
Method Article
Los enfoques genéticos y bioquímicos para
En este artículo
Resumen
Oligomerization of the ryanodine receptor, a homo-tetrameric ion channel mediating Ca2+ release from intracellular stores, is critical for skeletal and cardiac muscle contraction. Here, we present complementary in vivo and in vitro methods to detect protein self-association and determine homo-oligomer stoichiometry.
Resumen
Oligomerization is often a structural requirement for proteins to accomplish their specific cellular function. For instance, tetramerization of the ryanodine receptor (RyR) is necessary for the formation of a functional Ca2+ release channel pore. Here, we describe detailed protocols for the assessment of protein self-association, including yeast two-hybrid (Y2H), co-immunoprecipitation (co-IP) and chemical cross-linking assays. In the Y2H system, protein self-interaction is detected by β-galactosidase assay in yeast co-expressing GAL4 bait and target fusions of the test protein. Protein self-interaction is further assessed by co-IP using HA- and cMyc-tagged fusions of the test protein co-expressed in mammalian HEK293 cells. The precise stoichiometry of the protein homo-oligomer is examined by cross-linking and SDS-PAGE analysis following expression in HEK293 cells. Using these different but complementary techniques, we have consistently observed the self-association of the RyR N-terminal domain and demonstrated its intrinsic ability to form tetramers. These methods can be applied to protein-protein interaction and homo-oligomerization studies of other mammalian integral membrane proteins.
Introducción
La contracción del músculo esquelético y cardíaco se activa por retículo sarcoplásmico Ca 2 + liberación mediada por RyR. Hay tres isoformas RyR de mamífero con el canal de función compuesta de cuatro subunidades idénticas. Cada subunidad RyR consiste en una porción de regulación citoplásmica gran N-terminal y una pequeña parte C-terminal que contiene los dominios transmembrana que forman una alta conductancia Ca 2 + poro. Intra anormal y entre la subunidad interacciones subyacen a la disfunción de canales RyR y dan lugar a trastornos neuromusculares y cardíacos 1. La identificación y caracterización de los dominios específicos implicados en la estructura de RyR: función de relación es por lo tanto crucial para la comprensión de la fisiopatología RyR.
técnicas de interacción proteína-proteína bioquímicos tradicionales requieren cantidades sustanciales de proteína purificada, a menudo se producen en bacterias. Esto no es factible en el caso de la RyR, una membrana muy grande protein compone de ~ 5000 aminoácidos, mientras que sus fragmentos recombinantes no son fácilmente susceptibles a la expresión bacteriana y purificación. Por lo tanto, se requieren sistemas de expresión alternativos que implican células huésped eucariotas para las proteínas integrales de membrana de mamíferos. Hemos empleado previamente Y2H, co-IP y los ensayos de reticulación para demostrar colectivamente que tetramerización N-terminal es una característica estructural que se conserva en todos los tres mamíferos RyR isoformas 2,3. Es importante destacar que se ha encontrado que un único punto de mutación asociada con la enfermedad cardíaca arritmogénica interrumpe N-terminal de auto-asociación y los resultados en un canal de RyR disfuncional 4. También hemos aplicado estas técnicas para estudios de oligomerización de la RyR citoplásmica cola C-terminal 5, así como el N-terminal de la Ca canal de liberación homóloga intracelular 2 +, el inositol 1,4,5 trifosfato receptor 2.
En el ensayo de Y2H, la interactien entre dos proteínas (X e Y) se mide por la reconstitución de un factor de transcripción funcional (GAL4) y la activación subsiguiente de los genes indicadores de 6-9. Dos vectores de clonación diferentes se utilizan para generar fusiones de las dos proteínas probadas con los dos dominios físicamente separables, independientes de GAL4: DNA-Binding Domain (DNA-BD) / proteína de fusión X (cebo) y activación de dominio (AD) / proteína Y fusión (objetivo). El Y2H puede ser utilizado para probar si una proteína interactúa con sí mismo mediante la generación de GAL4 DNA-BD y fusiones AD de la misma proteína. Modificados genéticamente cepas Y2H se GAL4 y GAL80 deficiente (la proteína GAL80 es un represor de GAL4), así como TRP1 y LEU2 deficiente (para proporcionar la selección nutricional para el cebo y presa plásmidos, respectivamente). En el núcleo de levadura, los péptidos DNA-BD y AD recombinantes se ponen en estrecha proximidad física para producir un factor híbrido de transcripción GAL4 sólo a través de X sus fusiones ': int Yeraction. Este enfoque permite la detección genética rápida para detectar interacciones proteína-proteína a través de la activación de la transcripción en paralelo de prototrophic (codificación HIS3 para una enzima necesaria para la biosíntesis de histidina) y cromógeno (codificación LacZ de β-galactosidasa (β-Gal)) genes informadores. La ventaja principal de la Y2H es que es un ensayo in vivo que detecta interacciones proteína-proteína, incluso débiles o transitorios. Además, la detección implica el uso sencillo de selección de crecimiento (en medios que carecen de histidina) o de una colorimétrico de ensayo (β-Gal) sin necesidad de purificación de las proteínas de cebo y de destino o la generación de anticuerpos específicos. Además, el Y2H se puede utilizar para escrutar una colección de clones desconocidos aleatorios (clones biblioteca de ADNc fusionado a GAL4 AD) para nuevas parejas de unión de una proteína cebo, también dan acceso directo al ADNc de la proteína de la biblioteca.
Para extender las observaciones Y2H, indeent técnicas bioquímicas se pueden emplear. Ensayos de reticulación en combinación con inmunotransferencia Co-IP y son métodos utilizados para detectar asociaciones de proteínas en mezclas de muestras complejas, por ejemplo., Lisados de células enteras 10. Su principal ventaja es que se informan en las interacciones proteína-proteína a partir de tejido nativo, a diferencia de otros métodos que requieren el uso de proteínas recombinantes. Las proteínas recombinantes también se pueden utilizar, por lo general se expresa en una línea celular de mamífero, en el que es probable que tengan sus correcta conformación y modificaciones post-traduccionales, así como la localización subcelular. Sin embargo, desde co-IP y reticulación están en ensayos in vitro haciendo uso de células homogeneizadas, es necesario confirmar si los dos socios de proteína se co-localizan en la célula intacta 11. Utilizamos rutinariamente transfección de células HEK293 de mamífero para expresar transitoriamente integral de membrana de mamíferos y proteínas citosólicas usando el método de precipitación de fosfato de calcio 2-4,12-14, Descrito aquí en detalle. Esta es una manera económica de entregar eficientemente el plásmido de ADN dentro de las células, pero depende de la línea particular de células usada y la confluencia celular, así como la pureza del ADN plasmídico 11,15.
El ensayo de co-IP implica el aislamiento de la proteína nativa o recombinante de interés a partir de lisados de células en condiciones no desnaturalizantes que permiten la co-purificación de parejas de interacción putativo 10,16. Se requiere el uso de dos anticuerpos independientes, el anticuerpo inmunoprecipitación para el aislamiento de la proteína X, y el anticuerpo inmunotransferencia para la detección de Y. acompañante de la proteína Se puede utilizar para probar si una proteína interactúa con sí mismo mediante la generación de dos fusiones diferentes con dos epítopo diferente etiquetas (por ejemplo., HA y cMyc). El anticuerpo inmunoprecipitación se une a través de su región Fc de la proteína-A (o proteína-G, en función de la especie animal en la que se planteen la de anticuerpos), queestá conjugado a agarosa (o sefarosa) de resina. Protein X es precipitado por el anticuerpo: Proteína-A de resina después de la incubación con el lisado de células, a saber, la fracción soluble en detergente de células homogeneizadas. Proteínas inmuno-complejos se eluyen con tampón que contiene SDS y posteriormente se analizaron por SDS-PAGE e inmunotransferencia usando un anticuerpo para detectar la presencia de la proteína Y 17. Co-IP debe llevarse a cabo con proteínas detergente soluble para evitar excesiva unión no específica. La elección del detergente y su concentración, así como el número de lavados, deben optimizarse para cada X: Y par 10,16,18.
se emplea reticulación para determinar la estequiometría del complejo de proteína oligomérica. Se basa en una reacción química para crear enlaces covalentes entre protómeros que interactúan adyacentes, y por lo tanto, permite la conservación del estado oligomérico de la proteína durante la separación SDS-PAGE. Hay numerosos reage entrecruzamientonts de diferentes longitudes y química dirigidos a diferentes grupos reactivos en las proteínas, aminas primarias, típicamente carboxílico o grupos tiol. A continuación, describimos el uso de glutaraldehído (OHC (CH 2) 3 CHO), un agente de reticulación homo-bi-funcional con dos grupos aldehído en ambos extremos que reaccionan con los grupos amino libres presentes en los residuos de lisina 19,20. La reticulación es seguido de una manera de la concentración o dependiente del tiempo que resulta en la formación de aductos. Reacción glutaraldehído se detiene con hidrazina (H 2 NNH 2) y muestras de proteínas son luego analizados por SDS-PAGE e inmunotransferencia 17 para evaluar su estado de oligomerización. Debemos tener en cuenta que la reticulación no induce oligomerización, sino que simplemente crea puentes estables entre complejos de proteínas preexistentes. Consideraciones importantes cuando se llevan a cabo experimentos de entrecruzamiento incluyen la elección del agente de reticulación, su concentración y tiempo de reacción 19,20.
Protocolo
1. La levadura de dos híbridos
- Transformación de levadura
- Preparar los siguientes medios y tampones:
- Preparar levadura (YPD) medio completo extracto de levadura-peptona-dextrosa mediante la mezcla de 20 g / L de peptona, 10 g / L de extracto de levadura, 2% w / v de glucosa (añadido después de la esterilización en autoclave) y 20 g / L de agar (para placas solamente) ; esterilizar en autoclave y usar fresco en el día del experimento.
- Preparar medio definido levadura mínimo sintético (SD) (leucina carente y triptófano para mantener la presión selectiva en ambos plásmidos de cebo y de destino) por mezcla de 6,7 g / L de levadura Base de nitrógeno, 1,6 g / L Dropout suplemento leucina carente y triptófano, 2% w v de glucosa / (añadido después de la esterilización en autoclave) y 20 g / L de agar (para placas solamente); esterilizar en autoclave y usar fresco en el día del experimento.
- Preparar 50% w / v de PEG (polietilenglicol) 3350; esterilizar a través de un filtro de 0,2 micras y se almacena a temperatura ambiente.
- Preparar mM Tris / EDTA 10 mM (10x TE) 100, ajusteel pH a 7,5, esterilizar a través de un filtro de 0,2 micras y se almacena a temperatura ambiente.
- Preparar acetato 1 M de litio (10x LiAc); ajustar el pH a 7,5 con CH 3 COOH, esterilizar a través de un filtro y tienda de 0,2 micras, con RT.
- Revivir la cepa Y2H (por ejemplo, Y190) rayando una pequeña cantidad del stock de glicerol congelado en una placa de YPD. Incubar a 30 ° C hasta que las colonias de levadura alcanzan ~ 2 mm de diámetro (generalmente 3 - 5 días, dependiendo de la cepa de levadura).
- Inocular 0,5 ml de YPD (en un tubo de 1,5 ml) con una sola, grandes (2 - 3 mm de diámetro) colonia. Vortex vigorosamente durante ~ 2 min para dispersar los grumos.
- Transferir la suspensión celular en un matraz de 500 ml que contiene 50 ml de medio YPD. Incubar a 30 ° C durante 16 - 18 horas con agitación a 250 rpm para la levadura para llegar a la fase estacionaria.
- Transferencia de 4-5 ml del cultivo de una noche en 200 ml de YPD (en un matraz cónico de 1 L) para producir una densidad óptica a 600 nm (OD 600, mideutilizando un espectrofotómetro) de 0,2 - 0,3 (200 ml será suficiente para 20 transformaciones).
- Incubar a 30 ° C con agitación a 250 rpm hasta que las células están en fase mid-log, es decir, OD 600 = 0,5-0,6 (por lo general 2 - 3 h).
- levadura cosecha por centrifugación (en tubos de 50 ml) a 1500 xg durante 5 min a TA. Descartar el sobrenadante, resuspender cada pellet en 5 ml de H2O estéril y la piscina juntos.
- Re-centrifugar a 1500 xg durante 5 min a RT y desechar el sobrenadante. Resuspender pellet de levadura en 1 ml de recién preparada, 1x LiAc / TE estéril.
NOTA: Utilice las células de levadura competentes en el plazo de 1 hora de preparación. - Preparar muestras de plásmidos (en tubos de 1,5 ml) mediante la adición de 200 ng de ADN de plásmido para las transformaciones individuales, o 0,5 a 1 g de cada ADN plásmido para los compañeros de transformaciones, y 100 g de testículos de arenque ADN portador (hervida durante 20 min y se enfrió en hielo justo antes de su uso).
NOTA: Incluir un control positivo, por ejemplo, la levadura.co-transformada con pVA3 (codificación para la fusión GAL4 DNA-BD con la proteína p53) y pTD1 (codificación para la fusión GAL4 AD con el antígeno T grande de SV40). - Añadir 100 l de la recién preparada, suspensión de levadura competente (paso 1.1.8) y 600 l de solución de 1x LiAc / PEG (8 ml de existencias de PEG 3350, 1 ml de Stock TE, 1 ml de Stock LiAc), y agitar durante ~ 30 seg. Incubar a 30 ° C durante 30 min con agitación a 200 rpm.
- Añadir 80 l de sulfóxido de dimetilo (10% v / v de concentración final) y se mezcla bien por inversión suave. El choque térmico durante 15 minutos en un baño de agua C 42 ° mientras se mezcla cada 2 - 3 min.
- Chill suspensión de células en hielo durante 2 min y se centrifuga a 14.000 xg durante 15 seg a temperatura ambiente para recuperar la levadura.
- Resuspender pellet celular en 100 l de 1x TE y la placa sobre placas de medio SD mínimas apropiadas para el crecimiento selectivo (medio que carecía de leucina y triptófano para mantener una presión selectiva sobre ambos plásmidos cebo y de destino).
- Incubar las placas hasta boca abajo a 30° C hasta que las colonias son ~ 2 mm de diámetro (generalmente 4 - 5 días). Las placas se pueden almacenar a 4 ° C durante 2 - 3 semanas; para el almacenamiento ya que las existencias de glicerol y se almacena a -80 ° C.
NOTA: Verificar que las proteínas de cebo y de destino se expresan en levadura por inmunotransferencia 17, y que no tienen la activación del gen reportero autónomo separado cuando se expresa en la levadura (por el ensayo de β-Gal como se describe en la Sección 1.3).
- Preparar los siguientes medios y tampones:
- Elevadoras colonia papel de filtro de β-Gal Ensayo
- Preparar los tampones siguientes:
- Prepare el buffer Z que contiene 100 mM de Na 2 HPO 4, 40 mM NaH 2 PO 4, KCl 10 mM, 1 mM de MgSO4; ajustar el pH a 7,4. Esterilizar en autoclave y almacenar a temperatura ambiente.
- Preparar tampón de X-Gal mediante la disolución de 5-bromo-4-cloro-3-indolil-β-D-galactopiranósido en 20 mg / ml en N, N-dimetilformamida y almacenar en la oscuridad a -20 ° C. Preparar la solución tampón Z / X-Gal. hacer tampónantes de su uso mediante la mezcla X-Gal a 0,33 mg / ml y β-mercaptoetanol a 0,27% v / v en tampón Z. Use 2,5 ml por muestra.
- Añadir 2,5 ml de solución tampón Z / X-Gal recién preparada en una placa de 100 mm limpio y colocar dentro de un papel de filtro de celulosa.
- Colocar un nuevo papel de filtro sobre la superficie de la placa con las colonias de levadura a ensayar. Frote suavemente el papel de filtro sobre la placa con pinzas y dejar actuar durante ~ 5 min de colonias para adjuntar.
NOTA: Procesar el control positivo en paralelo, es decir, la levadura co-transformada con pVA3 y pTD1.. - Levante el papel de filtro y sumergirlo (con pinzas) en un depósito de nitrógeno líquido durante 30 segundos (nitrógeno líquido debe ser manejado con cuidado, siempre use guantes gruesos y gafas). Deje que el deshielo de papel de filtro congelado a temperatura ambiente durante ~ 2 min.
- Colocar el papel filtro (lado de las colonias hacia arriba) en la parte superior del papel de filtro previamente remojado el interior de la placa de 100 mm, y se incuba a 30 ° C.
- Compruebe periódicamente(Cada ~ 30 min) para la aparición de colonias de color azul. La levadura Y190 co-transformada con los plásmidos de control positivo (+ pVA3 pTD1) se vuelve azul dentro de 60 min (observaciones no publicadas).
NOTA: Cebo Débil: interacciones objetivo pueden tardar varias horas para producir una señal de color azul positivo (evitar la incubación prolongada (> 8 h) que pueden dar resultados falsos positivos). Para obtener los mejores resultados, utilice colonias recién co-transformadas, es decir, que se cultiva a 30 ° C durante 4 -. 5 días, ~ 2 mm de diámetro.
- Preparar los tampones siguientes:
- Líquido de cultivo β-Gal Ensayo
- Preparar los tampones siguientes:
- Prepare el buffer Z que contiene 100 mM de Na 2 HPO 4, 40 mM NaH 2 PO 4, KCl 10 mM, 1 mM de MgSO4; ajustar el pH a 7,4, esterilizar en autoclave y almacenar a temperatura ambiente.
1 M Na 2 CO 3; almacenar a temperatura ambiente. - Preparar tampón Z / β-mercaptoetanol. Hacer tampón antes de su uso mediante la adición de β-mercaptoetanol a 0,27% v /v en tampón Z; utilizar 700 l por muestra. Prepare el buffer Z / solución ONPG. Hacer tampón antes de su uso mediante la mezcla de ONPG (o-nitrofenil-β-D-galactopiranósido) a 4 mg / ml y β-mercaptoetanol a 0,27% v / v en tampón Z; utilizar 160 l por muestra.
- Prepare el buffer Z que contiene 100 mM de Na 2 HPO 4, 40 mM NaH 2 PO 4, KCl 10 mM, 1 mM de MgSO4; ajustar el pH a 7,4, esterilizar en autoclave y almacenar a temperatura ambiente.
- Utilice una sola colonia para inocular 5 ml de medio SD mínima (que carece de leucina y triptófano para mantener la presión selectiva en ambos plásmidos de cebo y de destino) y se incuba a 30 ° C durante 16 - 18 horas con agitación a 250 rpm.
NOTA: Ensayo de cinco colonias distintas co-transformaron con el mismo cebo y de destino plásmidos. - Transferencia suficiente del cultivo de una noche en 10 ml de medio SD fresco para producir una DO 600 = 0,2-0,3. Incubar a 30 ° C con agitación a 250 rpm hasta que las células están en fase semilogarítmica (OD 600 = desde 0,5 hasta 0,6).
- Transferir 0,5 ml de cultivo de levadura en un tubo de 1,5 ml y centrifugar a 14.000 xg durante 2 min a TA. Registre el OD 600 exacta cuando se cosecha el ceLLS. Resuspender pellet en 100 pl de tampón Z; esto dará lugar a un factor de concentración 5 veces.
- Colocar el tubo en nitrógeno líquido durante ~ 1 min (nitrógeno líquido debe ser manejado con cuidado, siempre use guantes gruesos y gafas) y luego en un baño de agua a 37 ° C durante 3 min se descongele. Repita este ciclo de congelación-descongelación dos veces más para asegurarse de células se rompen.
- Configurar un tubo blanco con 100 l de tampón Z.
- Añadir 700 l de tampón Z / β-mercaptoetanol y 160 l de tampón Z / ONPG a la muestra y los tubos en blanco; iniciar el temporizador y el lugar en una incubadora a 30 ° C.
- Comprobar periódicamente si hay color amarillo se desarrolle. Añadir 400 l de 1 M de Na 2 CO 3 para detener el desarrollo del color y registrar el tiempo transcurrido en minutos. La levadura Y190 co-transformada con los plásmidos de control positivo (+ pVA3 pTD1) se volverá amarilla dentro de 60 min ..
NOTA: Cebo Débil: interacciones objetivo pueden tardar varias horas para producir una señal amarilla positiva Para un mejor reresultados, utilice colonias recién co-transformadas, es decir, que se cultiva a 30 ° C durante 4 -. 5 días, ~ 2 mm de diámetro. - Centrifugar a 14.000 xg durante 5 min a temperatura ambiente para sedimentar los restos celulares y transferir el sobrenadante a una cubeta limpia.
- El uso de un espectrofotómetro, se mide la absorbancia a 420 nm (A 420) de las muestras con respecto a los valores en blanco (debe ser de entre 0,02 a 1,0).
- Calcular unidades de β-galactosidasa, con 1 unidad se define como la cantidad que hidroliza 1 mmol de ONPG a o-nitrofenol y D-galactosa por min por célula, de acuerdo con la siguiente fórmula:
 = unidades β-galactosidasa
= unidades β-galactosidasa
Donde: t: tiempo transcurrido de incubación (en minutos); . cf: el factor de concentración de la etapa 1.4.8, es decir, cf = 5; OD 600: cuando se recogieron las células.
- Preparar los tampones siguientes:
2. Expresión de la proteína en una línea celular de mamífero
- La transfección de células de mamífero
- Preparar los siguientes medios y tampones:
- Preparar medio de crecimiento mediante la mezcla de DMEM con 4,5 g / l de glucosa, 10% v / v de suero bovino fetal y 2 mM L-glutamina; esterilizar a través de un filtro y tienda de 0,2 micras a 4 ° C.
- Preparar solución salina tamponada con Hepes 2x (2x HBS) mezclando NaCl 280 mM, KCl 10, 1,5 mM Na 2 HPO 4, glucosa 12 mM, Hepes 50 mM; ajustar el pH a 7,05, esterilizar a través de un filtro y tienda de 0,2 micras, con -20 ° C. Preparar 2,5 M CaCl 2. Esterilizar a través de un filtro de 0,2 micras y almacenar a -20 ° C.
- Un día antes de la transfección, las semillas de 1 - 2 x 10 6 células HEK293 células en una placa Petri de 100 mm con el fin de ser un 60-70% de confluencia al día siguiente. La cultura para el 16 - 18 horas a 37 ° C en una incubadora humidificada con 5% de CO2.
- En el día de la transfección, eliminar las células viejas de media y de alimentación con 10 ml de medio de crecimiento fresco.
NOTA: Los antibióticos se omiten en el medio de cultivo durante la transfección, porque pueden aumentar la muerte celular. - Diluir 24 g de ADN plásmido (para los co-transfecciones, una relación molar igual de los dos plásmidos) y 60 l de 2,5 M CaCl 2 en 600 l volumen total (hecho con H desionizada estéril 2 O) dentro de un tubo de 1,5 ml; vórtice para mezclar.
NOTA:. Para conseguir la máxima eficiencia de la transfección, el ADN plásmido debe ser de la más alta pureza, es decir, tener una relación de Abs 260 / Abs 280 = ~ 1,8. - Añadir la solución de ADN del plásmido / calcio a gota en un tubo de 50 ml que contiene 600 l de 2x HBS mientras que constantemente y mezclándolo vigorosamente por agitación. Incubar durante 20 min a RT para permitir la formación de complejos de fosfato / plásmido de ADN de calcio.
- Después de la 20 min de incubación, brevemente vórtice y añadir la gota solución sabia sobre las células para cubrir toda la superficie de la placa de Petri de 100 mm.
- Se incuban las células a 37 ° C en 5% CO 2. Después de ~ 6 hr cambiar el medio de crecimiento y colocar de nuevo en la incubadora.
- Recoger las células 24 horas después de la transfección por centrifugación a 1000 xg durante 3 min a RT y descartar el sobrenadante. Los sedimentos celulares se pueden almacenar a -80 ° C hasta que se necesite.
NOTA: La expresión alcanza su más alto 24 de - 72 horas después de la transfección.
- Preparar los siguientes medios y tampones:
- La homogeneización de la célula
- Preparar los tampones siguientes:
- Preparar tampón de homogeneización Co-IP mediante la mezcla de NaCl 150 mM, Tris 20 mM; ajustar el pH a 7,4 y se almacena a 4 ° C.
- Prepare Entrecruzamiento de tampón de homogeneización mezclando mM Hepes 5, 0,3 M de sacarosa; ajustar el pH a 7,4 y se almacena a 4 ° C (filtro antes de su uso para eliminar cualquier partícula). Suplemento con inhibidores de proteasa antes de su uso.
- Añadir 250 l de perlas de vidrio (425 - 600 micras) dentro de un tubo de 1,5 ml y se lava con 500 l de tampón de homogeneización. perlas de vidrio de pellets de una breve centrifugaciónpulso (1.000 xg durante 5 seg) y eliminar el sobrenadante. Repita este paso de lavado una vez más.
- Resuspender pellet celular (de la etapa 2.1.8) en 500 l de tampón de homogeneización enfriado con hielo y transferir la suspensión de células en el tubo de perlas de vidrio que contiene.
- Homogeneizar células en hielo por 20 pasos a través de una aguja fina (23 G, 0,6 x 30 mm) unida a una jeringa de 1 ml. Con la tapa del tubo cerrado, perfore a través de ella con la aguja y se dispersan suspensión celular vigorosamente a través de las cuentas de vidrio.
- Centrifugar a 1500 xg durante 10 min a 4 ° C para eliminar los núcleos y las células intactas y desechar el sedimento. Guardar el sobrenadante representa la fracción post-nuclear y proceder directamente a co-IP o de reticulación según el caso.
- Preparar los tampones siguientes:
3. Los métodos in vitro bioquímicos
- Co-inmunoprecipitación
- Preparar los tampones siguientes:
- Preparar tampón de homogeneización Co-IP como se describe en sección 2.2.1.1. Preparar tampón de IP mediante la mezcla de Tris 20 mM, NaCl 150 mM, 0,5% (w / v) de CHAPS, ditiotreitol 2 mM (opcional; ditiotreitol puede ser incluido para reducir los agregados de proteínas que se puedan haber formado debido a la oxidación del aire); ajustar el pH a 7,4 y se almacena a 4 ° C.
- Preparar 2% w / v de CHAPS en tampón de homogeneización co-IP; almacenar a 4 ° C.
- Preparar tampón de proteína de carga mediante la mezcla de Tris 60 mM, 2% w / v de SDS, 10% v / v de glicerol, EDTA 5 mM, 0,01% w / v azul de bromofenol, 2% v / v β-mercaptoetanol (opcional); ajustar el pH a 6,8 y se almacena a temperatura ambiente.
- Homogeneizar las células de un mm placa de Petri confluentes 100 (~ 8 x 10 6 células HEK293 si, contó con el uso de un hemocitómetro) como se describe en la Sección 2.2.
- Solubilizar la fracción subcelular post-nuclear con 0,5% de CHAPS (utilizando el 2% de valores) e incubación durante ≥2 horas a 4 ° C con agitación constante. Centrifugar a 20.000 xg durante 10 min a 4 ° C para sedimentar las materi insolublecol y desechar el sedimento. Guardar el sobrenadante denominado lisado celular.
NOTA: La inclusión de un detergente y la eliminación del material insoluble es absolutamente necesario para minimizar la unión no específica. Detergentes. Intermedios, por ejemplo, CHAPS o Triton X-100, a una concentración de 0,2 - 1%, son los más comúnmente utilizados. - Preparar dos distintos tubos de 1,5 ml y añadir ~ 20 l (dependiendo de la capacidad de unión a IgG) de 6 mg / ml de proteína-A o Proteína G-agarosa (o de sefarosa) perlas. Se lava con 200 l de buffer de propiedad intelectual.
NOTA: Elija la resina de unión a Ig adecuada en función de las especies animales utilizadas para producir el anticuerpo inmunoprecipitación. Protein-G se une a un rango más amplio de subtipos de Ig en comparación con proteína-A. - Recuperar perlas por centrifugación a 1500 xg durante 2 min a 4 ° C. Repetir el lavado una vez más con tampón IP, a continuación, volver a suspender los granos en 200 l de tampón de IP.
- Añadir 1 g (5 ng / l) del anticuerpo inmunoprecipitación o IgG no inmune (paraservir como control negativo) en los / G tubos que contenían sendos Proteína-A. Incubar durante ≥2 horas a 4 ° C con agitación constante.
NOTA: El proceso siempre un control negativo con el uso de IgG no inmune criado en la misma especie animal que el anticuerpo inmunoprecipitación. - Recuperar perlas por centrifugación a 1500 xg durante 2 min a 4 ° C y desechar cuidadosamente el sobrenadante con la pipeta.
- Transferir 200 l de lisado celular (de la etapa 3.1.3) en cada uno de los dos tubos con anticuerpos y / G cuentas de Proteína-A. Incubar durante ≥2 horas a 4 ° C con mezcla constante para permitir la unión antígeno-anticuerpo.
- Recuperar los granos y se lava con 200 l de buffer de propiedad intelectual; incubar durante 10 min a 4 ° C con mezcla. Recuperar los granos y lavar dos veces más (múltiples lavados evitar que reduzcan tanto la unión no específica y específica). deseche con cuidado el sobrenadante con la pipeta.
- Añadir 30 l de tampón de proteína de carga para eluir immunoprecipitATED proteínas. Se centrifuga a 1.500 xg durante 2 min a 4 ° C, tirar las bolas y guardar el sobrenadante que contiene la muestra co-IP eluido.
- Para verificar la precipitación de la proteína X éxito, cargar una pequeña cantidad (1/10, 3 l) de la muestra co-IP en un gel de SDS-PAGE para ser analizados por inmunotransferencia 17 con un anticuerpo específico para la proteína X. Incluye una alícuota de la lisado celular para verificar la expresión de la proteína X en la muestra.
- Para probar la presencia de la proteína co-precipitado Y, cargar el resto (9/10, 27 l) de la muestra co-IP en un gel de SDS-PAGE por separado para ser analizados por inmunotransferencia con un anticuerpo específico 17 para Y. proteínas incluir una alícuota del lisado celular para verificar la expresión de proteínas Y en su muestra.
- Preparar los tampones siguientes:
- Reticulación química
- Preparar los tampones siguientes:
- tampón de homogeneización de entrecruzamiento tal como se describe en el apartado 2.2.1.1.
- Preparar 5x ptampón rotein de carga mediante la mezcla de Tris 300 mM, 10% w / v de SDS, 50% v / v de glicerol, EDTA 25 mM, 0,05% w / v azul de bromofenol, 10% v / v β-mercaptoetanol (opcional); ajustar el pH a 6,8 y se almacena a temperatura ambiente; caliente a 50 ° C antes de su uso.
- Homogeneizar las células de un mm placa de Petri confluentes 100 (~ 8 x 10 6 células HEK293 si, contó con el uso de un hemocitómetro) como se describe en la Sección 2.2.
NOTA: Sacarosa (a 0,3 M) se utiliza para crear un tampón iso-osmótica. Sal, por ejemplo, 120 - 150 mM NaCl o KCl se pueden utilizar en lugar, dependiendo de la proteína oligomérica complejo de interés. - Se centrifuga la fracción subcelular post-nuclear a 20.000 xg durante 10 min a 4 ° C para sedimentar los agregados de proteínas y guardar el sobrenadante. Tomar ocho alícuotas de 20 l cada uno (típicamente ~ 20 g de proteína) en distintos tubos de 0,5 ml.
- Añadir 0,0025% v / v de glutaraldehído a todas las muestras e iniciar el temporizador. Que cada uno de los ocho muestras reaccionan con glutaraldehyde a TA durante: 0, 2, 5, 10, 15, 20, 30, 60 min.
NOTA: El glutaraldehído tiene dos grupos aldehído que reaccionan con aminas libres. Evitar el uso de tampones de pH u otras sustancias con grupos amino primarios porque van a enfriar la reacción glutaraldehído. - Detención de la reacción glutaraldehído a la hora especificada con un 2% v hidracina / v y añadir 5 l de tampón de carga de proteína-5x para desnaturalizar las proteínas.
- Proceder a SDS-PAGE e inmunotransferencia 17.
- Preparar los tampones siguientes:
Resultados
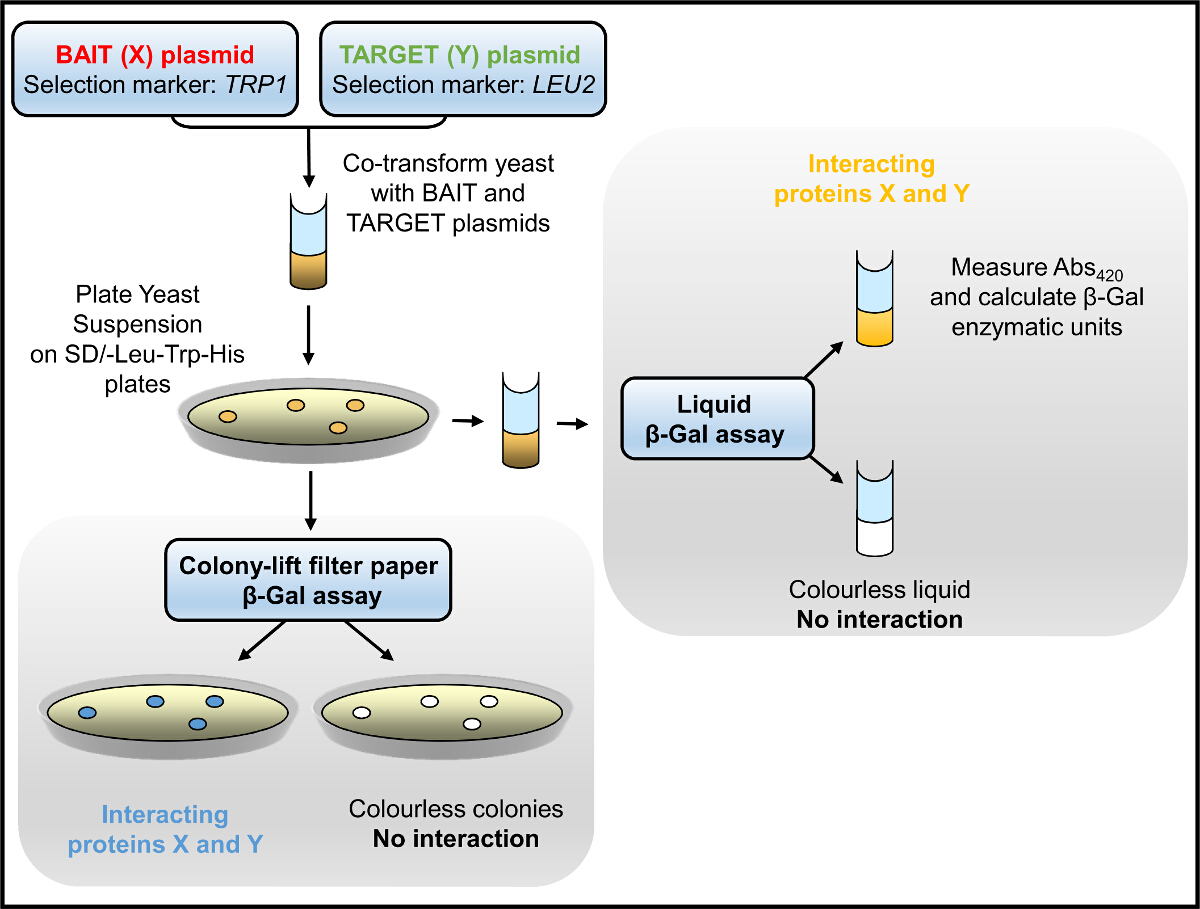
En el sistema de Y2H, el cebo: presa interacción se prueba inicialmente por la selección de levadura que carece de crecimiento en medio (triptófano, leucina e) histidina y posteriormente evaluada por β-Gal ensayos de actividad enzimática (Figura 1). La levadura se cultivaron en medio que carece de histidina tiene tasa de crecimiento lento, que depende de la fuerza del cebo: la interacción de proteínas presa. El ensayo de β-Gal se lleva a cabo en la levadura (cultivaron en medio que carece de triptófano y leucina solamente) o bien crece en soporte sólido (placas de agar) o en cultivo líquido, con los últimos resultados cuantitativos rendimiento. Hemos utilizado con éxito la Y2H para identificar las interacciones de dominio de dominio dentro del RyR2, así como nuevos socios de proteínas 2-4,12,21,22. Por ejemplo, hemos examinado una serie de construcciones que abarcan toda la longitud de la secuencia del péptido RyR2 para la interacción con un fragmento N-terminal de solapamiento (AD4L: residuos RyR2 1-906 fusionada con GAL4 AD). 3 ensayos de filtros de colonias de elevación del papel β-Gal producen colonias vivas de color azul levadura única para el BT4L: par AD4L (Figura 2), lo que demuestra que AD4L interactúa consigo misma, a saber, la BT4L construir (residuos RyR2 1-906 fusionado con GAL4 DNA-BD). Se detectaron colonias de color azul claro para el BT8: par AD4L lo que sugiere una asociación más débil secundaria con el dominio C-terminal extrema (BT8), mientras que la levadura co-transformada con cualquier otro constructo permanecieron blancas y por lo tanto negativo como cebo: la interacción de proteínas presa. Los resultados cuantitativos, obtenidos mediante ensayos de β-Gal líquidos (Figura 2B), indicaron BT4L robusto: equivalente interacción AD4L en fuerza a la conocida asociación entre la proteína p53 (pVA3) y grande de SV40 T antígeno (pTD1), mientras que el BT8: interacción AD4L fue considerablemente más débil (<10% en comparación con el control).
Rutinariamente realizamos experimentos de co-IP (Figura 3) fespués de expresión transitoria en una línea de células de mamífero (HEK293), como un ensayo bioquímico independiente para reforzar los hallazgos Y2H 2-4,12-14,21-24. Para verificar RyR2 N-terminal libre interacción, dos plásmidos separados que codifican para los residuos de RyR2 1-906 etiquetados, ya sea con el péptido cMyc o epítopo HA (BT4L y AD4L, respectivamente), se co-transfectaron en células HEK293 utilizando el método de precipitación con fosfato cálcico 3. La fracción post-nuclear de las células homogeneizadas se solubilizó con los CHAPS detergente, y el material insoluble se separó por centrifugación para producir el lisado celular. El lisado celular, tratados con el agente reductor ditiotreitol, se incubó con Ab HA y cuentas de Proteína-A Sepharose para inmunoprecipitar AD4L marcada con HA. Muestras de Co-IP, eluidas con tampón que contenía SDS, se cargaron en dos geles de SDS-PAGE separados (1/10 y 9/10 TH divididas) y se analizaron por inmunotransferencia con Ab HA y Ab cMyc, respectively. El éxito directo IP de AD4L (~ 100 kDa) por Ab HA fue verificada mediante inmunotransferencia, pero no en el control negativo utilizando IgG de conejo no inmune (Figura 4A). Es importante destacar que, de etiquetado cMyc BT4L (~ 100 kDa) se recuperó sólo en el HA IP Ab, y no en el control negativo en ausencia de AD4L inmunoprecipitada (Figura 4B).
Y2H ensayos y co-IP proporcionan evidencia consistente para RyR2 N-terminal libre interacción, pero no informan sobre el estado de oligomerización del dominio N-terminal, es decir si se forma solamente dímeros o complejos mayores. Cabe señalar que los complejos se disocian y solamente las subunidades que comprenden será detectada por SDS-PAGE a causa de SDS y desnaturalización de la proteína inducida por calor se suprime las interacciones proteína-proteína. Para superar esto, utilizamos el entrecruzamiento (Figura 5) que se conjuga químicamente y de manera estable preexistente protein oligómeros, cuya masa molecular puede entonces ser examinado por SDS-PAGE separación por tamaños 2-5. Por ejemplo, HEK293 homogenado celular que expresa cMyc-BT4L, se trata con el agente reductor ditiotreitol, se hizo reaccionar con glutaraldehído y se analizó mediante SDS-PAGE e inmunotransferencia usando Ab cMyc (Figura 6). Además del monómero (~ 100 kDa), una banda de proteína de alta masa molecular de ~ 400 kDa se detectó en una manera dependiente del tiempo, lo que indica RyR2 N-terminal formación tetrámero 3. En particular, tetrámero fue la especie predominante oligómeros, con dímero mínima y bandas trimer observados. Para determinar su masa molecular aparente, el oligómero BT4L se separó al 4 - 15% de geles de gradiente de SDS-PAGE 3. Hemos producido la curva estándar retraso masa / gel molecular usando patrones de proteína con un rango de 30 - 460 kDa, y se calculó el oligómero ser 358 kDa 15 (n = 4). Esta masa molecular aparente es consistente con un tetrámero BT4L dispuestos en unade forma circular cerrada en lugar de en forma lineal, como se espera de la disposición de las cuatro subunidades dentro del canal RyR2 nativo.

Figura 1. Diagrama de flujo Y2H. La levadura, co-transformada con cebo y de destino plásmidos, se selecciona para el crecimiento en un medio que carece de histidina y / o ensayó la actividad enzimática β-Gal. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. Y2H Sugiere RyR2 N-terminal de dominio de la libre interacción. (A) Esquema que representa la serie (cebo) de RyR2 fragmentos de proteínas superposición humanos probados en el sistema de Y2H interacción con el constructo AD4L RyR2 N-terminal (presa). Los resultados cualitativos obtenidos mediante ensayos de papel β-Gal filtros de lavado de colonia se indican en múltiplos "+" o "-" para la interacción negativa ensayos (B) de líquido cuantitativa β-Gal se normalizaron con el control positivo (pVA3 codifica para GAL4 DNA-BD. fusión con la proteína p53; pTD1 codifica para la fusión GAL4 AD con el antígeno T grande de SV40). Modificado de 3. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. Co-IP Diagrama de flujo. Las células de mamífero, co-transfectadas con plásmidos que X e Y, se homogeneizan y detergente solubilizadas para producir el lisado celular utilizada en el ensayo de co-IP, seguido de SDS-PAGE e inmunotransferencia.f = "https://www.jove.com/files/ftp_upload/54271/54271fig3large.jpg" target = "_ blank"> Haga clic aquí para ver una versión más grande de esta figura.

Figura 4. Co-IP indica RyR2 N-terminal de dominio libre interacción en células de mamífero. HEK293 células se co-transfectaron durante transitoria co-expresión de cMyc-tagged (BT4L) y HA-tagged dominio (AD4L) RyR2 N-terminal ( los residuos 1 a 906). AD4L se inmunoprecipitó con Ab HA de solubilizadas en CHAPS y lisado HEK293 ditiotreitol tratados, mientras que los ensayos de control, co-IP como negativos se realizaron con IgG de conejo no inmune. Las proteínas inmunoprecipitadas se calentaron a 85 ° C durante 5 min y se resolvieron en 20 mA durante 3 horas a través de 6% en geles de SDS-PAGE separados cargados de 1/10 o 9/10 de las muestras de IP. Después de la transferencia de proteínas a 80 V durante 2 horas a polyvinylidene membrana de difluoruro, análisis de inmunotransferencia se llevó a cabo usando (1: 1000 dilución) Ab HA (A) o Ab cMyc (B), respectivamente, seguido de peroxidasa de rábano conjugada anti-IgG de ratón (dilución 1: 10.000) y el aumento de la detección de quimioluminiscencia (1 min de exposición). Una alícuota del lisado celular, 1/50 del volumen procesado en muestras de IP, también se incluyó para servir como patrón de masa molecular. Modificado de 3. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5. Diagrama de flujo de reticulación. Las células de mamífero transfectadas con el plásmido X, se homogeneizan y se somete a reacción con glutaraldehído en el ensayo de reticulación, seguido de SDS-PAGE e inmunotransferencia. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6. La reticulación Indica RyR2 N-terminal de dominio de tetrámeros Formación Se transfectaron células HEK293 para la expresión transitoria de dominio (BT4L) RyR2 N-terminal marcada con cMyc (residuos 1 - 906).. homogeneizado celular, se trata con el agente reductor ditiotreitol, se incubó con glutaraldehído para los puntos de tiempo indicados. Las muestras se calentaron a 85 ° C durante 5 min y se resolvieron por SDS-PAGE (6% de gel) a 20 mA durante 3 horas. Después de la transferencia de proteínas a 80 V durante 2 horas a una membrana de difluoruro de polivinilideno, el análisis de inmunotransferencia se llevó a cabo usando (1: 1000 dilución) Ab cMyc, seguido de peroxidasa de rábano picante conjugada con IgG anti-ratón (1:10.000 dilución) y la detección de quimioluminiscencia (1 min de exposición). Monomérico (M: ~ 100 kDa) y las formas tetrámera (T) se indican por las flechas. Modificado de 3. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
La formación de proteínas homo-oligómeros es un proceso biológico fundamental que regula la actividad de factores de transcripción, enzimas, canales iónicos y los receptores 25,26. Es importante destacar que, oligomerización proteína tiene también implicaciones patológicas incluyendo la neurodegeneración y la enfermedad cardiaca arritmogénica 4,27. Las metodologías descritas en este artículo se utilizan para identificar dominio de dominio interacciones proteína que median la auto-asociación y oligomerización. A continuación, señalamos en las etapas decisivas dentro de cada protocolo, y se discute consideraciones importantes, limitaciones y solución de problemas.
El sistema Y2H puede ser empleado por primera vez para la detección de potenciales socios de interacción de proteínas, debido a su relativamente cribado de alto rendimiento, facilidad de uso y resultados altamente reproducibles. Y2H procedimientos se llevan a cabo en un laboratorio de microbiología con incubadoras estándar (placa o agitador) y las instalaciones de contención habitación. El uso de frLas células competentes preparadas eshly (paso 1.1.8) es fundamental para la transformación de levadura de alta eficiencia, mientras que para obtener los mejores resultados en los ensayos de β-Gal, colonias de levadura recién crecido (hasta 5 días de edad) se debe utilizar (paso 1.2.3). Los sistemas basados en factores de transcripción distintos de GAL4 y / o genes indicadores adicionales / alternativas están disponibles, y por lo tanto plásmidos cebo y presa deben corresponderse con la cepa Y2H apropiado.
Algunas cepas deben ser cultivadas en presencia de 3-amino-1,2,4-triazol, un inhibidor competitivo de la proteína HIS3, con el fin de apagar cualquier expresión constitutiva del gen indicador HIS3 7,8. La expresión de proteínas de fusión de cebo y de destino debe ser verificada mediante inmunotransferencia 17. En caso de cebo / proteínas de fusión de presa son tóxicos para la levadura, los niveles de proteína inferior tolerables podrían alcanzarse mediante la clonación de vectores en diferentes donde cebo / presa expresión es conducida por un promotor diferente. Además, es esencial para asegurar tLas proteínas de fusión de cebo / presa sombrero no muestran la actividad del gen reportero autónomo. la activación del gen indicador autónoma puede ser rescatado mediante la modificación de la construcción para eliminar la región responsable, o mediante el canje de la GAL4 DNA-BD y AD fusiones de las dos proteínas de la prueba. Por otra parte, los dominios transmembrana están mejor omiten de construcciones cebo / presa, ya que pueden inducir o mal plegamiento de proteínas de fusión mislocalization en compartimentos intracelulares de membrana. De hecho, la principal desventaja del sistema de Y2H es que las proteínas de cebo y de destino se localizan en el núcleo de levadura lejos de su localización subcelular fisiológica y, potencialmente, que carecen de modificaciones post-traduccionales específicas, lo que resulta en las interacciones falsos positivos o falsos negativos 6-9 .
De mamíferos Los sistemas de expresión heterólogos son más adecuados para el estudio de proteínas integrales de membrana de mamíferos en los términos de conformación, modificaciones post-traduccionales y la localización subcelular.Uno de los métodos de transfección celular más ampliamente utilizada es la precipitación con fosfato cálcico, principalmente debido a los equipos mínimos y reactivos requeridos 11,15. Los métodos alternativos, a saber, la electroporación, liposomas, lípidos catiónicos y polímeros, pueden ofrecer una mayor eficiencia de la transfección dependiendo de la línea celular y construcción utilizada. En general, los factores principales que afectan la eficiencia de transfección son la calidad del ADN de plásmido y células de la salud / viabilidad. Los mejores resultados se obtienen cuando se utilizan ADN de plásmido de la más alta pureza (relación de absorbancia 260 nm / 280 nm de ~ 1,8) y las células que se dividen activamente. Por lo tanto, las células deben ser transfectadas a no más de 60-70% de confluencia (etapa 2.1.2), debido a que la capacidad de absorción de ADN extraño se relaciona con el área de superficie de la célula expuesta al medio 11. Además, la inclusión de antibióticos en el medio de cultivo durante la transfección (etapa 2.1.3) no se recomienda debido al aumento de la muerte celular 15.
Fo precipitación de fosfato de calcio, en particular, una cuidadosa preparación y ajuste del pH (a 7,05 precisamente) de la solución de 2x HBS (etapa 2.1.1) y la formación adecuada de plásmido de ADN / calcio / complejos de fosfato por mezcla vigorosa (etapa 2.1.5) son pasos críticos para la transfección de alta eficiencia. Por lo general, la expresión de proteínas por picos de transfección transitoria dentro de 24 - 72 h.
Una vez que se recogieron las células, los procedimientos subsiguientes deben llevarse a cabo a 4 ° C para minimizar la actividad de proteasa, y se recomienda la adición de inhibidores de la proteasa. la homogeneización de la célula debe ser seguido por una etapa de centrifugación para eliminar los núcleos porque el ADN cromosómico puede aumentar la viscosidad de solución y mejorar la unión no específica. Por lo tanto, se prefiere la homogeneización por medios mecánicos en un tampón iso-osmótica, por lo general en sacarosa (0,3 M) o (150 mM) NaCl. En general, se conocen tampones a base de sacarosa para mejorar la estabilidad de proteínas y reducir el potencial de agregación de proteínas no nativas, pero debido a phidratación de referencia en la superficie de la proteína, interacciones proteína-proteína electrostáticos son favorecidos. Por el contrario, los tampones a base de sal influyen en la carga neta de los grupos laterales de aminoácidos cargados en la superficie de la proteína, por lo tanto tener un sesgo hacia las interacciones más basado en hidrófobas 28.
Co-IP es el ensayo bioquímico más comúnmente empleado para evaluar las interacciones proteína-proteína especialmente de tejido nativo. Su principal inconveniente es la necesidad de anticuerpos altamente específicos validados para su uso en IP e inmunotransferencia 10,16,18. Así, las proteínas recombinantes a menudo son etiquetados con un epítopo de péptido, por ejemplo., Hemaglutinina de la gripe (YPYDVPDYA) o cMyc humana (EQKLISEEDL), para los que de alta afinidad anticuerpos específicos están disponibles comercialmente. Si se desea, el anticuerpo inmunoprecipitación puede conjugarse químicamente a la Proteína-A de resina para evitar su elución y la detección durante la etapa de inmunotransferencia que pueden oscurecer el co-precipitado prOtein 16; Para lograr esto, hemos utilizado con éxito el reticulador químico 3,3'-ditiobis (sulfosuccinimidilpropionato) 24. Es imperativo que un detergente apropiado se incluye en el tampón de IP y el material insoluble de las células homogeneizadas se retira por centrifugación para minimizar la unión no específica (etapa 3.1.3). La elección y la concentración de detergente son consideraciones importantes: los detergentes fuertes y / o concentraciones más altas reducirán sustancialmente la unión no específica, pero también pueden abolir X: la interacción proteína Y, mientras que las concentraciones más bajas o detergentes más suaves puede permitir una interacción débil para ser observado, pero puede resultar en exceso de la unión no específica. Se prefieren los detergentes de fuerza intermedios, por ejemplo, Triton X-100 al 0,5 -. Concentración 1%. Para reducir aún más la unión no específica, una proteína neutro (por ejemplo, albúmina de suero bovino en 100 g / ml) puede ser incluido en el tampón de IP, y / o el lisado celular se puede-cleare pred con la incubación previa con resina de Proteína-A sola. El número de lavados se debe optimizar para la X: Y ensayó par, por lo general tres lavados de 10 min con tampón IP (paso 3.1.9). En cualquier caso, una muestra co-IP con IgG no inmune como el anticuerpo de inmunoprecipitación siempre debe ser procesado en paralelo para servir como control negativo (etapa 3.1.6).
La principal ventaja de la reticulación química es que informa sobre la composición estequiométrica de la proteína homo-oligómero. El glutaraldehído es un reticulante de uso común, ya que no requiere equipo especializado y que genera térmica y químicamente estables enlaces cruzados entre las proteínas que interactúan 19,20. Los compuestos con grupos amino libres pueden faltar en tampones de ensayo (paso 3.2.1) porque van a enfriar la reacción química. concentración de glutaraldehído y el tiempo de reacción (paso 3.2.4) deben ser optimizados para el complejo de la proteína oligomérica de interés. El principal inconveniente de esta técnica, especialiado cuando se realiza en preparaciones de células enteras, es la no especificidad de la reacción química que podría producir agregados de proteínas artificiales que carecen de importancia biológica.
Alternativa en vivo (por ejemplo., Transferencia de energía por resonancia de fluorescencia, fluorescencia bi-molecular o complementación de luminiscencia) y las técnicas in vitro (por ejemplo., Cromatografía de exclusión por tamaño, ultracentrifugación analítica, titulación isotérmica calorimetría) están disponibles para la caracterización de la proteína libre asociación y la evaluación estequiometría de oligomerización 29,30. Cada método tiene sus propias ventajas y desventajas, y puede ser más adecuado para el estudio de una proteína específica en función de la proteína de purificación / estabilidad y disponibilidad de los equipos / reactivo. Los tres métodos complementarios que se describen aquí en detalle, a saber Y2H, co-IP y la reticulación, se han utilizado en combinación para proporcionar evidencia convincente de RyR2 homo-oligomeformación de r en el aislamiento y dentro de una célula viva.
Divulgaciones
Los autores no tienen nada que declarar.
Agradecimientos
Este trabajo fue apoyado por becas de la Fundación Británica del Corazón a SZ (FS / 08/063 y FS / 15/30/31494).
Materiales
| Name | Company | Catalog Number | Comments |
| PART 1 yeast two-hybrid | |||
| Plate incubator | Hereaus | B6120 | Used at 30 °C |
| Orbital shaker incubator | New Brunswick Scientific | INNOVA 4300 | Used at 30 °C with shaking at 230 - 250 rpm |
| Spectrophotometer | Perkin Elmer | Lambda Bio+ | To measure OD600 of yeast culture; to measure Absorbance at 420 nm in liquid b-Gal assay |
| Matchmaker Two-Hybrid System 2 Kit | Takara Clontech | K1604-1 | Contains bait vector pGBKT7, prey vector pACT2 and yeast strain Y190 |
| Yeast Nitrogen Base | Sigma-Aldrich | Y0626 | To prepare minimal SD growh medium |
| Dropout supplement lacking leucine and tryptophan | Sigma-Aldrich | Y0750 | To prepare minimal SD growh medium |
| Dropout supplement lacking leucine, tryptophan, histidine | Sigma-Aldrich | Y2001 | To prepare minimal SD growh medium |
| Herring testes carrier DNA | Takara Clontech | 630440 | For yeast transformation |
| Whatman filter paper Grade 5 | Sigma-Aldrich | WHA1005070 | for use in colony-lift filter paper b-Gal assay |
| X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) | Sigma-Aldrich | B4252 | for use in colony-lift filter paper b-Gal assay |
| ONPG (o-nitrophenyl-β-D-galactopyranoside) | Sigma-Aldrich | N1127 | for use in liquid b-Gal assay |
| PART 2. Protein expression in a mammalian cell line | |||
| HEK293 cell line | ATCC | ATCC® CRL-1573™ | |
| Humidified incubator (5% CO2, 37 °C) | SANYO | MCO-18AIC | To culture mammalian cells |
| DMEM (Dulbecco's Modified Eagle's medium) | Invitrogen (ThermoFisher) | 41966 | To prepare growth medium for mammalian cells |
| Fetal Bovine Serum | Invitrogen (ThermoFisher) | 10500 | To prepare growth medium for mammalian cells |
| Glass beads | Sigma-Aldrich | G8772 | To homogenise cells |
| Needle 23 G (0.6 x 30 mm) | BD Microlance | 300700 | To homogenise cells |
| Protease inhibitor cocktail (Complete) | Roche | 11873508001 | To prevent proteolysis |
| PART 3. In vitro biochemical methods | |||
| Mini-PROTEAN Tetra Cell (SDS-PAGE system) | Bio-Rad | 1658000EDU | For polyacrylamide gel electrophoresis |
| Trans-Blot SD (Semi-dry transfer system) | Bio-Rad | 1703940 | For electrophoretic transfer |
| Compact X-ray film processor | Xograph | X4 | For use in immunoblotting |
| Glutaraldehyde | Sigma-Aldrich | G5882 | For use in chemical cross-linking |
| Protein-A sepharose beads | GE Healthcare Life Sciences | 17-5280-01 | For use in co-IP assay |
| Anti-HA (Y-11 rabbit polyclonal IgG) | Santa Cruz Biotechnology | sc-805 | For use in co-IP assay |
| Non-immune rabbit IgG | Santa Cruz Biotechnology | sc-2027 | For use in co-IP assay |
| Anti-cMyc (9E10 mouse monoclonal IgG) | Santa Cruz Biotechnology | sc-40 | For use in immunoblotting |
| Anti-HA (16B12 mouse monoclonal IgG) | Covance | MMS-101P | For use in immunoblotting |
| Anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 | For use in immunoblotting |
| Hyperfilm ECL | GE Healthcare Life Sciences | 28906836 | For use in immunoblotting |
| ECL Western Blotting Substrate | Pierce (ThermoFisher) | 32106 | For use in immunoblotting |
Referencias
- Seidel, M., Lai, F. A., Zissimopoulos, S. Structural and functional interactions within ryanodine receptor. Biochem Soc Trans. 43 (3), 377-383 (2015).
- Zissimopoulos, S., Marsh, J., Stannard, L., Seidel, M., Lai, F. A. Amino-terminus oligomerisation is conserved in intracellular calcium release channels. Biochem J. 459 (2), 265-273 (2014).
- Zissimopoulos, S., et al. N-terminus oligomerization regulates the function of cardiac ryanodine receptors. J Cell Sci. 126 (Pt 21), 5042-5051 (2013).
- Seidel, M., Thomas, N. L., Williams, A. J., Lai, F. A., Zissimopoulos, S. Dantrolene rescues aberrant N-terminus inter-subunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc Res. 105 (1), 118-128 (2015).
- Stewart, R., Zissimopoulos, S., Lai, F. Oligomerization of the cardiac ryanodine receptor C-terminal tail. Biochem J. 376, 795-799 (2003).
- Deane, C. M., Salwinski, L., Xenarios, I., Eisenberg, D. Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol Cell Proteomics. 1 (5), 349-356 (2002).
- Fashena, S. J., Serebriiskii, I., Golemis, E. A. The continued evolution of two-hybrid screening approaches in yeast: how to outwit different preys with different baits. Gene. 250 (1-2), 1-14 (2000).
- Stynen, B., Tournu, H., Tavernier, J., Van Dijck, P. Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol Mol Biol Rev. 76 (2), 331-382 (2012).
- Yang, M., Wu, Z., Fields, S. Protein-peptide interactions analyzed with the yeast two-hybrid system. Nucleic Acids Res. 23 (7), 1152-1156 (1995).
- Elion, E. A. Chapter 8, Detection of protein-protein interactions by coprecipitation. Curr Protoc Immunol. , (2007).
- Kingston, R. E., Chen, C. A., Rose, J. K. Chapter 9, Calcium phosphate transfection. Curr Protoc Mol Biol. , (2003).
- Lam, A. K., Galione, A., Lai, F. A., Zissimopoulos, S. Hax-1 identified as a two-pore channel (TPC)-binding protein. FEBS letters. , (2013).
- Zissimopoulos, S., Lai, F. Interaction of FKBP12.6 with the cardiac ryanodine receptor C-terminal domain. J Biol Chem. 280, 5475-5485 (2005).
- Zissimopoulos, S., Thomas, N. L., Jamaluddin, W. W., Lai, F. A. FKBP12.6 binding of ryanodine receptors carrying mutations associated with arrhythmogenic cardiac disease. Biochem J. 419 (2), 273-278 (2009).
- Jordan, M., Wurm, F. Transfection of adherent and suspended cells by calcium phosphate. Methods. 33 (2), 136-143 (2004).
- Kaboord, B., Perr, M. Isolation of proteins and protein complexes by immunoprecipitation. Methods Mol Biol. 424, 349-364 (2008).
- Sambrook, J., Fritsch, E. F., Maniatis, T. . Molecular cloning: a laboratory manual. , (1989).
- Yang, W., Steen, H., Freeman, M. R. Proteomic approaches to the analysis of multiprotein signaling complexes. Proteomics. 8 (4), 832-851 (2008).
- Migneault, I., Dartiguenave, C., Bertrand, M. J., Waldron, K. C. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques. 37 (5), 790-796 (2004).
- Wine, Y., Cohen-Hadar, N., Freeman, A., Frolow, F. Elucidation of the mechanism and end products of glutaraldehyde crosslinking reaction by X-ray structure analysis. Biotechnol Bioeng. 98 (3), 711-718 (2007).
- Zissimopoulos, S., Lai, F. Central domain of the human cardiac muscle ryanodine receptor does not mediate interaction with FKBP12.6. Cell Biochem Biophys. 43, 203-220 (2005).
- Zissimopoulos, S., West, D., Williams, A., Lai, F. Ryanodine receptor interaction with the SNARE-associated protein snapin. J Cell Sci. 119, 2386-2397 (2006).
- Zissimopoulos, S., Docrat, N., Lai, F. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem. 282, 6976-6983 (2007).
- Zissimopoulos, S., Seifan, S., Maxwell, C., Williams, A. J., Lai, F. A. Disparities in the association of the ryanodine receptor and the FK506-binding proteins in mammalian heart. J Cell Sci. 125. 125 (Pt 7), 1759-1769 (2012).
- Ali, M. H., Imperiali, B. Protein oligomerization: how and why. Bioorg Med Chem. 13 (17), 5013-5020 (2005).
- Hashimoto, K., Panchenko, A. R. Mechanisms of protein oligomerization, the critical role of insertions and deletions in maintaining different oligomeric states. Proc Natl Acad Sci U S A. 107 (47), 20352-20357 (2010).
- Haass, C., Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 8 (2), 101-112 (2007).
- Chi, E. Y., Krishnan, S., Randolph, T. W., Carpenter, J. F. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm Res. 20 (9), 1325-1336 (2003).
- Gell, D. A., Grant, R. P., Mackay, J. P. The detection and quantitation of protein oligomerization. Adv Exp Med Biol. 747, 19-41 (2012).
- Kaczor, A. A., Selent, J. Oligomerization of G protein-coupled receptors: biochemical and biophysical methods. Curr Med Chem. 18 (30), 4606-4634 (2011).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados