), um eine saubere und flache Oberfläche zu schaffen. Bei der Inkubation von HDL-LDL-Partikeln auf Glimmer muss der Verdünnungsfaktor (and/oder die Inkubationszeit) angepasst werden, um einzelne Partikel zu beobachten. Cluster erlauben keine Bestimmung von Teilchenmaßen. HDL-Partikel sind auf Glimmer mobil. Bei der Verwendung von herkömmlichem AFM anstelle von HS-AFM muss das Wegfahrprotokoll entsprechend angepasst werden (Puffer, Oberflächenbeschichtung), um die seitliche Partikelmobilität zu reduzieren. Beim Scannen der Probe muss die Bildkraft niedrig gehalten werden (Abtastmodus), um eine Verformung der Partikel zu vermeiden, was die Messwerte beeinflusst. Für die Datenanalyse wurden Partikel über einen Schwellenalgorithmus (z.B. in Gwyddion: Körner > Mark nach Schwelle) erkannt und ihre Höhe in Bezug auf die Glimmeroberfläche bestimmt. Die Messung der Partikelhöhe ist die präziseste Methode, um die Partikelgrößen zu bestimmen, da die scheinbaren Seitenabmessungen durch die Spitzform erweitert werden (siehe exemplarische Bilder in Abbildung 4). Für die statistische Auswertung und den Vergleich der Größenverteilungen der verschiedenen Lipoprotein-Teilchen wurden die Funktionen der Wahrscheinlichkeitsdichte (pdfs) berechnet. Ein Vergleich von nativen und miRNA-beschrifteten LDL-Partikeln, wie sie in Abbildung 4 gezeigt werden, ermöglicht es, die Hauptähnlichkeit zwischen beschrifteten und nicht beschrifteten (d.h. nativen) Lipoprotein-Partikeln (gekennzeichnet LDL-Tonchen ohne Zusatz von miRNA/ Spermienmischungen werden als Steuerung für das Beschriftungsverfahren selbst angezeigt). Das ganze Verfahren dauerte etwa 1 Tag.

Abbildung 4: Flowchart und repräsentative Ergebnisse von HS-AFM-Messungen. Die HDL/LDL-Partikelprobe in PBS (1:10 2-1:10 3)verdünnen und 5 min lang auf frisch gespaltenem Glimmer inkubieren, gefolgt von einer sorgfältigen Spülung mit PBS, um freie (nicht elektrostatisch adsorbierte) Partikel zu entfernen. Führen Sie die HS-AFM-Bildgebung durch und überprüfen Sie die Partikeldichte auf der Oberfläche. Führen Sie die Messungen in PBS bei Raumtemperatur durch. Das obere Bild dieser Abbildung zeigt eine zu hohe Partikeldichte; Das untere Bild eignet sich für die Analyse. Die Höhe der einzelnen Teilchen wurde nach Schwellen analysiert und einheimische (schwarze Kurve) und rekonstituierte/beschriftete (rote und grüne Kurve) Partikel wurden in einer statistischen Auswertung verglichen. Der Maßstab bar = 100 nm. Diese Figur wurde von Axmann et al. 19modifiziert. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
MiRNA-Extraktion, umgekehrte Transkription und qPCR
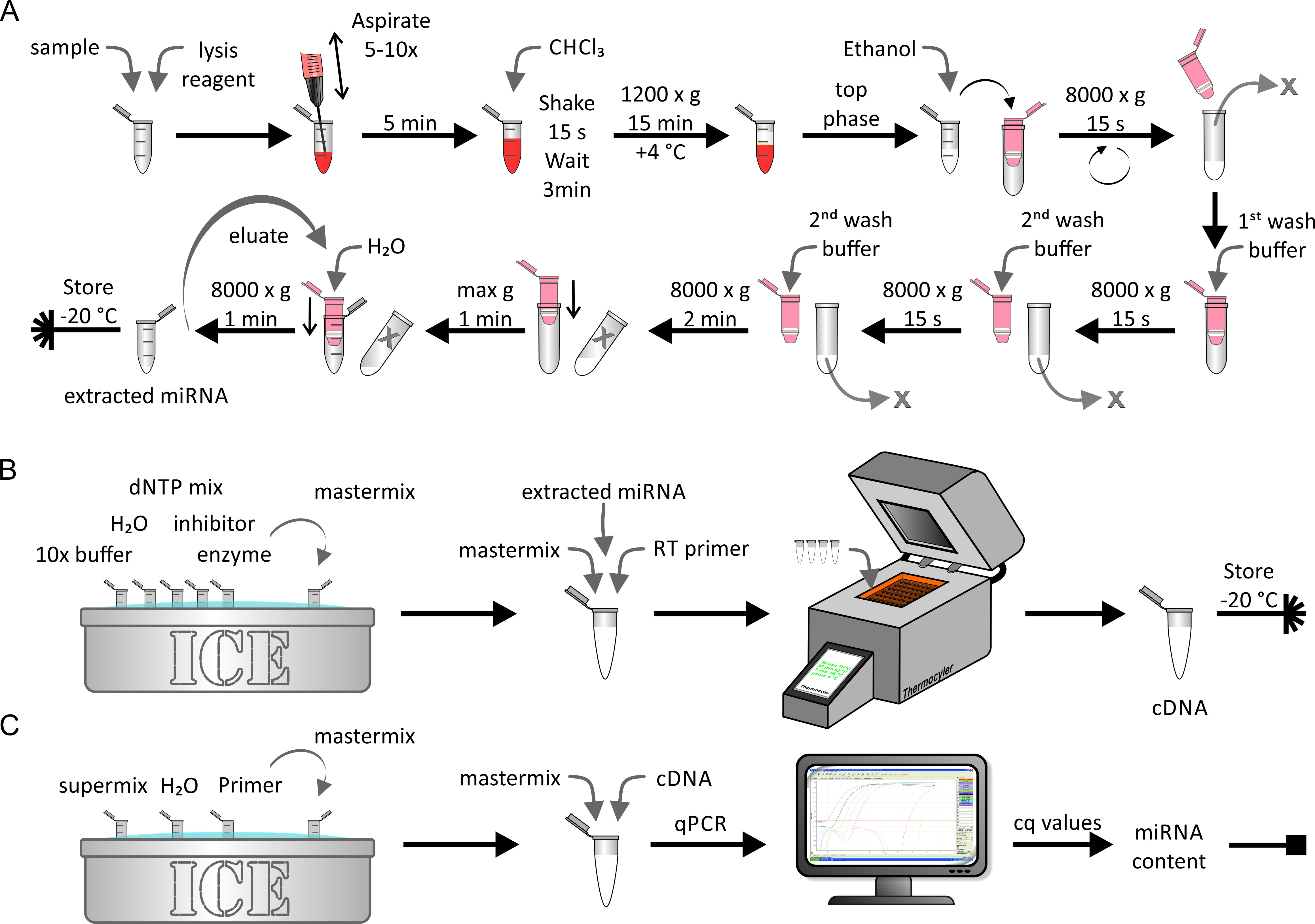
Die Extraktion von miRNA aus native/künstlich angereichertem Lipoprotein oder Zellproben wurde mit einem miRNA-Extraktionskit durchgeführt, wie in Abbildung 5Agezeigt. Dabei war ein rNase-freies Umfeld kritisch. Dieser Schritt dauerte etwa 1 Stunden. Die umgekehrte Transkription der extrahierten miRNA-Probe (Abbildung 5B) wurde mit Hilfe von biochemischen Standardverfahren durchgeführt, wie sie vom Hersteller beschrieben wurden. Dieser Schritt dauerte etwa 1,5 Stunden. Schließlich wurde die Menge der cDNA, die während des letzten Schrittes erreicht wurde, mit qPCR (Abbildung 5C) ermittelt. Eine Standardkurve, die die gewonnenen cq-Werte mit der absoluten miRNA-Strang-Nummer in Beziehung steht, ergab den absoluten miRNA-Gehalt der ersten Probe. Das dauerte etwa 2,5 Stunden.

Abbildung 5: Flowchart der miRNA-Extraktion, umgekehrte Transkription und qPCR. (A) miRNA-Extraktion: Die Probe mit Lyse-Reagenz vermischen und über Aspiration mit einer Spritze lysern. 5 min inkubieren und mit CHCl 3hinzufügen. Schütteln Sie kräftig für 15 s und inkubieren für 3 min. Nach der Zentrifugation bei 1.200 x g für 15 min bei 4 ° C die obere Phase sammeln und mit Ethanol vermischen. Übertragen Sie die Lösung auf eine Spinnsäule (maximale Lautstärke & lt;700 μL) und Zentrifuge mit 8.000 x g für 15 s. Verwerfen Sie das Elut und wiederholen Sie den letzten Schritt mit dem Rest der Lösung. Den ersten Waschpuffer und die Zentrifuge mit 8.000 x g für 15 s dazugeben. Das Eluent ablegen, den zweiten Waschpuffer und die Zentrifuge mit 8.000 x g für 15 s. Wiederholen Sie den letzten Schritt mit einer Zentrifugationszeit von 2 min. Die Membran durch Zentrifugation mit maximaler Geschwindigkeit für 1 min weiter trocknen. Die miRNA mit H2 O undZentrifuge mit 80.000 x g für 1 min aufwerten. Die extrahierte miRNA-Probe bei-20 ° C aufbewahren. (B) Reverse-Transkription: Tauschen Sie den 10x-Puffer, H2 O,dNTP-Mix, Inhibitor und Enzym auf Eis und bereiten Sie den Master-Mix vor. Fügen Sie die extrahierte miRNA aus dem Panel A in den Master-Mix und die umgekehrte Transkription ein und führen Sie die umgekehrte Transkription mit einer Thermocycler-Maschine durch. Die cDNA-Probe bei-20 ° C aufbewahren. (C) qPCR: Den Supermix, H 2 OundGrundierung auf Eis auftürmen und den Master-Mix vorbereiten. Fügen Sie die cDNA aus dem Panel B in den Master-Mix ein und führen Sie qPCR durch. Analysieren Sie die Daten, um c q-Werte zu erhalten und berechnen Sie den absoluten miRNA-Gehalt der Probe (siehe Abbildung 6 und die repräsentativen Ergebnisse für Details). Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Absolute miRNA-Inhalte und Übertragungsrate
Der absolute miRNA-Gehalt von nativen und künstlich angereicherten HDL und LDL-Partikeln wurde aus den cq-Werten der Proben und einer Standardkurve der jeweiligen miRNA berechnet, wie in Abbildung6 gezeigt. Abbildung 6A zeigt Daten, wie sie von der Analysesoftware berechnet werden (mit aktivierter DynamicTube-Normalisierung [zur Kompensation verschiedener Hintergrundebenen mit dem zweiten Derivat jeder Probenspur] und einer Lärmabhänge Korrektur [Normalisierung auf den Geräuschpegel]). Die c q-Werte der Standardkurven wurden mit Hilfe der Auto-Fund-Schwellfunktion des Softwarepakets auf dem von der qPCR-Maschine gemessenen Normsignal-Signal ermittelt. Dabei maximierte die Software den R-Wert der Passform der Standardkurve. Der Schwellenwert wurde für jede spezifische miRNA-Probenanalyse konstant gehalten. Anschließend wurden die cq-Werte als Funktion der Anzahl der miRNA-Stränge dargestellt und eine Regressionslinie berechnet. Die Probenwertec q wurden mit dem gleichen Schwellenwert ermittelt, wie in Abbildung 6B dargestellt; Die Reaktionseffizienzunterschiede zwischen den verschiedenen QPCR-Läufen wurden von der Software automatisch mit einer zusätzlichen Kalibrierkurven-Probe kompensiert, die in jedem Lauf enthalten ist. Mit Hilfe der Regressionsleitungsgleichung konnte die unbekannte Menge der miRNA in der Probe berechnet werden. Die Lipoprotein-Partikelzahl wurde von der anfänglichen Proteinkonzentration und ihrem durchschnittlichen Molekülgewicht (MWHDL ~ 250 kDa) abgeschätzt. Dabei wurde kein Lipidbeitrag zum molekularen Gewicht angenommen — so wurde die Anzahl der miRNA-Stränge pro Lipoprotein-Teilchen leicht überschätzt. Darüber hinaus wurde eine 100-prozentige Rückgewinnungsrate von miRNA während des miRNA-Extraktionsschrittes angenommen. Darüber hinaus wurde der miRNA-Gehalt der Zellen vor und nach der Inkubation mit HDL-Partikeln ermittelt und die miRNA-Übertragungsrate berechnet, wie in Abbildung 6Cgezeigt.

Abbildung 6: Flowchart der Berechnung des absoluten miRNA-Inhalts und der Übertragungsrate. (A) Standardkurve für miR-155: Ein MiR-155 aliquot (100 μL, 10 μM) wurde seriell mit RNA-freiem Wasser verdünnt, wie angegeben. qPCR ergab für jede serielle Verdünnungsprobe (zweimal gemessen) c q Werte mit der Auto-Fundwellenfunktion des Softwarepakets. Negative Kontrollexperimente (ohne Zusatz von miRNA) ergaben c q-Werte von & gt;35. Die Datenpunkte der c q-Werte als Funktion der Anzahl der MiRNA-Stränge pro Probenvolumen (berechnet aus der Anfangskonzentration und den seriellen Verdünnungen) wurden mit der dargestellten Gleichung (rote Linie, rechtes Bild) versehen, die M =-3,36 ergibt und B = 42.12. Die ermittelte PCR-Effizienz betrug 0,98. Die Fehlerbalken wurden aus den Ergebnissen experimenteller Wiederholungen berechnet und waren kleiner als der Durchmesser des Datenpunktkreises. B) cq Werte von native/künstlich angereicherten HDL-Partikeln wurden mit dem gleichen Schwellenwert wie in der Gruppe A ermittelt und in die Anzahl der miRNA-Stränge im qPCR-Probenvolumen umgewandelt. Das absolute Verhältnis der miRNA der ursprünglichen Probe wurde aus der Anzahl (Konzentration) der HDL-Partikel im Probenvolumen (3,2 x 1011 Partikel) berechnet. (C) Zellproben (Zelllinie ldlA7-SRBI) wurden 16 h lang mit künstlich angereicherten HDL-Partikeln (50 μg/mL) inbratiliert und ähnlich analysiert. Die ermittelten c q-Werte lagen bei 22,5, 22,5 und 19,3 nur für Zellen, für Zellen, die mit nativen HDL inkubiert wurden, oder für Zellen, die mit rHDL-Partikellösung (beide 50 μg/mL) inkubiert wurden. Diese Werte wurden in die Anzahl der miRNA-Stränge umgewandelt, wie sie in der Gruppe B ausgeführt wurden. Die Anzahl der MiRNA-Stränge nach der Inkubation (7.3 x 106)wurde vor der Inkubation durch Abzug der Anzahl der MiRNA-Stränge korrigiert (8,6 x 105). Das Ergebnis wurde durch die Anzahl der Zellen im Probenvolumen (3.100), das miRNA-Partikel-Verhältnis (1,5 x 10-4) und die Inkubationszeit (16 h) geteilt. Dies ergab die Übertragungsrate von Lipoprotein-Partikeln über miRNA-Aufnahme (240 HDL-Teilchen-Auftaktereignisse pro Zelle und Sekunde). Diese Figur wurde von Axmann et al. 19modifiziert. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
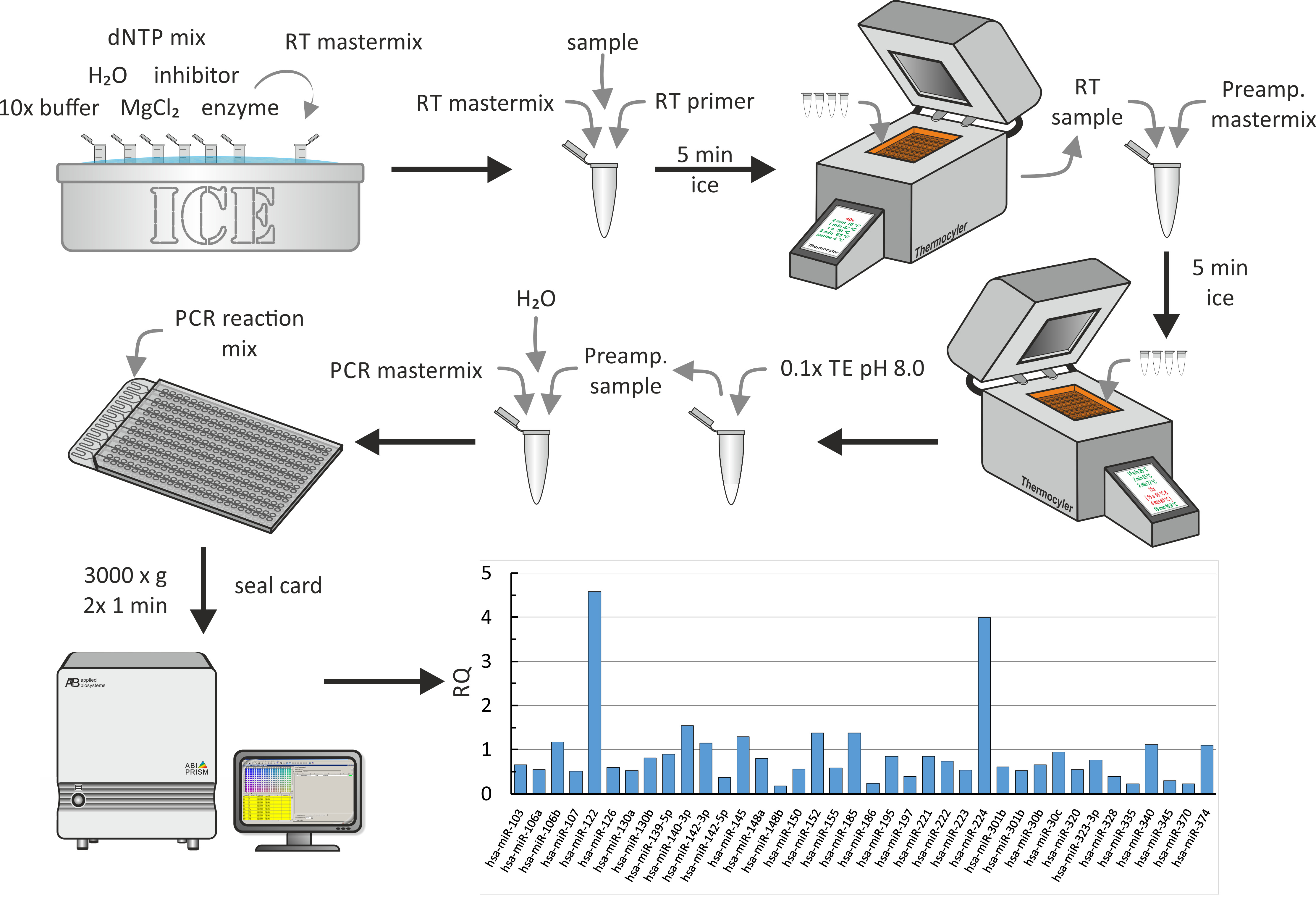
Multiwell mikrofluidisches Array
Aufgrund der geringen Erträge der miRNA-Extraktion folgte die umgekehrte Transkription der extrahierten miRNA mit einem Vorverstärkungsschritt. Schließlich wurde qPCR, wie in Abbildung6 gezeigt, durchgeführt. Für alle Schritte wurden die vom Hersteller beschriebenen Standardbiochemiemie verwendet. Hier wird ein Teil des globalen miRNA-Profils auf HDL-Partikeln von Uromizipatienten gezeigt, die für eine Studie über den Einfluss von CRF auf den Cholesterin-Abflussaus Makrophagen 18 rekrutiert wurden. In dieser Studie wurde die Cholesterinannapazität von HDL oder Serum in — neben others—17 jungen erwachsenen uremischen Patienten (CKD-Stadien 3-5) und 14 jungen erwachsenen Hämodialysepatienten ohne damit verbundene Erkrankungen und abgestimmte Kontrollen gemessen. Zur Analyse der Daten wurden Standardeinstellungen verwendet (zulässiger zulässiger CT-Wert: 40.0, inklusive maximaler CT-Werte bei Berechnungen und Ausschluss von Ausreißern unter den Replikaten). P-Werte wurden mit der falschen Entdeckungsrate von Benjamini-Hochberg (Korrektur des Auftretens falscher Positivwerte) angepasst, und als Normalisierungsmethode wurde die Globale Normalisierung gewählt, die die gängigen Assays unter allen findet. Proben, um sein Median CT für die Normalisierung zu verwenden. In den repräsentativen Ergebnissen sind einige RQs von miRNAs abgebildet, die von HDLs von uremischen Patienten isoliert sind (RQs der Kontrollen sind 1). Offensichtlich sind miR-122 und miR-224 in den HDLs der Harnröhren stark ausgedrückt. Dieser ganze Schritt dauerte ungefähr 1 Tag.

Abbildung 7: Flowchart und repräsentative Ergebnisse des multiwell mikrofluidischen Arrays. Nach der miRNA-Extraktion, wie sie in Abbildung 5A gezeigt wird, mischen Sie die miRNA-Probe mit der umgekehrten Transkription und einem Master-Mix mit 10x Puffer, H2O, dNTP-Mix, Inhibitor, MgCl2und Enzym. Nach der Inkubation auf Eis für 5 min, führen Sie die umgekehrte Transkription mit einer Thermozykler-Maschine. Fügen Sie den Vorverstärker-Master-Mix hinzu, inkubieren Sie 5 Minuten auf Eis und führen Sie die Vorverstärkung mit einer Thermocycler-Maschine durch. Fügen Sie 0,1x TE (pH 8,0) hinzu und mischen Sie einen Aliquot mit PCR-Master-Mix und H2O. Pipette den PCR-Reaktionsmix in den Füllport des multiwell-mikrofluidischen Arrays und drehen Sie zweimal bei 3.000 x g für je 1 min. Führen Sie qPCR mit einem PCR-System durch und analysieren Sie die Daten, um RQ-Werte zu erzeugen (hier zeigt die Abbildung RQ-Werte von HDL-Partikeln von Umia-Patienten im Vergleich zu einer gesunden Kontrollgruppe 18). Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Diskussion
Hier wird Schritt für Schritt die Isolierung von Lipoprotein-Teilchenfraktionen aus menschlichem Blut und die Bestimmung ihres individuellen MiRNA-Inhalts beschrieben. Es ist wichtig, in einer rNase-freien Umgebung zu arbeiten, während man mit isolierten und synthetisierten miRNA umgeht — die partikelembedded miRNA offensichtlich vor enzymatischen Abbaumaßnahmen geschützt ist. Da das miRNA/-Partikel-Verhältnis von einheimischen Lipoprotein-Partikeln eher gering ist, ist eine künstliche Anreicherung mit miRNA erforderlich, um die Holo-Partikelaufnahme von Zellen zu untersuchen. Dabei wird die Rekrekonstitution von HDL-Partikeln, wie sie zuvorbeschrieben wurde, in miRNA-Stränge modifiziert. Darüber hinaus ermöglicht die Trennung der Lipid-und Proteinfraktion während dieses VerfahrensdenWissenschaftlern, Lipid-und Eiweißbestandteile des Lipoprotein-Teilchens 19 zu untersuchen. In ähnlicher Weise wird der Beschriftungsvorgang von LDL-Teilchen angepasst. Interessanterweise hat die Zugabe von Sperma — einem natürlichen Stabilisator von Nukleotiden — das miRNA/Partikelverhältnis nicht beeinflusst. Es ist zu beachten, dass die Methode grundsätzlich die Einfaltung von anderen Stoffen als miRNA innerhalb eines Lipoprotein-Teilchens erlaubt. Natürlich gibt es eine Grenze in Bezug auf die physikalische Größe des Stoffes, basierend auf der Gesamtgröße von HDL (Durchmesser: 5-12 nm) und LDL-Partikeln (Durchmesser: 18-25 nm).
Was die Qualitätskontrolle von rekonstituierten/beschrifteten Lipoprotein-Partikeln betrifft, so ist HS-AFM eine anwendbare Methode, um HDL/LL-Partikel auf der einzelnen Partikelebene zu charakterisieren. Im Vergleich zu EM ermöglicht es kürzere Vorbereitungszeiten und nahezu physiologische Bedingungen (Nass, Raumtemperatur).
Aufgrund seiner inhärenten Empfindlichkeit und Verstärkung ist qPCR die Methode der Wahl, um niedrige miRNA-Konzentrationen zu erkennen. Alternativ wäre die einmolekulare empfindliche Fluoreszenzmikroskopie, die auch einzelne Moleküle erkennen kann, aufgrund der geringen Konzentrationen beispielsweise fluoreszierend als miRNA-Stränge pro Partikel nicht geeignet. So wurde das Verhältnis der miRNA-Stränge pro einheimischem Lipoprotein-Teilchen auf 10-8 gefunden. Künstliche Anreicherung erhöht das Verhältnis um den Faktor 10.000, was die Schätzung der zellulären Lipoprotein-Auftaktrate erleichtert (kein signifikanter Unterschied wird mit einheimischen Lipoprotein-Partikeln 19 erkannt). Die hohe Empfindlichkeit von qPCR ermöglicht es, diese Aufführungsrate zu ermitteln, indem die Anzahl der miRNA-Stränge nach der Inkubationszeit und dem miRNA/Partikelverhältnis gemessen wird. Es ist zu beachten, dass der berechnete Wert den zellulären Abbau und die Freisetzung von miRNA ignoriert und somit mindestens eine niedrigere Grenze für das Verhältnis der Lipoprotein-Partikel darstellt.
In Zukunft kann die Methode angepasst werden, um pharmazeutische Substanzen (insbesondere auch lipophile) in Zellen zu übertragen und ihre biologische Wirkung mit der intrazellulären Konzentration (die über die Auftaktrate von Lipoprotein-Partikeln bestimmt wird) zu korrelieren.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Unterstützt wurde diese Arbeit vom Wissenschaftsfonds Projekt P29110-B21, der Hochschuljubiläumsstiftung der Stadt Wien zur Förderung der Wissenschaft, dem Europäischen Fonds für regionale Entwicklung (EFRE, IWB2020), dem Land Oberer Österreich, und die "Land OÖ Basisfinanzierung".
Materialien
| Name | Company | Catalog Number | Comments |
| Ultracentrifuge | Beckman | Optima L-100 XP | Lipoprotein isolation |
| Ultracentrifuge rotor | Beckman | TI 55.2 | Lipoprotein isolation |
| Vacutainer (EDTA) | Becton Dickinson | 366643 | Lipoprotein isolation |
| Kaliumbromid | Sigma Aldrich | 2110 | Lipoprotein isolation |
| Ultracentrifuge tubes | Beckman | 342414 | Lipoprotein isolation |
| Ultracentrifuge tube sealer | Beckman | 342428 | Lipoprotein isolation |
| Sodiumchlorid | Carl Roth GmbH | P029.2 | Lipoprotein isolation |
| EDTA | Fisher Scientific | D/0650/50 | Lipoprotein isolation |
| Dialysis tubes, MWCO 12 - 14k | Spectra | 132700 | Lipoprotein isolation |
| synthetic miRNA | microSynth | - | synthetic miRNA |
| TRIS buffer pH 7 | Ambion | AM9850G | synthetic miRNA |
| TRIS buffer pH 8 | Ambion | AM9855G | synthetic miRNA |
| sterile tubes | Carl Roth GmbH | ENE8.1 | synthetic miRNA |
| Photometer | Eppendorf | Eppendorf Biophotometer | Bradford assay |
| Cuvette | Carl Roth GmbH | XK20 | Bradford assay |
| Coomassie G-250 Stain | BioRad GmbH | 161-0786 | Bradford assay |
| Sodiumchlorid | Carl Roth GmbH | 3957.1 | Delipitation |
| EDTA | Fluka Analytical | 03690-100ML | Delipitation |
| TRIS buffer pH 8.0 | Ambion | AM9855G | Delipitation |
| Ethanol 100% | AustrAlco | Ethanol Absolut 99.9% | Delipitation |
| Diethyl ether | Carl Roth GmbH | 3942.1 | Delipitation |
| Centrifuge | Thermo Scientific | Multifuge X3R | Delipitation |
| Chloroform | Carl Roth GmbH | 3313.1 | Reconstitution |
| Methanol | Carl Roth GmbH | 4627.1 | Reconstitution |
| Phosphatidylcholine | Sigma Aldrich GmbH | P3556-25mg | Reconstitution |
| Cholesterol oleate | Sigma Aldrich GmbH | C-9253-250mg | Reconstitution |
| cholesterol | Sigma Aldrich GmbH | C8667-500mg | Reconstitution |
| glass tubes | Carl Roth GmbH | K226.1 | Reconstitution |
| spermine | Sigma Aldrich GmbH | S3256-1G | Reconstitution |
| Thermomixer | Eppendorf | Thermomixer comfort | Reconstitution |
| Sodium deoxycholate | Sigma Aldrich GmbH | 3097-25G | Reconstitution |
| Magnetic stir bar | Carl Roth GmbH | 0955.2 | Reconstitution |
| 100µL micropipettes | Carl Roth GmbH | A762.1 | Reconstitution |
| PBS | Carl Roth GmbH | 9143.2 | Dialysis |
| Amberlite XAD-2 beads | Sigma Aldrich GmbH | 10357 | Dialysis |
| Slide-A-Lyzer casette (cut-off 20 kDa) | Thermo Scientific | 66003 | Dialysis |
| Syringe | Braun | 9161406V | Dialysis |
| Syringe needle | Braun | 465 76 83 | Dialysis |
| EGTA | Carl Roth GmbH | 3054.2 | Labeling of LDL particles |
| DMSO | life technologies | D12345 | Labeling of LDL particles |
| Atomic Force Microscope | RIBM | SS-NEX | Quality control of reconstituted/labeled lipoprotein particles |
| Muscovite Mica (V-1 Grade) | Christine Gröpl | G250-1/V1 | Quality control of reconstituted/labeled lipoprotein particles |
| AFM Cantilever | Nanoworld | USC-F1.2-k0.15 | Quality control of reconstituted/labeled lipoprotein particles |
| Gwyddion 2.49 | Czech Metrology Institute | http://gwyddion.net | Quality control of reconstituted/labeled lipoprotein particles |
| Chamber slides, Nunc Lab-Tek | VWR | 734-2122 | Cell culture |
| HBSS | Carl Roth GmbH | 9117.1 | Cell culture |
| Cell counter | Omni Life Science GmbH | Casy | Cell culture |
| miRNeasy Mini Kit | QIAGEN | 217004 | miRNA extraction |
| Centrifuge | Eppendorf | 5415R | miRNA extraction |
| 20G needle | Braun | Sterican 465 75 19 | miRNA extraction |
| 5ml syringe | Becton Dickinson | 309050 | miRNA extraction |
| PCR cabinet | Esco | PCR-3A1 | Reverse Transcription |
| TaqMan Reverse Transcription Kit | Thermo Scientific | 4366596 | Reverse Transcription |
| Rnase-free water | Carl Roth GmbH | T143 | Reverse Transcription |
| 0.2 ml tubes | Brand | 781305 | Reverse Transcription |
| Centrifuge | Carl Roth GmbH | Microcentrifuge AL | Reverse Transcription |
| Thermocycler | Sensoquest | Labcycler | Reverse Transcription |
| TaqMan Primer | Thermo Scientific | - | qPCR |
| iTaq Universal probe supermix | BioRad GmbH | 1725131 | qPCR |
| PCR machine | Corbett | Rotor-Gene RG-6000 | qPCR |
| TaqMan MicroRNA Reverse Transcription Kit | Applied Biosystems | 4366596 | TaqMan Array |
| Megaplex RT Primers, Human Pool A v2.1 | Applied Biosystems | 4399966 | TaqMan Array |
| TaqMan PreAmp Master Mix | Applied Biosystems | 4391128 | TaqMan Array |
| Megaplex PreAmp Primers, Human Pool A v2.1 | Applied Biosystems | 4399933 | TaqMan Array |
| TaqMan Universal Master Mix II, no UNG | Applied Biosystems | 4440040 | TaqMan Array |
| TaqMan Advanced miRNA Human A Cards | Applied Biosystems | A31805 | TaqMan Array |
| Nuclease-free water | Thermo Scientific | R0581 | TaqMan Array |
| MgCl2 (25mM) | Thermo Scientific | R0971 | TaqMan Array |
| TE, pH 8.0 | Invitrogen | AM9849 | TaqMan Array |
| 7900HT Fast Real-Time PCR System | Applied Biosystems | 4351405 | TaqMan Array |
| TaqMan Array Card Sealer | Applied Biosystems | - | TaqMan Array |
Referenzen
- Vickers, K. C., Palmisano, B. T., Shoucri, B. M., Shamburek, R. D., Remaley, A. T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature Cell Biology. 13 (4), 423-435 (2011).
- Tabet, F., et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nature Communications. 5, (2014).
- Wahid, F., Shehzad, A., Khan, T., Kim, Y. Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochimica et Biophysica Acta - Molecular Cell Research. 1803 (11), 1231-1243 (2010).
- Carthew, R. W., Sontheimer, E. J. Origins and Mechanisms of miRNAs and siRNAs. Cell. 136 (4), 642-655 (2009).
- Fabian, M. R., Sonenberg, N., Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annual Review of Biochemistry. 79 (1), 351-379 (2010).
- Filipowicz, W., Bhattacharyya, S. N., Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight?. Nature Reviews Genetics. 9 (2), 102-114 (2008).
- Jonas, A. Reconstitution of High-Density Lipoproteins. Methods in Enzymology. , 553-582 (1986).
- Ando, T., Uchihashi, T., Kodera, N. High-Speed AFM and Applications to Biomolecular Systems. Annual Review of Biophysics. 42 (1), 393-414 (2013).
- Bustin, S. A., et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry. 55 (4), 611-622 (2009).
- Patsch, J. R., Patsch, W. Zonal ultracentrifugation. Methods in Enzymology. 129 (1966), 3-26 (1986).
- Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 72 (1-2), 248-254 (1976).
- Yamamoto, D., et al. High-Speed Atomic Force Microscopy Techniques for Observing Dynamic Biomolecular Processes. Methods in Enzymology. 475, 541-564 (2010).
- Stangl, H., Cao, G., Wyne, K. L., Hobbs, H. H. Scavenger receptor, class B, type I-dependent stimulation of cholesterol esterification by high density lipoproteins, low density lipoproteins, and nonlipoprotein cholesterol. Journal of Biological Chemistry. 273 (47), 31002-31008 (1998).
- Hochberg, B. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 57 (1), 289-300 (1995).
- Mestdagh, P., et al. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biology. 10 (6), (2009).
- Schumaker, V. N., Puppione, D. L. 6] Sequential Flotation Ultracentrifugation. Methods in Enzymology. 128 (C), 155-170 (1986).
- Wagner, J., et al. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arteriosclerosis, Thrombosis, and Vascular Biology. 33 (6), 1392-1400 (2013).
- Meier, S. M., et al. Effect of chronic kidney disease on macrophage cholesterol efflux. Life Sciences. 136, 1-6 (2015).
- Axmann, M., et al. Serum and Lipoprotein Particle miRNA Profile in Uremia Patients. Genes. 9 (11), 533 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten