
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
In vivo Bildgebung von vollaktivem Hirngewebe bei wachen Zebrafischlarven und Jungtieren durch Schädel- und Hautentfernung
In diesem Artikel
Zusammenfassung
Hier stellen wir eine Methode vor, um das embryonale Gehirn des Zebrafischs in vivo bis hin zu Larven- und Juvenilenstadien abbilden zu können. Dieses mikroinvasive Verfahren, das von elektrophysiologischen Ansätzen adaptiert wurde, bietet Zugang zu zellulären und subzellulären Details reifer Neuronen und kann mit optogenetischen und neuropharmakologischen Studien zur Charakterisierung der Gehirnfunktion und der medikamentösen Intervention kombiniert werden.
Zusammenfassung
Das Verständnis der ephemeren Veränderungen, die während der Entwicklung und Reifung des Gehirns auftreten, erfordert eine detaillierte hochauflösende Bildgebung in Raum und Zeit mit zellulärer und subzellulärer Auflösung. Fortschritte in molekularen und bildgebenden Technologien haben es uns ermöglicht, zahlreiche detaillierte Einblicke in zelluläre und molekulare Mechanismen der Gehirnentwicklung im transparenten Zebrafischembryo zu gewinnen. In jüngster Zeit sind Prozesse der Verfeinerung neuronaler Konnektivität, die in späteren Larvenstadien mehrere Wochen nach der Befruchtung auftreten, die beispielsweise die Kontrolle des Sozialverhaltens, die Entscheidungsfindung oder motivationsgetriebenes Verhalten sind, in den Fokus der Forschung gerückt. In diesen Stadien stört die Pigmentierung der Zebrafischhaut das Eindringen von Licht in das Hirngewebe, und Lösungen für Embryonalstadien, z. B. pharmakologische Hemmung der Pigmentierung, sind nicht mehr realisierbar.
Daher wird eine minimalinvasive chirurgische Lösung für den mikroskopischen Zugang zum Gehirn von wachen Zebrafischen bereitgestellt, die aus elektrophysiologischen Ansätzen abgeleitet ist. Bei Teleosten können Haut- und weichschädeliger Knorpel vorsichtig entfernt werden, indem diese Schichten mikroschält werden, wodurch darunter liegende Neuronen und axonale Bahnen ohne Schäden freigelegt werden. Dies ermöglicht die Erfassung der neuronalen Morphologie, einschließlich synaptischer Strukturen und ihrer molekularen Inhalte, sowie die Beobachtung physiologischer Veränderungen wie Ca2+ Transienten oder intrazelluläre Transportereignisse. Darüber hinaus ist eine Abfrage dieser Prozesse mittels pharmakologischer Hemmung oder optogenetischer Manipulation möglich. Dieser Gehirnexpositionsansatz liefert Informationen über strukturelle und physiologische Veränderungen in Neuronen sowie die Korrelation und Interdependenz dieser Ereignisse im lebenden Hirngewebe im Bereich von Minuten oder Stunden. Die Technik eignet sich für die In-vivo-Bildgebung von Zebrafischlarven bis zu 30 Tage nach der Befruchtung, dem neuesten bisher getesteten Entwicklungsstadium. Es bietet somit Zugang zu so wichtigen Fragen wie synaptischer Verfeinerung und Skalierung, axonaler und dendritischer Transport, synaptisches Targeting von Zytoskelettladung oder lokale aktivitätsabhängige Expression. Daher ist mit einem breiten Einsatz für diesen Montage- und Imaging-Ansatz zu rechnen.
Einleitung
Der Zebrafisch (Danio rerio) hat sich in den letzten Jahrzehnten zu einem der beliebtesten Wirbeltiermodellorganismen für Embryonal- und Larvenentwicklungsstudien entwickelt. Die große Fruchtbarkeit der Zebrafischweibchen gepaart mit der schnellen Ex-utero-Entwicklung des Embryos und seiner Transparenz in frühen embryonalen Entwicklungsstadien sind nur einige Schlüsselfaktoren, die Zebrafische zu einem leistungsfähigen Modellorganismus für adressierte Entwicklungsfragen machen1. Fortschritte in molekulargenetischen Technologien in Kombination mit hochauflösenden In-vivo-Bildgebungsstudien ermöglichten es, zellbiologische Mechanismen anzugehen, die Entwicklungsprozessen zugrundeliegen 2. Insbesondere auf dem Gebiet der neuronalen Differenzierung, Physiologie, Konnektivität und Funktion hat Zebrafisch das Zusammenspiel von molekularer Dynamik, Gehirnfunktionen und organismischem Verhalten in bisher unerreichter Detailgenauigkeit beleuchtet.
Die meisten dieser Studien beschränken sich jedoch auf embryonale und frühe Larvenstadien in der ersten Entwicklungswoche, da die Transparenz des Gewebes des Nervensystems allmählich verloren geht. In diesen Stadien wird hirngewebe durch hochauflösende Mikroskopieansätze am Zugang gehindert, die durch Schädeldifferenzierung und Pigmentierung abgeschirmt werden3.
Daher sind Schlüsselfragen der neuronalen Differenzierung, Reifung und Plastizität wie die Verfeinerung der neuronalen Konnektivität oder die synaptische Skalierung schwer zu untersuchen. Diese zellulären Prozesse sind wichtig, um zelluläre Mechanismen zu definieren, die beispielsweise sozialverhalten, Entscheidungsfindung oder motivationsbasiertes Verhalten antreiben, Bereiche, zu denen die Zebrafischforschung an mehreren Wochen alten Larven kürzlich wichtige Erkenntnisse auf der Grundlage von Verhaltensstudien beigetragen hat4.
Pharmakologische Ansätze zur Hemmung der Pigmentierung bei Zebrafischlarven über mehrere Wochen sind kaum realisierbar oder können sogar schädliche Auswirkungen haben5,6,7,8. Doppel- oder dreifach mutierte Stämme mit spezifischen Pigmentdefekten, wie Casper9 oder Crystal10, sind zu enorm wertvollen Werkzeugen geworden, sind aber mühsam in der Zucht, liefern wenig Nachkommen und stellen die Gefahr dar, genetische Fehlbildungen aufgrund übermäßiger Inzucht anzuhäufen.
Hier ist alternativ ein minimalinvasives Verfahren vorgesehen, das auf jeden Zebrafischstamm anwendbar ist. Dieses Verfahren wurde aus elektrophysiologischen Studien adaptiert, um die neuronale Aktivität bei lebenden und wachen Zebrafischlarven aufzuzeichnen. Bei Teleosten können Haut und weicher Schädelknorpel durch Mikropeeling dieser Schichten vorsichtig entfernt werden, da sie nicht eng mit dem Gehirngefäß verwoben sind. Dies ermöglicht es, Hirngewebe, das Neuronen und axonale Bahnen enthält, ohne Schädigung freizulegen und die neuronale Morphologie einschließlich synaptischer Strukturen und deren molekularem Inhalt zu erfassen, was wiederum die Beobachtung physiologischer Veränderungen wie Ca2+ Transienten oder intrazellulärer Transportereignisse für bis zu mehreren Stunden umfasst. Darüber hinaus ermöglicht der direkte Zugang zum Hirngewebe über deskriptive Charakterisierungen hinaus die Abfrage reifer neuronaler Funktionen mittels neuropharmakologischer Substanzgabe und optogenetischer Ansätze. Daher können mit dieser Gehirnexpositionsstrategie wahre Strukturfunktionsbeziehungen im jugendlichen Zebrafischgehirn aufgedeckt werden.
Access restricted. Please log in or start a trial to view this content.
Protokoll
Alle hier beschriebenen Tierarbeiten entsprechen den gesetzlichen Bestimmungen (EU-Richtlinie 2010/63). Die Pflege und Handhabung von Fischen wurde von den örtlichen Behörden und vom Tierschutzbeauftragten der Technischen Universität Braunschweig genehmigt.
1. Herstellung von künstlicher Zerebro-Rückenmarksflüssigkeit (ACSF), niedrigschmelzender Agarose und scharfen Glasnadeln
- Bereiten Sie die ACSF vor, indem Sie die aufgeführten Chemikalien in folgenden Konzentrationen in destilliertem Wasser auflösen. 134 mM NaCl (58,44 g/mol), 2,9 mM KCl (74,55 g/mol), 2,1 mM CaCl2 (110,99 g/mol), 1,2 mM MgCl2 6xH2O (203,3 g/mol), 10 mM HEPES (238,31 g/mol) und 10 mM d-Glucose (180,16 g/mol).
HINWEIS: FürMgCl2,CaCl2und KCl werden 1 M Stammlösungen in entsalzenem sterilem Wasser hergestellt und bei 4 °C gelagert, um anschließend frisches ACSF zuzubereiten. Glukose, HEPES und NaCl werden als feste Verbindungen in der frischen ACSF-Lösung gelöst. Zum Auflösen von Chemikalien befolgen Sie die Anweisungen des Herstellers. - Stellen Sie den pH-Wert des ACSF auf 7,8 mit 10 M NaOH ein. Die Herstellung von ACSF erfordert eine präzise Messung von Chemikalien und eine Feineinstellung des pH-Werts, da sie die Zerebro-Rückenmarksflüssigkeit ersetzt und die physiologischen Bedingungen aufrechterhält, die für die volle Funktionsfähigkeit der Neuronen erforderlich sind, da sie sonst zu Fehlfunktionen des Gehirns und neuronalem Tod führen kann.
- Lagern Sie den frisch zubereiteten ACSF bei 4 °C für maximal 4 Wochen. Für die Arbeitsbedingungen das erforderliche ACSF-Volumen für den Tag/das Experiment aliquotieren und bei 25-28 °C vorwärmen (und gegebenenfalls mit Sauerstoff versorgen, Schritt 2.5)
HINWEIS: Frisch zubereiteter ASCF ist für 1 Tag in Ordnung. Wenn Sie planen, es über mehrere Tage zu verwenden, muss ACSF steril gefiltert werden. - Für eine spätere Anästhesie der Larven eine 50 mM Stammlösung von d-Tubocurarin in destilliertem Wasser vorbereiten und die Lösung bei -20 °C als 100 μL Aliquots im Gefrierschrank lagern, bis sie benötigt wird.
- Um den Fisch einzubetten, bereiten Sie 2,5% niedrigschmelzende (LM) Agarose vor, indem Sie 1,25 g LM-Agarose (Table of Materials) in 50 mL ACSF auflösen und kochen, bis die Agarose vollständig gelöst ist.
HINWEIS: Alternativ können je nach Versuchsaufbau höhere oder niedrigere Konzentrationen von LM-Agarose verwendet werden. Wenn die Agarose jedoch zu weich ist, kann sie den Fisch beim Öffnen des Schädels nicht in Position halten. - Lagern Sie die Agarose bei 37 °C Wasserbad, um eine Erstarrung zu vermeiden und weil diese Temperatur den Larven auch beim Einbetten nicht schadet. Nachdem die gekochte Agarose im Wasserbad auf 37 °C abgekühlt ist, fügen Sie die notwendige Menge d-Tubocurarin zu der aliquoteierten Agarose hinzu, die für den Tag benötigt wird, um eine Arbeitskonzentration von 10 μM zu erreichen. Für den zukünftigen Gebrauch die übrig gebliebene Agarose bei 4 °C lagern, um eine Kontamination zu vermeiden.
- Bereiten Sie scharfe und dünne Glasnadeln aus Glaskapillaren vor (Ergänzende Abbildung 1) mit einem Mikropipettenzieher mit den folgenden Einstellungen.
- Puller I, Kapillartyp 1: Heat 1: 65,8; Hitze 2: 55,1; 2-stufiges Ziehen
Puller II, Kapillartyp 2: Wärme = 700; Fil = 4; Vel = 55; Entf = 130; Pul = 55; 1-stufiges Ziehen.
HINWEIS: Die Einheiten sind spezifisch für jeden hier verwendeten Abzieher bzw. jede Glaskapillare (siehe Materialtabelle). Andere Kapillaren und Puller können auch verwendet werden, um die Glasnadeln vorzubereiten. Die Glasnadeln sollten jedoch nicht zu dünn sein, da sie bei Kontakt mit dem Schädel brechen können. Kapillare: Länge: 100 mm (4 Zoll); OD: 1,5 mm; Kennung: 0,84 mm; Filament: Ja
- Puller I, Kapillartyp 1: Heat 1: 65,8; Hitze 2: 55,1; 2-stufiges Ziehen
2. Anästhesie von Larven und Präparate zur Einbettung
- Wenn Sie das Experiment für den Tag beginnen, übertragen Sie die Tiere, die mit einer Pasteur-Pipette aus Kunststoff benötigt werden, in eine Petrischale mit 90 mm Durchmesser, die entweder mit Danieau (für Larven, die noch in einer Petrischale mit Danieau gehalten werden) oder Wasser aus der Fischanlage (für Larven, die älter als 7 dpf sind und in der Fischanlage gehalten werden) gefüllt ist.
- Wenn Sie Fische pipettieren, die älter als 2 Wochen sind, stellen Sie sicher, dass die Öffnung der Pipette groß genug ist, um zu vermeiden, dass die Fische beim Übertragen verletzt werden. Verwenden Sie kein Netz, da es vor allem die jüngeren Larven physisch schädigt.
- Fügen Sie Rotiferen oder Artemia-Nauplien hinzu, die für die Größe der in der Petrischale gehaltenen Larven geeignet sind, um den freien Zugang zu Nahrung und den maximalen Gesundheitszustand der Larven zu gewährleisten und Stress abzubauen.
- Zur Einbettung die ausgewählten Larven in eine mit ACSF gefüllte Petrischale mit 35 mm Durchmesser überführen. Fügen Sie das notwendige Volumen von d-Tubocurarin hinzu, um eine Arbeitskonzentration / effektive Dosis von 10 μM zu erreichen, und warten Sie einige Minuten, bis die Larven vollständig immobilisiert sind11.
HINWEIS: Wenn die Fische älter werden oder wenn eine schnellere Vollnarkose erforderlich ist (unter 5 min), ist es möglich, die Konzentration von d-Tubocurarin zu erhöhen (LD50 für Mäuse beträgt 0,13 mg / kg intravenös12). Es ist auch möglich, ein anderes Anästhetikum wie α-Bungarotoxin (Arbeitskonzentration: 1 mg / ml) zu verwenden, das die gleiche Wirkung wie Curare hat und auch das Gehirn voll aktiv hält13. Wenn ein voll aktives Gehirn für das interessierende Thema nicht notwendig ist, ist Tricain in einer nicht-tödlichen Dosis (0,02%) auch eine Option, um die Larven vollständig zu betäuben. Tricain blockiert jedoch Natriumkanäle und beeinträchtigt dadurch die Gehirnaktivität14. - Bereiten Sie die Montagekammer vor, indem Sie den Deckel der Petrischale mit 35 mm Durchmesser nehmen, den Deckel auf den Kopf stellen und einen quadratischen Glasdeckel (24 x 24 mm) auf die Unterseite des Deckels legen. Eine schematische Beschreibung dieser Schritte finden Sie in Abbildung 1 (oberer Teil). Die glattere Oberfläche des Glases verhindert ein Abrutschen des Agaroseblocks, der die Larven während des Schädelöffnungsvorgangs enthält.
- Aliquotieren Sie die für den Tag benötigte Menge an ACSF in einer geeigneten Durchstechflasche (z. B. 50 ml Tube, Becherglas, Schottflasche usw.) und mit Carbogen (5% CO2,95% O2)mit Sauerstoff. Wenn nur Morphologie (z. B. Fluoreszenzmuster) abbildet, ist ACSF immer noch notwendig, um die Integrität des Gehirns zu gewährleisten und dass Zellen nicht negativ durch Osmolaritätseffekte beeinflusst werden, sondern keine Sauerstoffversorgung des ACSF erforderlich ist. Dieser Schritt muss nur durchgeführt werden, wenn die volle Gehirnaktivität für die Bildgebung erforderlich ist.
HINWEIS: Für eine optimale Sauerstoffsättigung des Mediums fügen Sie einen Luftstein am Ende des Carbogenrohrs hinzu. Um einen ausreichend hohen Sauerstoffgehalt zu gewährleisten, ist es notwendig, den ACSF in den Bildgebungskammern alle 20-60 Minuten mit frisch sauerstoffreichem ACSF auszutauschen, abhängig von der Anzahl und dem Alter der Larven, die in dieselbe Bildgebungskammer eingebettet sind (z. B. für eine einzelne eingebettete Larve ist acSF-Austausch jede Stunde ausreichend). Für sechs Larven, die älter als 14 dpf sind, die parallel eingebettet sind, ist ein Austausch von ACSF alle 20 Minuten erforderlich), also planen Sie die notwendige Menge an sauerstoffsättigten ACSF gemäß dem geplanten Experiment.
3. Einbettung der Larven
- Die voll betäubten Larven mit einer Kunststoff-Pasteur-Pipette in die (in Schritt 2.4) vorbereitete Montagekammer geben. Entfernen Sie dann vorsichtig das überschüssige Medium, um eine Verdünnung der LM-Agarose zu vermeiden. Alle folgenden Schritte sollten unter einem Stereomikroskop mit ausreichender Vergrößerung durchgeführt werden.
HINWEIS: Das Kippen der Montagekammer kann helfen, das Medium vollständig zu entfernen. - Fahren Sie sofort mit dem nächsten Schritt fort, indem Sie einen ausreichend großen LM-Agarose-Tropfen auf die Larven (ca. 1 ml, abhängig von der Größe der Larven) geben, um die Tiere vor dem Austrocknen zu schützen und unnötigen Stress zu reduzieren.
- Orientieren Sie die Larven in Position, bevor die Agarose erstarrt. Stellen Sie sicher, dass der dorsale Teil der Larven nach oben gerichtet ist. Achten Sie auch darauf, die Larven so nah wie möglich an der Oberfläche der Agarose einzubetten.
HINWEIS: Abhängig von der Größe und Anzahl der Larven, die gleichzeitig eingebettet werden sollen, ist es möglich, die Agarosekonzentration anzupassen. Zum Beispiel wird für 1-3 Larven, die 30 dpf alt sind, eine Konzentration von 1,8% -2% LM-Agarose empfohlen. Für 1-4 Larven, die 7 dpf alt sind, ist es am praktikabelsten, 2,5% LM-Agarose zu verwenden, während für 5-8 Larven 2% besser geeignet sind. Wenn ein voll aktives Gehirn benötigt wird, wird empfohlen, nur drei Fische gleichzeitig einzubetten, um die Zeit für die Arbeit der Larven zu reduzieren. Generell wird empfohlen, niedrigere Konzentrationen (1,8%-2%) zu verwenden, je älter die Larven werden oder je mehr Larven gleichzeitig eingebettet werden sollen. - Wenn Bilder mit einem invertierten Mikroskop aufgenommen werden, schneiden Sie den Agaroseblock, der die Larven enthält, in eine kleine Quaderform. Dies ist wichtig, um die Larven später in die Bildkammer zu übertragen. Bei Verwendung eines aufrechten Mikroskops ist ein solches Trimmen nicht notwendig, da die Montagekammer auch als Bildkammer verwendet werden kann. In Abbildung 1 (oberer Teil) finden Sie eine schematische Beschreibung dieser Schritte.
4. Das Gehirn entlarven
HINWEIS: Alle folgenden Schritte sollten mit größter Sorgfalt durchgeführt werden, um die Larven nicht unnötig zu verletzen. Wenn ein voll aktives Gehirn für das Experiment erforderlich ist, denken Sie daran, dass mit jeder Sekunde, die vergeht, während der Fisch noch vollständig in Agarose montiert ist und einen offenen Schädel ohne sauerstoffreiche ACSF hat, das Gehirn unter Sauerstoffmangel leidet und auch austrocknet. Die Auswirkungen von Sauerstoffmangel werden noch dramatischer, je älter die eingebetteten Larven sind. Daher ist es wichtig, die Operation nicht nur innerhalb kürzester Zeit, sondern auch mit maximaler Präzision durchzuführen, um keine mechanischen Hirnschäden mit der Nadel hervorzurufen. Wenn trainiert, sollten die Schritte 4.2-4.4 nicht mehr als 30 s pro Fisch dauern.
- Beginnen Sie mit der Operation, sobald sich die Agarose verfestigt hat. Schneiden Sie zunächst die gesamte überschüssige Agarose über der interessierenden Gehirnregion ab, um freien Zugang zum Kopf und einen klaren Arbeitsbereich zu erhalten. Wenn der dorsale Teil des Kopfes bereits aus der Agarose herausragt, überspringen Sie diesen Schritt.
- Wählen Sie je nach Interessenregion einen Platz, um mit der Operation zu beginnen. Nehmen Sie die Glasnadel und machen Sie einen kleinen Schnitt durch die Haut, ohne jedoch zu tief in das Gewebe einzudringen. Dies ist der Ausgangspunkt für das Abziehen der überlagernden Haut.
HINWEIS: Für optimale Ergebnisse sollten Sie niemals direkt über dem interessierenden Bereich beginnen, um das Risiko einer Beschädigung wichtiger Strukturen zu verringern. Bei Bedarf ist es möglich, sogar nach vorne bis zum Hinterhirn zu beginnen und von dort aus vorwärts zu arbeiten, bis der unerwünschte Hautbereich abgezogen ist. - Fahren Sie mit sehr kleinen Schnitten entlang des Hautteils fort, die darauf abzielen, die Nadel zu entfernen, indem Sie die Nadel kaum direkt unter der Oberfläche bewegen. Meistens ist es nicht notwendig, sich vollständig um das Gehirn zu bewegen und ein kreisartiges Stück Haut und Schädel auszuschneiden, sondern nur zwei Schnitte entlang des Kopfes zu machen und dann die Haut auf die eine oder andere Seite wegzudrücken. Abbildung 2 zeigt eine schematische Darstellung der optimalen Schneidstrategie, um freien Zugang zum Kleinhirn zu erhalten.
HINWEIS: Diese Mikrooperation ist ein heikles Verfahren und es wird höchstwahrscheinlich etwas Training benötigen, um die Haut perfekt zu entfernen, ohne das darunter liegende Gehirn zu schädigen. Es wird auch empfohlen, die optimale Schnittstrategie für die interessierende Gehirnregion herauszufinden und für die Dauer des Experiments dabei zu bleiben. - Unmittelbar nachdem Sie die Haut von allen eingebetteten Larven entfernt haben, gießen Sie (sauerstoffhaltiges) ACSF über die Agarose, um unerwünschte Hautpartikel und Blut zu entfernen und das Gehirn voll aktiv zu halten und es vor dem Austrocknen zu schützen.
HINWEIS: Wenn für das Experiment ein gesundes Gehirn benötigt wird, wird empfohlen, maximal drei Fische gleichzeitig zu essen. - Wenn Sie ein aufrechtes Mikroskop verwenden, beginnen Sie direkt mit der Bildgebung.
- Wenn Sie ein invertiertes Mikroskop verwenden, schieben Sie einen kleinen Spatel unter den quaderförmigen Agaroseblock (Schritt 3.4).
- Fügen Sie einen kleinen Tropfen LM-Agarose auf den Boden der Bildkammer (z. B. Glasbodenschale) hinzu und drehen Sie den Agaroseblock, der die Larven enthält, sofort mit dem Spatel um 180 ° um und drücken Sie ihn vorsichtig auf den Boden der Bildkammer, während der flüssige Agarosetropfen als Klebstoff wirkt.
- Wenn die Agarose erstarrt ist, füllen Sie die Bildgebungskammer mit (sauerstoffhaltigem) ACSF und beginnen Sie dann mit der Bildgebung. Eine schematische Beschreibung finden Sie in Abbildung 1 (unterer Teil).
- Wenn für das Experiment volle Gehirnaktivität erforderlich ist, stellen Sie immer sicher, dass ACSF in der Bildgebungskammer einen ausreichend hohen Sauerstoffgehalt auftut. Um dies zu gewährleisten, tauschen Sie das Medium nach Möglichkeit alle 20-60 Minuten vorsichtig mit frisch sauerstoffreichem ACSF aus (abhängig von der Anzahl und Größe des Fisches, der Größe und Oberfläche der Bildgebungskammer und der Bildgebungsdauer).

Abbildung 1: Schematisches Verfahren zur schrittweisen Vorbereitung von Zebrafischen mit offenem Schädel für die In-vivo-Bildgebung. Die Arbeitsanweisungen für die verschiedenen Schritte finden Sie in der Grafik selbst. Grafik gestaltet von Florian Hetsch und adaptiert von Paul Schramm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Detaillierte schematische Darstellung der Mikrochirurgie, die durchgeführt wurde, um Haut- und Schädelstücke über der interessierenden Hirnregion zu entfernen. Der rote Kreis markiert die Stelle, an der der erste Schnitt gemacht werden muss. Die rot gepunktete Linie beschreibt den optimalen Weg, um zusammen mit der Nadel zu schneiden, um freien Zugang zum Kleinhirn zu erhalten, ohne es zu beschädigen. Der grüne Pfeil markiert die Richtung, in die die überschüssigen Haut- und Schädelstücke leicht weggeschoben werden können. Stellen Sie sicher, dass Sie während des gesamten Eingriffs niemals in das Gehirngewebe eindringen. Nach erfolgreichem Abschälen der Haut wird die interessierende Hirnregion (hier Kleinhirn) für jede Art von hochauflösender In-vivo-Bildgebung frei zugänglich sein. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
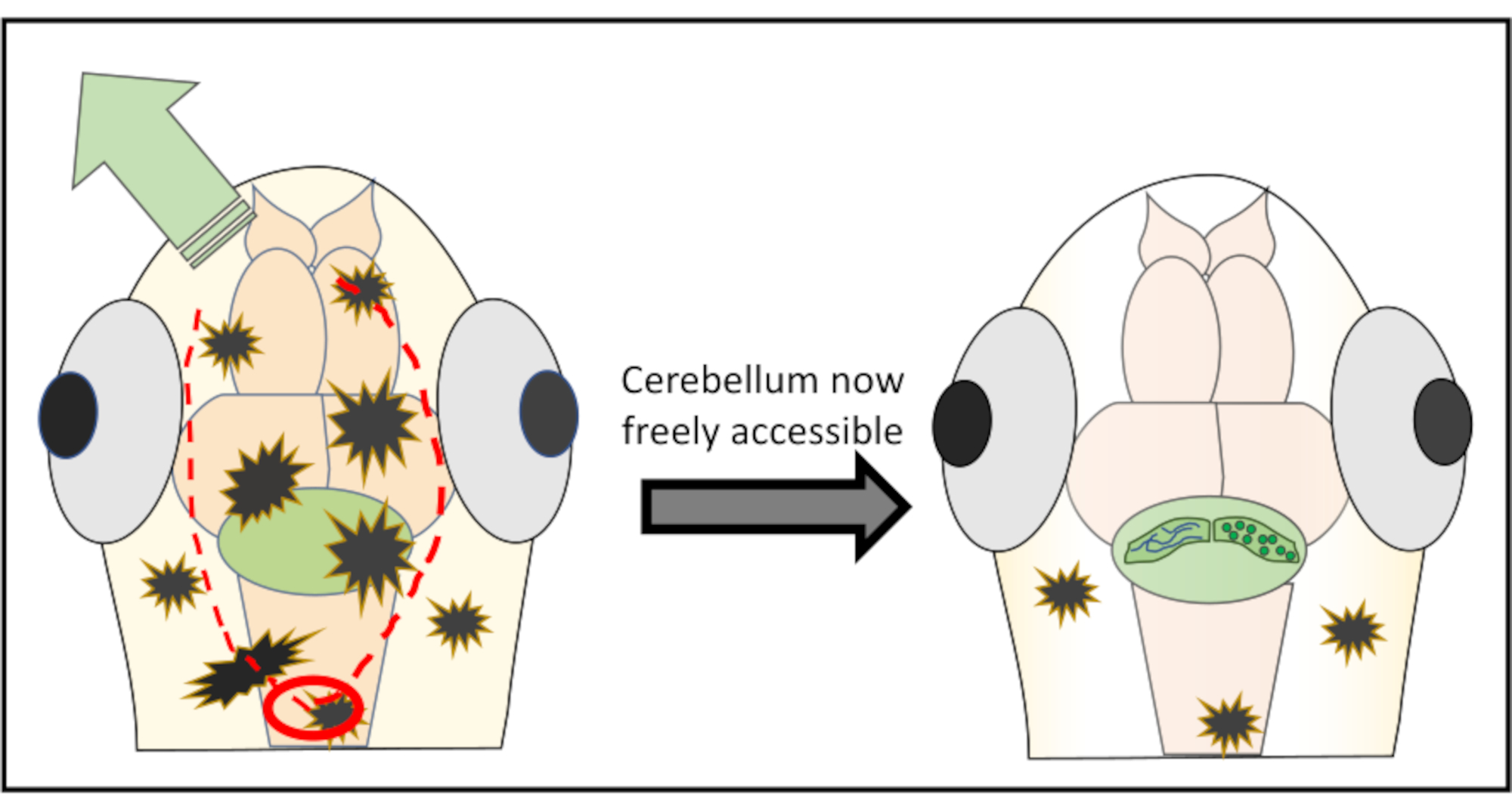{kind=link}
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Abbildung 3A,C zeigen eine 14 dpf Larve der transgenen Linie Tg[-7.5Ca8:GFP]bz12[15] mit dem Schädel noch intakt. Die Pigmentzellen in der überlagernden Haut sind über den ganzen Kopf verteilt und stören das Fluoreszenzsignal im interessierenden Bereich (hier Kleinhirn). Mit der Larve in diesem Zustand ist es nicht möglich, hochauflösende Bilder des Gehirns zu erhalten. ...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Die vorgestellte Methode bietet einen alternativen Ansatz zur Gehirnisolierung oder zur Behandlung von Zebrafischlarven mit pigmenthemmenden Arzneimitteln zur Aufnahme hochauflösender Bilder von Neuronen in ihrer In-vivo-Umgebung. Die Qualität der mit dieser Methode aufgenommenen Bilder ist vergleichbar mit Bildern aus explantierten Gehirnen, jedoch unter natürlichen Bedingungen.
Weiterhin wird ein Intensitätsverlust der Fluoreszenz vermieden, da keine Behandlung mit Fixiermitteln...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Wir danken insbesondere Timo Fritsch für die hervorragende Tierpflege und Hermann Döring, Mohamed Elsaey, Sol Pose-Méndez, Jakob von Trotha, Komali Valishetti und Barbara Winter für die hilfreiche Unterstützung. Wir danken auch allen anderen Mitgliedern des Köster-Labors für ihr Feedback. Das Projekt wurde zum Teil von der Deutschen Forschungsgemeinschaft (DFG, KO1949/7-2) gefördert, 241961032 (zu RWK) und dem Bundesministerium für Bildung und Forschung (BMBF; Era-Net NEURON II CIPRESS Projekt 01EW1520 zu JCM) wird anerkannt.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Calcium chloride | Roth | A119.1 | |
| Confocal Laser scanning microscope | Leica | TCS SP8 | |
| d-Glucose | Sigma | G8270-1KG | |
| d-Tubocurare | Sigma-Aldrich | T2379-100MG | |
| Glass Capillary type 1 | WPI | 1B150F-4 | |
| Glass Capillary type 2 | Harvard Apparatus | GC100F-10 | |
| Glass Coverslip | deltalab | D102424 | |
| HEPES | Roth | 9105.4 | |
| Hoechst 33342 | Invitrogen (Thermo Fischer) | H3570 | |
| Imaging chamber | Ibidi | 81156 | |
| Potassium chloride | Normapur | 26764298 | |
| LM-Agarose | Condalab | 8050.55 | |
| Magnesium chloride (Hexahydrate) | Roth | A537.4 | |
| Microscope Camera | Leica | DFC9000 GTC | |
| Needle-Puller type 1 | NARISHIGE | Model PC-10 | |
| Needle-Puller type 2 | Sutter Instruments | Model P-2000 | |
| Pasteur-Pipettes 3ml | A.Hartenstein | 20170718 | |
| Sodium chloride | Roth | P029.2 | |
| Sodium hydroxide | Normapur | 28244262 | |
| Tricain | Sigma-Aldrich | E10521-50G | |
| Waterbath | Phoenix Instrument | WB-12 | |
| 35 mm petri dish | Sarstedt | 833900 | |
| 90 mm petri dish | Sarstedt | 821473001 |
Referenzen
- Hill, M. A. Embryology. Zebrafish Development. , Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Zebrafish_Development (2020).
- Sassen, W. A., Köster, R. W. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. Dove Medical Press. 2015 (5), 151-163 (2015).
- Singh, A. P., Nüsslein-Volhard, C. Zebrafish stripes as a model for vertebrate colour pattern formation. Current Biology. 25 (2), 81-92 (2015).
- Kalueff, A. V., et al. Time to recognize zebrafish 'affective' behavior. Brill: Behaviour. 149 (10-12), 1019-1036 (2012).
- Karlsson, J., von Hofsten, J., Olsson, P. -E. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology. 3, 522-527 (2001).
- Bohnsack, B. L., Gallina, D., Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PloS One. 6, 22991(2011).
- Elsalini, O. A., Rohr, K. B. Phenylthiourea disrupts thyroid function in developing zebrafish. Development Genes and Evolution. 212, 593-598 (2003).
- Baumann, L., Ros, A., Rehberger, K., Neuhauss, S. C. F., Segner, H. Thyroid disruption in zebrafish (Danio rerio) larvae: Different molecular response patterns lead to impaired eye development and visual functions. Aquatic Toxicology. 172, 44-55 (2016).
- White, R., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2, 183-189 (2008).
- Antinucci, P., Hindges, R. A crystal-clear zebrafish for in vivo imaging. Scientific Reports. 6, 29490(2016).
- Burr, S. A., Leung, Y. L. Curare (d-Tubocurarine). Encyclopedia of Toxicology (3rd Edition). , 1088-1089 (2014).
- Gesler, H. M., Hoppe, J. 3,6-bis(3-diethylaminopropoxy) pyridazine bismethiodide, a long-acting neuromuscular blocking agent. The Journal of Pharmacology and Experimental Therapeutics. 118 (4), 395-406 (1956).
- Furman, B. Alpha Bungarotxin. Reference Module in Biomedical Sciences. , (2018).
- Attili, S., Hughes, S. M. Anaesthetic tricaine acts preferentially on neural voltage-gated sodium channels and fails to block directly evoked muscle contraction. PLoS One. 9 (8), 103751(2014).
- Namikawa, K., et al. Modeling neurodegenerative spinocerebellar ataxia type 13 in zebrafish using a Purkinje neuron specific tunable coexpression system. Journal of Neuroscience. 39 (20), 3948-3969 (2019).
- Hennig, M. Theoretical models of synaptic short term plasticity. Frontiers in Computational Neuroscience. 7 (45), (2013).
- Wang, Y., et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 137, 3119-3128 (2010).
- Hobro, A., Smith, N. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Knogler, L. D., Kist, A. M., Portugues, R. Motor context dominates output from purkinje cell functional regions during reflexive visuomotor behaviours. eLife. 8, 42138(2019).
- Hsieh, J., Ulrich, B., Issa, F. A., Wan, J., Papazian, D. M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Frontier in Neural Circuits. 8 (147), (2014).
- Scalise, K., Shimizu, T., Hibi, M., Sawtell, N. B. Responses of cerebellar Purkinje cells during fictive optomotor behavior in larval zebrafish. Journal of Neurophysiology. 116 (5), 2067-2080 (2016).
- Harmon, T. C., Magaram, U., McLean, D. L., Raman, I. M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife. 6, 22537(2017).
- Zehendner, C. M., et al. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8 (12), 82823(2013).
- Dhabhar, F. S. The short-term stress response - mother nature's mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Frontiers of Neuroendocrinology. 49, 175-192 (2018).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten