
Method Article
Análisis en tiempo real del Factor de transcripción vinculante, transcripción, traducción y rotación para mostrar los eventos globales durante la activación celular
* Estos autores han contribuido por igual
En este artículo
Resumen
Este protocolo describe el uso combinatorio de ChIP-seq, 4sU-seq, total RNA-seq y perfiles para las líneas celulares y células primarias del ribosoma. Permite el seguimiento de cambios en el factor de transcripción vinculante, novo de transcripción, procesamiento de RNA, rotación y traducción con el tiempo y viendo el curso general de los acontecimientos en las células activadas o cambiantes.
Resumen
Después de la activación, las células cambian rápidamente sus programas funcionales y, por tanto, su perfil de expresión génica. Ocurren grandes cambios en la expresión génica, por ejemplo, durante la diferenciación celular, morfogénesis y estimulación funcional (por ejemplo, la activación de células inmunes), o después de la exposición a las drogas y otros factores del entorno local. Dependiendo del tipo de estímulo y de la célula, estos cambios ocurren rápidamente y en cualquier nivel de regulación génica. Mostrando todos los procesos moleculares de una célula responde a un determinado tipo de estímulo y las drogas es una de las tareas más difíciles en biología molecular. Aquí, describimos un protocolo que permite el análisis simultáneo de múltiples capas de regulación génica. Comparar, en particular, factor de transcripción vinculante (cromatina-inmunoprecipitación-secuenciación (ChIP-seq)), transcripción de novo (4-thiouridine-secuenciación (4sU-seq)), procesamiento de mRNA y facturación, así como traducción (ribosoma profiling). Mediante la combinación de estos métodos, es posible visualizar un plan de acción detallado y genoma.
Secuencia de RNA recién transcrito es especialmente recomendada cuando se analizan rápidamente adaptar o cambiar sistemas, ya que esto representa la actividad transcripcional de los genes durante el tiempo de exposición 4sU (independientemente de si son para arriba - u o). La combinatoria total RNA-seq y perfiles de ribosoma además permite el cálculo de las tasas de rotación y traducción de RNA. Análisis Bioinformático de los resultados de secuenciación de alto rendimiento permite muchos medios para el análisis e interpretación de los datos. Los datos generados también permite seguimiento co-transcriptional y alternativa de empalme, por citar algunos resultados posibles.
El enfoque combinado descrito aquí puede ser aplicado para organismos diferentes modelos o tipos de células, incluyendo células primarias. Además, ofrecemos protocolos detallados para cada método utilizado, incluyendo controles de calidad y discutir posibles problemas y dificultades.
Introducción
En los últimos años, la secuenciación del RNA (RNA-seq) se ha convertido en la herramienta estándar para analizar todo expresado ARN dentro de una célula o un organismo1. Sin embargo, para entender todo el proceso de las células en respuesta a una estímulo específica y las drogas, es necesario determinar completamente todos los procesos subyacentes, desde la transcripción de mRNA a procesamiento, rotación y traducción. Cambios a corto plazo en la transcripción del RNA no pueden medirse por RNAseq total, ya que cambios de ARN total dependen de factores por ejemplo RNA vida media y actividad transcripcional, que son una plantilla pobre para reflejar la adaptación de las células efectos ambientales2,3. De hecho, han desarrollado una variedad de nuevas técnicas de secuenciación que permiten un análisis de los diferentes pasos en el proceso de gene Reglamento4 cuando se combinan en la forma correcta. Este protocolo describe cómo combinar algunas técnicas de secuenciación más fácilmente aplicables que permitan el seguimiento de la regulación de las capas esenciales de mRNA de manera comparativa. Para el análisis de actividad transcripcional, se han descrito una variedad de métodos, como el análisis de la tapa de gene expresión (jaula)5, alargamiento natural transcripción secuencia (NET-seq)6y genoma nuclear precisado (GRO-seq)7 , 8, así como secuenciación del bromouridine (Bru-seq) y 4-thiouridine-secuenciación (4sU-seq), que utilizan metabolitos que se incorporan en recién transcripción RNA9,10, sólo por mencionar algunos. Mientras jaula identifica el sitio de inicio de transcripción exacta, red-seq y GRO-seq entregan información más precisa acerca de las direcciones de lectura y 4sU-seq (que es el método descrito aquí) detecta sólo recién transcrito RNA. Sin embargo, 4sU seq es muy sensible y puede ser aplicado en diferentes marcos de tiempo para medir la actividad transcripcional cuantitativa en activamente cambiando las células, así como cambios cuantitativos en el procesamiento del mRNA (que ocurre dentro de minutos)9, 11,12. Además, es ideal para combinar con RNA-seq para calcular tasas de rotación de RNA genes94sU-seq. La transcripción del mRNA se lleva a cabo por ARN polimerasa II (RNAPII), que a su vez está influenciada por una multitud de factores, tales como factores de transcripción, modificaciones de las histonas y general activadores/represores que puede incluso ser parte del transcripcional complejo. Para comprobar cuántas regiones de potenciador de promotor de genes están vinculadas por un factor, se ha desarrollado ChIP-seq, que ahora es el método estándar para este propósito, debido a muchos anticuerpos comercialmente disponibles13. Sin embargo, aunque ChIPseq da información clara sobre donde regula factores de atar, no refleja si en efecto conduce a cambios en la transcripción14. Por lo tanto, realizar ChIP-seq con 4sU seq es la combinación ideal para este tipo de preguntas biológica. Regulación de la expresión génica puede también ocurrir en una etapa posterior, ya que los niveles de mRNA y proteína no necesariamente se correlacionan15,16, indicando potencialmente significativo regulación traduccional o poste-de translación nivel, dependiendo del contexto. En el año 2011, ribosome profiling había sido primero combinado con RNA-seq y ahora es el método de elección para cuantificar los cambios que se producen rápidamente en la proteína, puesto que todavía hay algunos límites de sensibilidad con espectrometría de masa17. De hecho, las tasas de traducción obtenidas de tales métodos han demostrado entregar una relativamente buena estimación de cambios en los niveles de proteína (menos medidos para cambios a largo plazo) y permite una visión más detallada sobre el proceso de traducción, por ejemplo, la determinación de traducción alternativa y el inicio Marcos17. El uso de la combinatorio de los cuatro métodos puede utilizarse en estado estacionario, entre los diversos tipos de células, o en una serie de tiempo el experimento de un cambiante celda11. Este uso proporciona un resumen de todo el genoma de los cambios en el atascamiento del factor de transcripción que influyen en la transcripción de ARN, procesamiento y traducción.
Protocolo
Todos los métodos están de acuerdo y conformidad con los lineamientos institucionales, estatales y federales de Helmholtz Zentrum München.
1. preparación
- Hacer un plan detallado de la instalación experimental incluyendo un calendario, cuando añadir 4sU al medio de cultivo celular y cuando cosechar las células de cada método. Dependiendo de la cuestión biológica, considerar cuidadosamente los puntos del tiempo para el dibujo de muestra, etiquetado 4sU tiempo y concentración (tabla 1).
Nota: Verificar el impacto de 4sU en viabilidad celular y estrés respuesta por adelantado (véase "Comprobar etiquetado 4sU condiciones óptimas" representante y figura 1). Se recomienda realizar una prueba preliminar de cada método con al menos una muestra. Verificar si la calidad y la cantidad de ARN/ADN es suficiente para secuenciación profunda (véase dedicado partes del Protocolo) y práctica rápida, pero un manejo suave de las células durante el experimento. - Calcular el número de células necesarias para cada momento de cada método (véase tabla 2 para evaluar al utilizar las células T primarias). También, considere secuenciación menos muestras de ARN total que 4sU RNA (p. ej., sólo para tarifas de traducción de punto de tiempo o volumen de RNA se calculará las tasas). Para confirmar y comprobar el significado de los resultados (recomendado), crear al menos una repetición biológica.
Nota: Importante, para todos los métodos y puntos de tiempo consecutivos, las muestras deben ser de la misma agrupación a partir de las células. Se recomienda por lo menos un investigador dedicado para cada método. - Preparar todo lo necesario por adelantado (por ejemplo, 4sU alícuotas, cicloheximida, mamíferos tampón de lisis y formaldehído al 1%). Después de la adición de 4sU, Evite exponer las células a la luz, ya que puede producirse entrecruzamiento de ARN marcado con 4sU a proteínas celulares18.
- Piscina de todas las células de interés en un frasco justo antes del tratamiento (figura 2). Cuente las células (por ejemplo, con un hemocitómetro) y utilizar la cantidad necesaria para el control sin tratar de cada método (no olvide etiquetar también el control sin tratar para 4sU-seq con 4sU). Tratar las restantes células y dividir inmediatamente el número de células para cada punto del tiempo y método. Manejar las células tan pronto como sea posible para minimizar el estrés debido a los cambios de temperatura o niveles de CO2 .
Nota: por ejemplo, se tomaron muestras de 1 h, 2 h, 3 h después del tratamiento, luego una cuarta parte de las células es utilizado para el control sin tratamiento, y tres cuartos se utilizan para el control del tratamiento.
2. 4sU-etiquetado
Nota: Este protocolo se modifica Rädle, et al. 19 consulte su Protocolo para obtener más información sobre el etiquetado metabólico con 4sU. Para todos los métodos y puntos de tiempo consecutivos, las muestras deben proceder de la misma agrupación a partir de las células.
-
Inicio de etiquetado
- 4sU alícuotas de deshielo antes de uso. 4sU añadir en cada momento directamente en el medio que contiene las células de interés (ver tabla 2 para el número recomendado de células T, menos de 60 μg RNA, por el momento), mezclar suavemente y colocar en la incubadora. Deseche restante 4sU (no volver a congelar).
- Al final de las etiquetas, recoger las células (e.g., raspador de la célula) y centrifugar a 330 x g durante 5 min a 4 ° C en tubos de polipropileno (que resisten fuerzas g alta). Aspire el medio y añadir Reactivo para el aislamiento de RNA (≥1 mL por 3 x 106 células, ver materiales por recomendación que utilizar) a cada tubo. Resuspender el precipitado (≥1 mL por 3 x 106 células), incubar por 5 min a temperatura ambiente (RT) y congelar las muestras a-20 ° C. Las muestras pueden almacenarse a-20 ° C durante al menos 1 mes.
PRECAUCIÓN: Los reactivos utilizados para el aislamiento de RNA son extremadamente peligrosos al entrar en contacto con la piel o los ojos. Manejar con cuidado y tener en cuenta las instrucciones de seguridad.
-
Preparación de RNA usando modificado protocolo de aislamiento de RNA
- Añadir 0,2 mL de cloroformo por 1 mL de reactivo para el aislamiento del RNA y mezclar bien agitando durante 15 s. proceder como se ha mencionado en el protocolo de etiquetado metabólico (paso 1-12, 2. Preparación de RNA usando modificado el protocolo de trizol) de Rädle et al. 19
- Medir la concentración de RNA (véase Tabla de materiales), según las instrucciones del fabricante. Utilice este RNA para total RNA-seq, así (véase el paso 3. Total RNA-seq) o almacenar a-80 ° C durante al menos 1 mes.
-
Biotinilación de tiol-específicas del ARN recién transcrito
- Comenzar con 30-80 μg de ARN celular total. 60 μg RNA deben producir suficientes cantidades del RNA recién transcrito.
- Preparar el etiquetado de la reacción. Pipeta en el siguiente orden (por cada μg de RNA): 1 μl de 10 x Buffer de Biotinilación, 7 μl de RNA (que contenga 1 μg RNA diluido en libre de nucleasa H2O) y 2 μl biotina-HPDP (1 mg/mL). Añadir biotina HPDP última y mezclar inmediatamente mediante pipeteo. Envuelva los tubos con papel de aluminio para evitar la exposición a la luz. Ver la discusión de una alternativa a la biotina HPDP. Incubar a temperatura ambiente durante 1,5 h con rotación.
- Adecuado 2 mL los tubos de centrífuga (véase Tabla de materiales) a 15.000 x g durante 2 min pipetear todos biotinilado RNA en el tubo previamente centrifugada a 2 mL, añadir un volumen igual de cloroformo y mezclan vigorosamente. Incubar durante 2-3 min hasta que las fases comienzan a separarse y las burbujas comienzan a desaparecer.
- Centrifugar a 15.000 x g durante 15 min a 4 ° C. Cuidadosamente transferir la fase acuosa superior a un tubo nuevo.
- Repita los pasos del 2.3.3. y 2.3.4. una vez. Añadir el 10% del volumen de NaCl (5 M) y un volumen igual de isopropanol a la fase acuosa. Centrifugar a 20.000 x g durante 20 min a 4 ° C. Deseche el sobrenadante.
- Añadir que un volumen igual prepara el etanol del 75%. Centrifugue a 20.000 x g. descartar el sobrenadante centrifugado brevemente y eliminar el etanol restante. Resuspender completamente en 30-100 μl de H2O (use 1 μl H2O por 1 μg de RNA entrada de paso 2.3.1) mezclando la pipeta.
- Verificar integridad del RNA por análisis electroforético, o tomar una alícuota y verificar más adelante.
-
Separación de recién transcrito (etiquetado) y preexistente ARN (sin etiqueta)
- Quitar granos paramagnéticos (véase Tabla de materiales) de almacenamiento de 4 ° C y déjela reposar por al menos 30 min a temperatura ambiente. Tampón de lavado calor 4sU (3 mL por muestra) a 65 ° C.
- Preparar solución de Ditiotreitol (DTT) de 100 mM. Pesar TDT a escala ultra fina y añadir la cantidad necesaria de agua libre de nucleasa. Prepare siempre fresco. Utilizar 200 μL por muestra.
- Calentar muestras de RNA biotinilado (1 μg/μl) a 65 ° C por 10 min desnaturalizar y colocar inmediatamente en hielo. Añadir granos de estreptavidina de 100 μl a biotinilado RNA e incube a TA por 15 min con rotación.
- Colocar una columna correspondiente (véase Tabla de materiales para las recomendaciones) para cada muestra en el soporte magnético y las equilibre cada columna con tampón de lavado 1 mL temperatura 4sU.
Nota: Esto le llevará aproximadamente 5-10 minutos. Si alguna de las columnas no iniciar drenaje después de 5 minutos, presione suavemente sobre la columna con un dedo enguantado. - Aplicar una mezcla de granos de la RNA a la mitad de cada columna. Deseche el flujo a través de, a menos que el RNA necesita ser recuperado. Si es así, recoge el flujo a través y por lo menos el primer lavado. Realizar la recuperación del ARN según lo descrito por Rädle et al. (paso 1-7, 7. Recuperación de ARN sin etiqueta, sin consolidar)19.
- Lavar tres veces con tampón de lavado 4sU 0,9 mL (precalentado a 65 ° C de paso 2.4.2.) y 0,9 mL RT 4sU de tampón de lavado, respectivamente.
- Usar perlas paramagnéticas para recuperar el RNA recién transcrito. Pipetee 400 μL de bien dispersadas perlas paramagnéticas RT en un tubo de muestra y lugar debajo de cada columna. Eluir el RNA recién transcrito con 100 μl 100 mM DTT. Espere 3 minutos y realizar una segunda elución con 100 μl 100 mM DTT. (Opcional: realizar recuperación y elución descrito por Rädle et al.) 19
- Mezcla recién transcrito RNA cuentas completamente con la pipeta 10 tiempos de mezcla y proceder según pauta del fabricante. Eluir el RNA en 11 μl libre de nucleasa H2O.Quantify RNA usando un fluorómetro adecuado (véase Tabla de materiales). RNA se puede almacenar a-80 ° C durante al menos 1 mes.
Nota: Recién transcrito RNA puede usarse para preparar bibliotecas de cDNA para secuenciación de próxima generación (véase Tabla de materiales para una sugerencia sobre que kit a utilizar) o análisis aguas abajo. 100 - 500 ng RNA son suficientes para más kits de preparación de biblioteca (ver discusión).
3. total RNA-Seq
- Tomar directamente RNA de 4sU etiquetados RNA después de la preparación de RNA mediante el reactivo modificado para protocolo de aislamiento de RNA total RNA-seq (ver paso 2.2.2).
- Para la preparación de la biblioteca, diluir una alícuota del RNA a una concentración final de 50-100 ng / μl. utilizar el mismo kit en cuanto a preparación de la biblioteca del RNA recién transcrito. 100 - 500 ng RNA son suficientes para más kits de preparación de la biblioteca.
4. ribosoma perfiles
Nota: Para todos los métodos y puntos de tiempo consecutivos, las muestras deben ser de la misma agrupación a partir de las células. Recomendaciones sobre que equipo usar, consulte la Tabla de materiales.
- Preparación y aislamiento de ribosoma protegen fragmentos (PRSA):
- Utilizar cantidades adecuadas de células para cada punto del tiempo (ver Tabla 2 para el número recomendado de células T). Tratar células adherentes con cicloheximida como se describe en el protocolo del fabricante.
PRECAUCIÓN: La cicloheximida es altamente tóxico y puede provocar mutaciones. Evite el contacto con la piel y la inhalación. - Recoger y piscina no - o semi - adherent células de cada vez señalan en un tubo de polipropileno y ajustan la concentración final de 1 x 106 células por mL de medio de célula-específica (p. ej., RPMI suplementado para células T). Añadir cicloheximida con una concentración final de 0,1 mg/mL, mezclar por inversión del tubo polipropileno e incubar durante 1 minuto las células de la centrífuga durante 5 minutos a 330 x g a 4 ° C. Medio de aspirar y lavar las células con menos de 10 mL de PBS suplementado con cicloheximida (concentración final de 0,1 mg/mL).
- Células de la centrífuga durante 5 minutos a 330 x g a 4 ° C. Aspire el medio y añadir el tampón de lisis de células de mamífero de μl 100 por 10 x 106 células. Mezclar mediante pipeteo y expulsarla a través de una aguja estéril de calibre 25 22 Lyse las células completamente.
- Transferir el lisado celular a un tubo de 1,5 mL preenfriada. Incubar por 10 min en hielo con inversiones periódicas. Centrifugar 10 min a 20.000 x g a 4 ° C para aclarar el lisado. Transfiera el sobrenadante a un tubo de 1,5 mL preenfriada.
- Preparar un 1:10 dilución del lisado con agua libre de nucleasas y registro una A260lectura usando un espectrofotómetro. Usar agua libre de nucleasa como un espacio en blanco y un 1:10 dilución del tampón de lisis de células de mamífero como el estándar. Calcular la concentración A260/ml del lisado según la siguiente ecuación:
(Una260 célula lisada - un260 mamíferos tampón de lisis) x factor de dilución 10 =un/260ml - Crear 200 alícuotas μl del lisado en hielo y continuar con el tratamiento de la nucleasa.
Nota: Opcionalmente, preparar una alícuota de 100 μl de RNA total, añadir 10 μl de SDS 10% y mezclar. Almacenar a 4 ° C y proceder a 4.3.2. Se recomienda el uso de ARN total de RNA marcado con 4sU (consulte el paso 3. Total RNA-seq).
- Utilizar cantidades adecuadas de células para cada punto del tiempo (ver Tabla 2 para el número recomendado de células T). Tratar células adherentes con cicloheximida como se describe en el protocolo del fabricante.
- Huella de ribosoma
- Realizar tratamiento de nucleasa inmediatamente sin que se congele el lisado. Añadir 7,5 unidades de nucleasa (incluido en el kit recomendado) para cada A260 de lisado. Por ejemplo: 80 A260/ml lisado x 0,2 mL de lisado x 7,5 U/A260nucleasa = 120 nucleasa de U.
Nota: Opcionalmente, valorar la nucleasa para la digestión como es descrito por el fabricante. - Incubar la reacción de nucleasa por 45 min a temperatura ambiente con una mezcla suave. Congelar alícuotas de μl 200 del lisado con nitrógeno líquido y almacenar a-80 ° C, o detener la reacción de nucleasa mediante la adición de inhibidor de Rnasa de 15 μl a cada alícuota de 200 μl y continúe con el paso 4.3.2.
- Realizar tratamiento de nucleasa inmediatamente sin que se congele el lisado. Añadir 7,5 unidades de nucleasa (incluido en el kit recomendado) para cada A260 de lisado. Por ejemplo: 80 A260/ml lisado x 0,2 mL de lisado x 7,5 U/A260nucleasa = 120 nucleasa de U.
- Purificación de la PRSA
- Descongelar una muestra de la PRSA de nucleasas que digieren y agregar inhibidor de Rnasa de 15 μl. Mantener las muestras en hielo.
- Purificar la PRSA según protocolo del fabricante (se recomienda la purificación de la columna) y medir la concentración de RNA en un espectrofotómetro.
- rRNA agotamiento
- Utilice 5 μg de PRSA purificado de agotamiento del rRNA.
- Seguir protocolo del fabricante (paso 1-2, agotamiento del rRNA primaria) por agotamiento de rRNA. Concentración de RNA de medida del rRNA había agotado PRSA en un espectrofotómetro.
- Purificación de la página de la PRSA
- Uso 500 ng de rRNA había agotado PRSA para la purificación de la página.
- Preparar Control de RNA, muestras y escalera para la purificación de la página. Mezcla 5 μl Control de RNA y desnaturalización de 5 μl gel carga del tinte en un tubo de microcentrífuga de 0, 5 mL. Mezclar 10 μl de cada RPF con 10 μl de desnaturalización gel carga, respectivamente. Preparar una escalera alícuota (4 μL 20/100 escalera, agua libre de nucleasa de 1 μl y 5 μl desnaturalización gel colorante de carga). Carga entre cada muestra y control para prevenir la contaminación cruzada.
- Desnaturalizar las muestras y la escala por incubar a 95 ° C durante 5 minutos y colocar inmediatamente en hielo. Carga 20 μl de cada muestra (opcionalmente, cargar 10 μl y congelación restante las muestras a 20 ° C) separadas por 10 μl de escalera dispuesta en un gel de poliacrilamida urea 12% o 15%. Cargar 10 μl de RNA Control. Correr el gel hasta la banda de azul de bromofenol alcanza la parte inferior del gel (180 V, ~ 70 min) (figura 3).
- Tinción del gel según protocolo del fabricante a 4 ° C. Usar un transiluminador de campo oscuro que emite luz azul para visualizar el ARN. Impuestos especiales gel rebanadas para cada muestra corresponde a ~ 28 y 30 nt de longitud. Tomar el RNA Control como referencia y suprimirlo.
Nota: PRSA es apenas visible. Impuestos especiales-cortes en el tamaño indicado por el RNA Control - que contiene dos oligos de 28 y 30 nt de longitud - incluso si las muestras no son visibles. - Perfore un agujero en la parte inferior de los tubos de microcentrífuga de 0, 5 mL con una aguja estéril de calibre 20. Transferir cada rebanada de gel en un tubo separado y tubos lugar tapón en un tubo de 1,5 mL. Centrifugar durante 2 min a 12.000 x centrifugación de repetición g. Si rodajas de gel no se destruya completamente en el tubo de 1,5 mL.
- Eluir el RNA de rodajas de gel interrumpida con 400 μL de agua libre de nucleasa, acetato de amonio de 40 μl (5 M) y 2 μl de SDS (10%) cada noche a 4 ° C.
- Transferir la mezcla a tubos de filtro de 1,5 mL (suministrados con el kit recomendado) con una pipeta de 1 mL (Punta ancha o hecho a sí mismo 1 mL con extremo cortado). Centrifugar por 3 min a 2.000 x g para separar eluida RNA de rodajas de gel. Pipetee suavemente solución acuosa en un tubo de 1,5 mL. Añadir 2 glucógeno μl (suministrado con el kit recomendado) y 700 μl 100% isopropanol y almacenar a-20 ° C durante al menos 1 h.
- Centrifugar a 4 ° C por 20 min a 13.000 x g. descartar sobrenadante. Lavar el pellet con etanol de 80% recién preparada previamente enfriada a 4 ° C durante 10 min a 13.000 x descartar sobrenadante de g. y deje secar al aire. Resuspender cada muestra de 20 μl y el Control de la ARN en 8 μl de agua libre de nucleasa. Almacenar a-20 ° C si es necesario.
- Fragmentación, reparación final, 3' adaptador de la ligadura, transcripción inversa
- Realice el procedimiento descrito por protocolo del fabricante (fragmentación y reparación final, 3' adaptador ligadura y reversa de la transcripción).
- Página de purificación de cDNA
- Preparación de muestras, Control de RNA y escalera para la purificación de la página: mezclar 10 μl de cada muestra y Control de RNA con 10 μl desnaturalización gel carga colorante, respectivamente. Preparar una escalera alícuota (4 escalera μl 20/100, 1 μl nucleasa agua libre, 5 μl desnaturalización gel colorante de carga). Carga entre cada muestra y control para prevenir la contaminación cruzada.
- Desnaturalizar las muestras y la escala por incubar a 95 ° C durante 5 minutos y colocar inmediatamente en hielo. Carga 20 μl de cada muestra (opcionalmente, cargar 10 μl y congelación restante las muestras a 20 ° C) separadas por 10 μl de escalera dispuesta en un 10% de poliacrilamida/7 - 8 M gel de urea/TBE. Cargar 10 μl de RNA Control. Correr el gel hasta que el azul de bromofenol migra completamente sin el gel (180 V, ~ 60 min).
- Tinción del gel según protocolo del fabricante a 4 ° C. Usar un transiluminador de campo oscuro que emite luz azul para visualizar el RNA y suprimir las rodajas de gel para cada muestra corresponde a ~ 70-80 nt.
- Proceda como se describe en el paso 4.5.5 - 4.5.8 y resuspender cada muestra 10 μl de agua libre de nucleasa.
- cDNA circularización
- Preparar suficiente mezcla maestra de circularización para todas las reacciones mediante la combinación de los siguientes reactivos para cada muestra en hielo: μl 4.0 mezcla de reacción de circularización, 2.0 μL ATP, 2.0 μL MnCl2y 2.0 μL ligasa.
- Añadir 10 μl de la master mix a cada muestra. Mezclar suavemente y centrifugar brevemente. Incubar las muestras a 60 ° C por 2 h. inmediatamente lugar muestras en el hielo.
- Amplificación por PCR
- Seguir el protocolo del fabricante (pasos 1-3, amplificación por PCR) para la amplificación por PCR. Utilice 4 μL de cDNA circular para amplificación con 9 ciclos de PCR de las células T primarias, para lograr mejores resultados.
- Bibliotecas de purificar y controlar su distribución de tamaño, según protocolo del fabricante (paso 4-8, amplificación por PCR). El tamaño esperado de la biblioteca amplificado es 140-160 bp (ver figura 4).
- Para las bibliotecas de la secuencia, consulte protocolo del fabricante y la facilidad de la secuencia para mayor orientación.
5. chIP-Seq
Nota: Este protocolo se modifica Blecher-Gonen et al. 14 consulte su Protocolo para obtener más información sobre ChIP-seq. Para todos los métodos y puntos de tiempo consecutivos, las muestras deben ser de la misma agrupación a partir de las células.
- Reticulación y recolección de las células
- Número apropiado de la reticulación de las células (ver Tabla 2 para el número recomendado de células T) para cada punto del tiempo con una concentración final de 1% de formaldehído en un medio de cellspecific (p. ej., RPMI suplementado para las células de T) 10 min a temperatura ambiente con suave balanceo. La adición de glicina a una concentración final de 0,125 M para detener la reacción de reticulación.
- Centrifugar las células a 330 x g durante 5 min a 4 ° C. Deseche el sobrenadante y lave las células en PBS helado. Repita el paso 5.1.2 dos veces y congelar los pellets de células a-80 ° C. Pellets congelados se pueden almacenar durante al menos 6 meses.
- Lisis celular y sonicación
Nota: Durante todos la célula lysis y sonicación los pasos, las muestras deben guardarse en hielo o a 4 ° C para minimizar la degradación de proteína y revocación de reticulación.- Resuspender el pellet celular en tampón de lisis celular helada 1 mL con inhibidores de la proteasa recién agregado para aislar los núcleos (Añadir inhibidores de fosfatasas opcionalmente). Incubar por 10 min en hielo y centrifugar a 2.600 x g durante 5 min a 4 ° C.
- Aspirar el sobrenadante y resuspender el precipitado de núcleos en tampón de lisis de núcleos helados de 1 mL con inhibidores de la proteasa recién añadido (opcional: Añadir inhibidores de la fosfatasa). Incubar por 10 min en hielo. Someter a ultrasonidos las células para generar una fracción media de tamaño de DNA de 0,2 - 1,0 kb (ver figura 5).
Nota: Condiciones de sonicación condiciones deban ser optimizados según el tipo de la célula y más (p. ej., número de celular, volumen y buffer). Las células T primarias, sonicación durante ciclos de 20-25 se recomienda (ver materiales para descripción detallada). - Tomar alícuota de la cromatina esquilada y el calor durante 10 min a 95 ° C y 1.000 rpm agitación para realizar una retroceso rápido de la reticulación y verificar tamaño cromatina un 20-50 μl. Añadir 2-5 μl proteinasa K e incubar durante 20 min a 56 ° C y agitación de 1.000 rpm. Realizar inactivación calor durante 10 minutos a 95 ° C y agitación de 1.000 rpm. Purificar la cromatina con un kit adecuado (véase Tabla de materiales). Compruebe el tamaño de la cromatina en un gel de agarosa 1% y use 100 bp Plus marcador.
- Centrifugue cromatina esquilada con media fracción del tamaño de ADN de 0,2 - 1,0 kb durante 10 minutos a 20.000 x g y 4 ° C para pellets escombros y cromatina insoluble. Transferir el sobrenadante a un tubo nuevo y mantener en hielo.
- Mantener 5-10% de la cromatina sonicado como entrada. Congelar a-20 ° C (usado en el paso 5.5.2).
- Anticuerpos par a granos
- Anticuerpo de pareja 10 μg (p. ej., anti-ARN Pol II; H3K36me3 anti-Histon) en 220 μl PBS (con 0,5% BSA y 0.5% Tween 20) a 80 granos superparamagnético μl acoplados a proteína G (véase Tabla de materiales) durante al menos 1 h a temperatura ambiente con rotación.
- Colocar los tubos en un imán. Espere hasta que todos los granos están limitados al imán y eliminar el sobrenadante. Otro bloque con 6 μl sonicada esperma de salmón ADN en PBS (con 0,5% BSA y 0.5% Tween 20) durante 30 min a temperatura ambiente con rotación.
- Colocar los tubos en un imán. Espere hasta que todos los granos están limitados al imán y eliminar el sobrenadante. Lavar granos con tampón de IP ChIP tres veces.
- Inmunoprecipitación de cromatina
- Diluir la cromatina al volumen total de 1 mL de tampón de lisis de núcleos con inhibidores de la proteasa recién añadido (opcionalmente, añadir inhibidores de la fosfatasa). Agregar buffer ChIP IP con inhibidores de la proteasa recién añadido (opcionalmente, añadir inhibidores de fosfatasas) hasta un volumen final de 3 mL. Mantenga en hielo o a 4 ° C mientras que el anticuerpo se acopla a los granos.
- Añadir cromatina diluida al anticuerpo acoplado granos de paso 5.3.3 e incubar durante la noche a 4 ° C con rotación suave.
- Lavar con los siguientes buffers (1 mL cada uno, consulte la Tabla de materiales) a temperatura ambiente durante 5 minutos con rotación, coloque los tubos en el imán y eliminar sobrenadante: tampón de lavado I, tampón de lavado II, Buffer de lavado III y 2 x pH TE 8.0 respectivamente.
- Deseche el sobrenadante y secar al aire durante ~ 5 min.
- Reticulación inversa
- Extraer muestras del imán. Añadir 50 μl de tampón de elución y mezclar mediante pipeteo para eluir complejos de DNA de la proteína de los granos.
- Incluyen muestras de entrada de este paso hacia adelante. Añadir el tampón de elución para muestras para un volumen final de 50 μl (para mantener la composición del tampón similar a las muestras de ChIP) de entrada y de proceso junto con las muestras de ChIP.
- Mezcla 3 μl de tampón de elución y 2 μl de ARNasa (DNasa libre). Añadir 5 μl de la mezcla a cada muestra e incubar 30 min a 37 ° C.
- Mezcla 2.5 μl proteinasa K, 1 glucógeno μl y 1,5 μl de tampón de elución por muestra. Añadir 5 μl de la mezcla a cada muestra (1 U proteinasa K y 20 μg glucógeno por muestra) e incubar por 2 h a 37 ° C.
- Incubar las muestras a 65 ° C durante la noche (mínimo 4 h) con agitación para realizar crosslinking inversa.
- Colocar los tubos en el imán de al menos 30 s y transferencia el sobrenadante a un tubo nuevo. Las muestras pueden congelarse a-20 ° C hasta por 12 meses.
- Purificación de DNA
- Añadir 140 μl de perlas paramagnéticas bien dispersos a 60 μL de la muestra (ratio 2.3:1). Pipetee cuidadosamente hacia arriba y abajo 25 veces para homogeneizar. Asegúrese de que el líquido en cada tubo es homogéneo. Incubar a temperatura ambiente durante 2 min lugar los tubos en el imán 4 min, o hasta que todos los granos estén limitados al imán y descarte el sobrenadante.
- Dejar los tubos en el imán y añadir 200 μL de etanol al 70% recién preparada. Incubar los tubos durante 30 s sin molestar a los granos. Deseche el sobrenadante y repetir este paso una vez más. Aspirar completamente el etanol y dejar las paramagnéticas cuentas seque al aire durante 4 minutos.
Nota: Remoción incompleta del etanol puede seriamente reducir la recuperación de ADN y rendimiento. Secar el precipitado sólo hasta que se seque. Sobre-secar el sedimento puede reducir la recuperación de ADN y rendimiento. - Retirar los tubos del imán y añadir 20 μl 10 mM Tris-HCl (pH 8,0). Pipetee suavemente todo el volumen arriba y abajo 25 veces para homogeneizar. Incubar durante 2 min a temperatura ambiente. Coloque los tubos en el imán de 4 min y transferir el sobrenadante a otro tubo.
- Medir la cantidad de ADN con un fluorómetro adecuado (véase Tabla de materiales).
- Verifique que el ChIP era de qPCR (1 μl diluir en 100 μl de H2O y uso 2-5 μL para qPCR). Uso de iniciadores específicos para un positivo (sitio de Unión conocido de la proteína de interés) y control negativo (por ejemplo, un gen que es silencioso o no un objetivo de la proteína de interés).
Nota: Preparación de la biblioteca puede realizarse con 2 ng de DNA ChIP dependiendo el kit (véase Tabla de materiales para una sugerencia sobre que kit a utilizar).
Resultados
4 sU etiquetado: verificar óptimo 4 las condiciones de sU etiquetado (apoptosis, tensión nuclear, citoplasmático estrés), el tiempo y la concentración: 4sU elevada puede inhibir la producción y procesamiento del rRNA e inducen citoplásmicos como nucleares estrés30. Por lo tanto, las células de interés deben analizarse para 4sU inducida por estrés, así como apoptosis. Se recomiendan análisis de Western blot para la visualización de la acumulación de p53, que indica la tensión nuclear, aumentando los niveles de EIF2a fosfo que muestran tensión citoplasmática y celular activado por fluorescencia clasificación análisis (FACS) de apoptosis. Altos niveles y la larga exposición a 4sU o drogas como thapsigargin o arsenito se pueden utilizar para inducir estrés celular. Para inducir la apoptosis, muerte celular, las células fueron tratadas con BH3I-1 (500 ng/μL) o incubados por 5 min a 95 ° C (choque térmico). Anexina V/7-AAD tinción se utilizó para determinar apoptosis (anexina V) y las células muertos (7-AAD). Etiquetado de in vitro generados por las células Th1 primarias durante 0,5 h con 500 μm 4sU (concentración final) apoptosis (figura 1) sino provocar suficiente 4sU Constitución ni 1 h con 200 μm 4sU ni induce señales de estrés celular.
RNA etiquetado tiempo también puede ser acortado (≤5 min) que conduce a un aumento de breve duración intronic secuencias veces Etiquetadoras comparados más de largo. Para visualizar las tasas co-transcriptional de empalmes, etiquetado 4sU veces no deben exceder de 30 minutos. Para más detalles sobre 4sUlabeling, consulte Rädle et al. 19
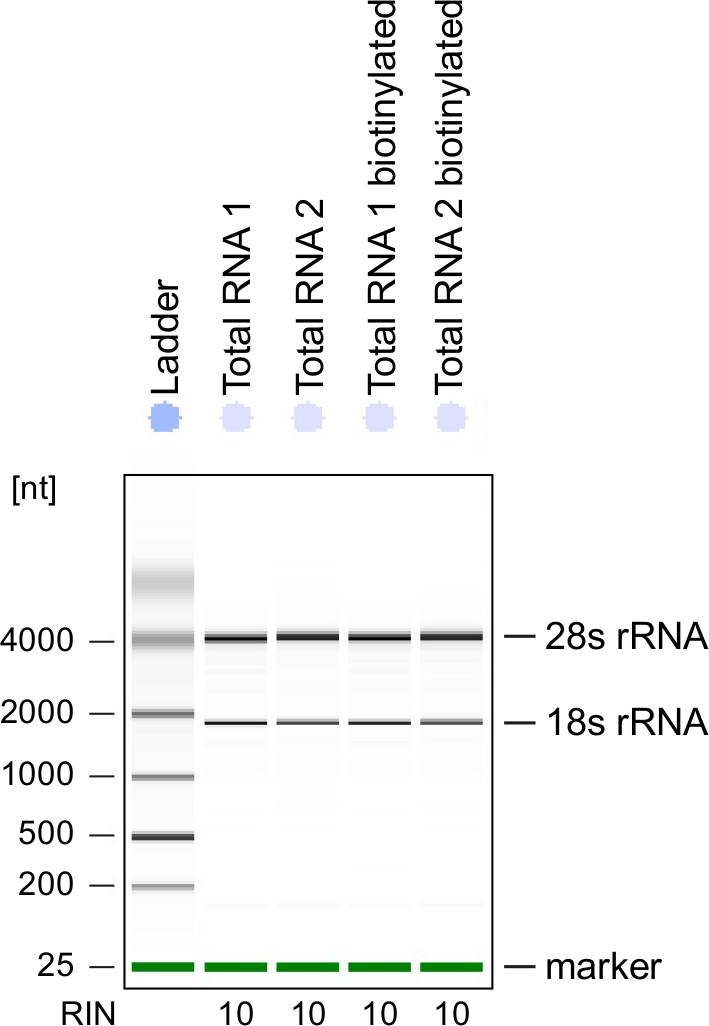
Control de calidad: Integridad del RNA es de gran importancia cuando se procesa el RNA. Es más conveniente para comprobar la calidad del RNA del RNA marcado con 4sU después de Biotinilación análisis electroforéticos (véase Tabla de materiales). Considerar la verificación de RNA aislado de paso 2.2.2, especialmente cuando se usa para la secuencia de ARN total. Número de integridad del RNA (RIN) debe ser ≥8 para asegurar la integridad del RNA para tratamiento posterior (figura 3).
Análisis electroforéticos pueden utilizarse también para verificar el RNA recién transcrito. Tenga en cuenta que el RNA recién transcrito contiene significativamente menos maduras rRNAs comparado con ARN total con las típicas bandas de rRNA es mucho menos prominente.
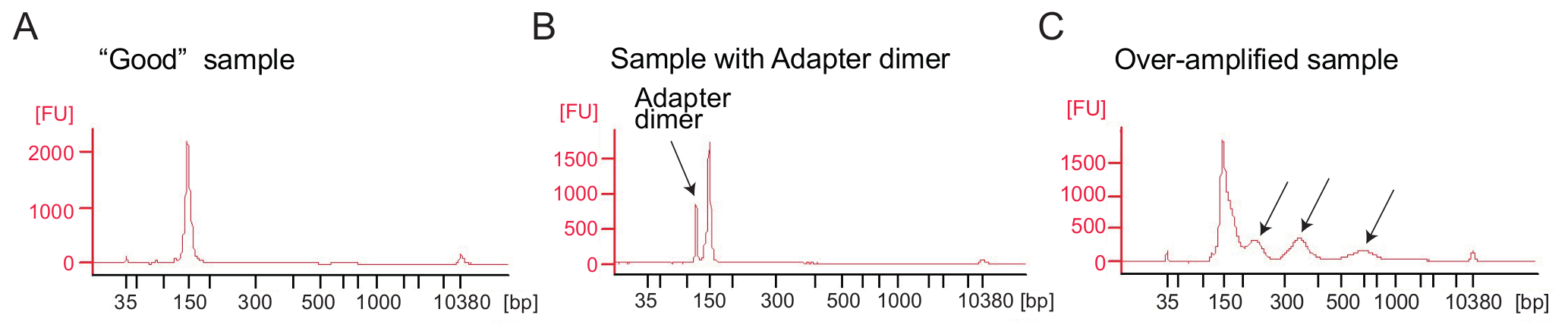
Ribosome profiling: amplificación por PCR de la biblioteca de cDNA: amplificación de cDNA (paso 4.9.1) es un paso crítico para asegurar buena secuencia resultados. Analizar las bibliotecas amplificadas por análisis electroforéticos. Una buena muestra de bibliotecas amplificadas muestra un pico alrededor de 140-160 bp (Figura 4A). Cantidades excesivas de adaptador dímeros deben ser evitado (Figura 4B) y las muestras deben ser purificadas más mediante el procedimiento de purificación de página según protocolo del fabricante (purificación de la página de productos de la polimerización en cadena). Pueden ocasionar demasiada plantilla o demasiados ciclos PCR amplificación excesiva caracterizada por la aparición de bandas de peso molecular mayor de lo esperado, untadas de PCR de productos y productos de dímero de adaptador (figura 4). Para la mayoría de las muestras 1-5 μl de cDNA circular y 9 ciclos PCR para la amplificación típicamente producirá cantidades suficientes del correcta del producto de PCR.
ChIP: cromatina de corte: Condiciones de corte óptimas deben ajustarse para cada tipo de célula. Determinar de antemano las condiciones de corte (por ejemplo, el número de ciclos de potencia alta o baja). Utilice el mismo número de células y el mismo volumen para probarlos, ya que una menor densidad celular aumenta la eficacia del corte. Trate de evitar la sobre - o bajo-corte la cromatina. Fragmentos de cromatina grande pueden afectar dramáticamente resultados ChIP por obstrucción y exceso de corte puede destruir epítopes de la proteína de interés, llevando a una menor eficacia vinculante por el anticuerpo. En este experimento, los mejores resultados se obtienen cuando la principal fracción de la cromatina esquilada fue alrededor de 1.000 bp o ligeramente inferior (figura 5A).
Verificación de ChIP por qPCR: Antes de empezar el ChIP, es recomendable comprobar si el anticuerpo utilizado es adecuado para el ChIP (si es posible, utilizar anticuerpos grado de ChIP) de ChIP-qPCR. Verificar el ChIP para la secuenciación por qPCR antes de iniciar la preparación de biblioteca (ver paso 5.6.5). Diseño de cebadores que se unen a un sitio de destino conocido de la proteína de interés. Si el sitio de destino exacto dentro de un gen es desconocido, varios pares de cartilla pueden utilizarse para analizar el gene y elementos reguladores asociados. Para RNAPII ChIP de Th1 células interferón, que es transcripcionalmente alza sobre el estímulo, y cartillas de actina pueden ser utilizados como control positivo. Sox9 e insulina sirven como un control negativo, dado que estos genes no se expresan en células Th1 (figura 5B). Recuerde no utilizar imprimaciones que abarca el exón, que normalmente se utilizan para qPCR de mRNA. También se puede usar un control de IgG para probar la especificidad del anticuerpo usado. Immunoprecipitated ADN puede medirse con un fluorómetro adecuado (véase Tabla de materiales). Cantidades de ADN hibridada por el control de IgG deben ser significativamente inferior comparados con la cantidad de ADN que el anticuerpo de interés.
Replica: prueba de importancia biológica: Es muy recomendable para realizar el experimento cinético para todos los métodos a partir de la misma agrupación de células para las células tengan la misma identidad de las muestras sin tratar y tratadas (figura 2). Sin embargo, se recomienda tomar pequeñas alícuotas de momentos principales de cada método comparar muestras biológica reproduce (por ejemplo, por qPCR, análisis FACS). Esto permite evaluar si el tratamiento para ambas repeticiones fue reproducible y podría continuar con la secuencia. Validación de las repeticiones debe realizarse mediante análisis de bioinformatical. Reproducibilidad de los resultados puede ser evaluada en términos de correlación entre los valores FPKM entre repeticiones y visualizados usando diagramas de dispersión (figura 6).

Figura 1: Verificación de las condiciones óptimas de etiquetado 4sU sin perturbar la fisiología de la célula (figura de Davari, et al. 11)
(A) detección de apoptosis de las células por el análisis FACS: In vitro genera Th0 células fueron tratadas con diferentes concentraciones de 4sU (indicado entre paréntesis) de 0.5 h, 1 h y 2 h, respectivamente. Tratamiento de BH3I-1 fue utilizado para inducir apoptosis determinada por anexina V, mientras que el choque del calor (a 5 min a 95 ° C) fue utilizado para inducir la muerte celular determinada por 7-AAD. (B) análisis de Western blot para p53 de 4sU tratados y las células T activadas: las muestras fueron etiquetadas con 200 μm 4sU para el tiempo indicado de la activación, excepto el punto de tiempo de 0.5 h que fue etiquetado con 500 μm 4sU. (C) análisis de Western blot de fosfo-EIF2a y total EIF2a en células Th1 activadas con las mismas condiciones de etiquetado como en (B). Thapsigargin fue utilizado como control positivo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Resumen esquemático de una instalación cinética para seguimiento de los cambios del genoma
Este esquema muestra la configuración para combinar 4sU-seq, total RNA-seq, Ribosome Profiling y ChIP-seq para estudiar cambios de genoma sobre el tratamiento de la célula. Las células de la piscina y apartar el número de células de control no tratados. Tratar células restantes y para cada punto de método y tiempo. Células de etiqueta sin tratamiento/tratada para 4sU-seq con 4sU como se describe. Puntos del tiempo y las muestras de cada método dependen de la pregunta biológica específica examinada. Tomar muestras para cada punto del tiempo y método y seguir el dedicado parte del protocolo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: control de calidad de ARN marcado con 4sU
Total RNA y biotinilado RNA de las células Th1 activadas se analizaron en un equipo Bioanalyzer. 18S rRNA y 28S rRNA se muestran y número de integridad del RNA (RIN) está dada por el instrumento para determinar la integridad del ARN. RIN debe ser ≥8 para asegurar la integridad del RNA. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Perfiles de equipo Bioanalyzer de ribosoma perfiles de bibliotecas
(A) A buena biblioteca: muestra un pico en el rango de tamaño esperado (140-160 PB) y purificación adicional no es necesario. (B) este ejemplo muestra excesiva adaptador dimer producto amplificado (120 bp) en relación con el producto deseado (bp 140-160). Esta biblioteca requiere más purificación. (C) una muestra demasiado amplificada: picos de peso molecular mayor de lo esperado y borrosos los amplicones PCR son accesibles (indicado por las flechas). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Tamaño de cromatina óptimo después de la esquila y la verificación de ChIP por qPCR
(A) Agarose gel imagen muestra el tamaño óptimo del fragmento de la cromatina esquilado de tres muestras que fueron esquiladas para 25 ciclos en un sonicador y purificado como se describe antes en el protocolo. (B) resultados de la Q-PCR de un ChIP de RNAPII total (anti-RNA Pol II, 8WG16, ab817) se representa como un porcentaje de entrada. IFNg y actina cartillas fueron utilizados como un positivo mientras que Sox9 y la insulina son controles negativos (ambos genes no se expresan en las células Th1 activadas). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Comparación de biológico Replica (figura de Davari et al. 11)
Diagrama de dispersión representante comparando valores de expresión (FPKM) entre réplicas de recién transcrito (4sU) RNA 4 h después de la estimulación de las células Th1 activadas. La línea verde indica valores iguales de FPKM y correlación de rango se indica en cada parcela.
| Duración de etiquetado (min) | 4sU recomendada concentración (μm) |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| < 10 | 500 - 2000 |
Tabla 1: Se recomienda concentraciones de 4sU (de Rädle, et al. 19)
El rango de concentraciones recomendadas 4sU está indicado para diferentes tiempos de etiquetado.
| Método | Número de células | Cantidad de RNA |
| etiquetado 4sU | ≥ 2 x 107 | ≥60µg |
| Ribosome profiling | ≥ 2 x107 | |
| ChIP-seq | ≥ 2 x 107 - 3 x 107 |
Tabla 2: Cantidad necesaria de las células T primarias
Cantidad mínima requeridas primarias de células de T para cada método. Cantidades pueden ser menos tratándose de otros tipos de células.
Discusión
Analizar todo el proceso de regulación génica es necesario comprender las adaptaciones celulares en respuesta a un estímulo específico o tratamiento. La combinación total de RNA-seq, 4sU-seq, ribosome profiling y ChIP-seq en diferentes puntos temporales conduce a un análisis exhaustivo de los principales procesos de regulación génica en el tiempo. Un profundo entendimiento de los procesos biológicos es necesaria para definir la configuración experimental así como los puntos de tiempo óptimo.
Ya que los métodos para el estudio de regulación génica mejoran rápidamente, estos protocolos puede adaptarse a los cambios rápidos. Sin embargo, proporcionan los medios más importantes para el estudio de mecanismos de regulación genética básica en cualquier tipo de célula. Aquí, discutimos algunos de los escollos y hechos que uno tiene que considerar cuando se utilizan estos métodos.
Células: Las células deben ser altamente viables y, si usando las células aisladas primarias, pureza de poblaciones celulares debe garantizarse (por ejemplo, el análisis FACS para las células T primarias). Células un poco estresadas pueden influir en los resultados de estos métodos de secuenciación muy sensibles y disminuir la cantidad de RNA recién transcrito o traducido y conducir a lecturas no deseadas de la respuesta de estrés en los resultados de la secuencia. La velocidad de centrifugación mencionada en este protocolo para que sedimenten las células está optimizada para las células T primarias. Por lo tanto, ajustar la velocidad según el tipo de célula.
4 sU efectos sobre la fisiología de la célula: Además de las opciones mencionadas para verificar la mínima perturbación a la fisiología celular a 4sU además, pueden realizarse análisis más o adicional, especialmente cuando números de celda son limitados. Efectos sobre la proliferación celular pueden ser probados verificando el tiempo de duplicación de células simplemente contando las células etiquetadas y sin etiquetar. Inducción de estrés nucleolares podría probarse también mediante el análisis de la morfología celular mediante tinción de inmunofluorescencia de nucleolin y núcleos. Para verificar más lejos el impacto de 4sU, expresión génica global alterada se podía medir correlacionando leer cuentas de etiquetado ARN ARN total sin etiqueta.
Números de celular: En vitro generados por las células T, se recomienda a partir de por lo menos la cantidad de las células indicadas en la tabla 2. Elegir números apropiados por método según el tipo de células. Puesto que las células T tienen menos citoplasma y el ARN en comparación con otras células, probablemente menos cantidad de otras células será suficiente. Para ChIP-seq, número de células altamente depende el anticuerpo utilizado y el nivel de expresión de la proteína de interés dentro de las células. Para histonas o ChIP RNAPII, pueden utilizarse células particulariz, mientras que el número de células necesita incrementarse cuando se utilizan factores de transcripción, sobre todo si se expresan en niveles bajos.
4 sU etiquetado y RNA Biotinilación: Cuando se utilizan células adherentes, 4sU etiquetado puede dirigir según lo descrito por Rädle et al. 19 desde 4sU incorporan muy rápidamente de las células, puede ser agregado directamente al medio de suspensión, adherente o células semi-adherentes.
Se recomienda comenzar la Biotinilación con 60-80 μg de ARN. Sin embargo, se puede utilizar menos cantidad de RNA, aunque no probamos por menos de 30 μg. Añadir un coprecipitant (e.g., GlycoBlue) cuando precipitando RNA si el precipitado es difícil de ver. Duffy et al también han demostrado que biotina activa methylthiosulfonate (MTS-biotina) reacciona más eficientemente con ARN marcados con 4sU que HPDP-biotina31. Por lo tanto, cabe considerar cambiar a MTS-biotina, en particular, para la recuperación de ARNs pequeños, que tienden a tener menos residuos de uridina (véase el protocolo de Biotinilación mencionado por sede de Duffy et al.; purificación del ARN marcado con 4sU, Procedimientos experimentales).
Para la recuperación del ARN recién transcrito, es posible utilizar granos paramagnéticos o granos de ARN limpieza de su elección. Siempre tener en cuenta que estos kits pueden o no pueden purificar para RNAs específicos. Por ejemplo, si usted está interesado en miRNAs, considere el uso de kits específicos para la secuencia y la captura de miRNA.
Cuantificación de RNA recién transcrito: Para cuantificar con precisión el RNA recién transcrito, la medición debe ser realizada por un fluorómetro adecuado (véase Tabla de materiales). Dentro de 1 h de exposición 4sU, RNA recién transcrito representa alrededor del 1-4% del ARN total. RNA recién transcrito de 1 h con la etiqueta, las células T activadas consiste en ~ 90-94% de ARNr11.
Ribosome Profiling: Cuando se establezca el método, se determinó que utilizando 1.5 x la cantidad de nucleasa que sugerido en el protocolo original garantiza una digestión apropiada. También, efectos adversos no se han divulgado para cantidades elevadas de nucleasa. Ya que es muy difícil de overdigest la PRSA mientras que forman parte del RNA por proteínas ribosomales, todavía un poco puede aumentar la cantidad para valorar la digestión óptima nucleasa.
Si es inferior a 500 ng de RNA del FPR fueron recuperados en paso 4.4.2, repita el agotamiento de los rRNA y piscina purificaron PRSA con ARN Clean & concentrador de 5 columnas. Por otra parte, carga de dos muestras idénticas al lado uno al otro en gel (paso 4.5.3) y láminas de gel de piscina durante la elución de RNA del gel (paso 4.5.6).
Se recomienda cortar la PRSA sobre un gel tan ajustadamente como sea posible a las bandas de nt 28 y 30. Esto ayuda a eliminar fragmentos no deseados de rRNAs y tRNAs, que más tarde se convertirá en parte de su biblioteca y reducir lee la secuencia de la PRSA.
También es aconsejable evitar la luz durante la purificación del gel UV. Esto puede crear muescas en los fragmentos de ARN, como dímeros de pirimidina, que al final pueden afectar seriamente la preparación de la biblioteca y los resultados de secuenciación.
Preparación de la biblioteca y la secuencia de datos: Ribosoma perfiles de protocolo permite para generar una biblioteca de cDNA adecuada para secuenciación. Las muestras generadas por 4sU etiquetado pueden ser utilizadas directamente para la preparación de la biblioteca con cualquier kit de secuenciación apropiada de RNA. Desde el RNA recién transcrito, especialmente cuando uso corto etiquetado veces, puede no ser aún cofia, ninguna selección de poly-A debe ser realizada. En su lugar, recomendamos agotamiento del rRNA para evitar reducir la profundidad de la secuencia de la muestra real. Usando las células de T, comenzamos con 400 ng de ARN recién transcrito y total (según el kit, ver materiales), realiza rRNA agotamiento y reduce los ciclos para la amplificación de la polimerización en cadena minimizar el sesgo de la polimerización en cadena. Preparación de la biblioteca puede realizarse con menos material de partida. Para tener en cuenta números de complejidad biblioteca de ciclos PCR debe ser optimizado.
Para ChIP-seq también hay muchos kits disponibles para la preparación de la biblioteca. En nuestra biblioteca de manos preparación trabajó bien a partir de 2 ng de DNA ChIP (ver materiales para una sugerencia sobre que kit a utilizar). Asegúrese de comprobar los índices de balance de color durante la secuencia. Se recomienda la secuencia de profundidad ≥ 40 x 106 Lee cada 4sU-seq, total RNA-seq, y muestras de ChIP-seq y ≥ 80 x 106 lecturas para perfiles muestras del ribosoma. La profundidad de la secuencia depende de la muestra y el análisis de aguas abajo de la Bioinformática y se debe considerar cuidadosamente. Analizar lecturas intronic de empalme cotranscriptional, 100 secuenciación del extremo apareado bp debe ser elegido.
Sesgo de secuencia: La secuencia se ha convertido en el estándar de oro para determinar cambios globales en la transcripción, traducción o atascamiento del factor de transcripción. Dentro de estos últimos años, los métodos existentes fueron empujados hasta sus límites o fueron desarrollando nuevas técnicas para secuenciar cada vez más pequeñas cantidades a partir de ARN. Esto requiere la amplificación del cDNA, que introduce ruido o sesgo. Recientemente, identificadores únicos moleculares (UMIs) fueron desarrollados para identificar experimentalmente duplicados introducidos por PCR. Recientemente, fue demostrado que UMIs sólo levemente mejoran el poder de la secuenciación y tarifa falsa del descubrimiento de expresión génica diferencial32. Sin embargo, considerar el uso de identificadores únicos de moleculares (UMIs) para todas las bibliotecas de la secuencia de control para la complejidad de la biblioteca, especialmente cuando a partir de pequeñas cantidades de ARN y se necesitan muchos ciclos PCR.
De buffer y soluciones: Todos los búferes para 4sU-seq y perfiles de ribosoma deben ser preparados bajo estrictas condiciones libre de ARNasa, con agua libre de nucleasa. Se recomienda comprar prefabricada NaCl libre de nucleasa, Tris-HCl, EDTA, citrato de sodio y agua. Para asegurar condiciones libres de nucleasas, una solución descontaminante de Rnasa se puede utilizar para limpiar pipetas o superficies. Todos los búferes para ChIP-seq deben ser menos libres de DNasa y puede almacenarse a temperatura ambiente. Siempre añadir inhibidores de la proteasa y, opcionalmente, los inhibidores de la fosfatasa justo antes de usar y mantener en hielo.
Bioinformática: Análisis de los datos de la secuencia (es decir, ChIP-seq, RNA-seq y ribsosome perfiles) implica el control de calidad (p. ej., con FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), ajuste del adaptador (por ejemplo, con cutadapt20) seguido de asignación para el genoma de referencia de las células bajo estudio. Para datos de RNA-seq (total y 4sU-seq), así como perfiles de datos del ribosoma, un asignador de RNA-seq empalmado se requiere, como ContextMap 221. Para alineaciones de datos unspliced ChIP-seq, usando BWA-MEM22 es suficiente. Expresión génica puede ser calculada usando el RPKM modelo (lecturas por Kilobase de exón por millón fragmentos asignados)1, después de determinar leer cuentas por gene usando un programa, por ejemplo, featureCounts23. Para llamar de pico desde datos de ChIP-seq, un número de programas está disponible, por ejemplo, Mac24 o GEM25. Más análisis descendentes pueden realizarse en R26, en particular mediante herramientas proporcionadas por el proyecto de Bioconductor27.
Aquí, un reto importante en la integración de niveles de ARN 4sU y total y actividad traslacional de ribosome profiling es normalización. Un enfoque clásico para resolver este problema es normalizar niveles de genes de mantenimiento de la casa. Para reducir el ruido debido a las fluctuaciones al azar de los genes individuales de mantenimiento de la casa, se recomienda usar no sólo unos cuantos genes de mantenimiento de la casa pero niveles mediana para un conjunto más amplio, por ejemplo la > 3.000 genes de mantenimiento elaborados por Eisenberg y Levanon28 . Para el cálculo de las tasas de facturación RNA de ratios de 4sU-de que ARN total, normalización se basa en volumen de ventas promedio de precios (por ejemplo, suponiendo un período de RNA de 5 h)29. Sin embargo, puesto que esto no supone ningún cambio general para genes de mantenimiento de la casa, se recomienda utilizar enfoques de análisis independiente de la normalización, por ejemplo, la agrupación de una serie de tiempo de los diferentes tipos de datos para identificar grupos de genes basados en correlación con un comportamiento distinto en la transcripción y traducción durante la activación. Para una descripción detallada sobre la bioinformatical integración de los diferentes tipos de datos, nos referimos a la publicación original11.
Análisis de la integración de tarifas y datos de facturación: Un trabajo recientemente publicado33 determinado por un control de gene multiplexados (MGC) de vida media en comparación con métodos globales podría demostrar que vida media correlaciona mejor con los obtenidos por métodos de etiquetado metabólicos en comparación con otros métodos (p. ej., inhibición general de la transcripción por las drogas). Sin embargo, cabe mencionar que las diferencias entre los cálculos de Half-Life pueden surgir y han descrito 15,34. Responsables de la mayoría de los problemas y diferencias que son introducidas por la respuesta de estrés debido a la exposición prolongada 4sU. Por lo tanto, es indispensable excluir la respuesta de estrés presentada por etiquetado 4sU. Para validar aún más tasas de rotación, se recomienda el uso de MGCs.
Además, un conjunto de datos generado aquí podría emplearse para un análisis (p. ej., Reglamento de ARN largo no codificante) datos más integrante35,36.
Divulgaciones
Los autores declaran que no tienen intereses financieros que compiten.
Agradecimientos
Damos las gracias a Lars Dölken Consejo establecer 4sU etiquetado para las células T primarias; Elisabeth Graf y Thomas Schwarzmayr ayuda crítica en las generaciones de la biblioteca y la secuencia; Dirk Eick y Andrew Flatley para proporcionar los anticuerpos RNAPII y la célula de T; N. Henriette Uhlenhaut y Franziska Greulich para ayuda en la preparación de la biblioteca de ChIP-seq; Caroline C. Friedel fue apoyado por becas de FR2938/7-1 y CRC 1123 (Z2) de la Deutsche Forschungsgemeinschaft (DFG); Elke Glasmacher fue apoyado por la beca GL 870/1-1 de la Deutsche Forschungsgemeinschaft (DFG) y del centro alemán de investigación de Diabetes (DZD), Helmholtz Zentrum München.
Materiales
| Name | Company | Catalog Number | Comments |
| 4sU-labeling | |||
| 4-thiouridine (100 mg) | Carbosynth | 13957-31-8 | Prepare 50 mM stock in sterile H2O/PBS; store at –20°C in aliquots of 50-500 µl; do not refreeze. |
| 1.5 ml safe-lock tubes | Eppendorf | 30121589 | Optional |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72692005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72694005 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| Agencourt RNAClean XP | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| Chloroform | Sigma Aldirch | 372978 | WARNING – HAZARDOUS TO HEALTH |
| Dimethylformamide | Sigma Aldrich | D4551 | |
| Dithiothreitol (DTT) | Roth | 6908.1 | Prepare as 100 mM DTT in nuclease-free H2O; always prepare fresh |
| Ethanol | Merck | 1.00983.1000 | |
| EZ-Link Biotin-HPDP (50 mg) | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg Biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4°C in aliquots of 1 ml. DMF dissolves some plastic materials. We recommend to use glass pipettes to transfer DMF from ist stock glass bottle to 50 ml Falcon tubes. |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| Isopropanol | Merck | 1.09634.1011 | |
| NaCl (5M) | Sigma Aldrich | 71386 | Stock solution |
| nuclease-free EDTA (500 mM ), pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| Nuclease-free H2O | Sigma Aldrich | W4502 | Stock solution |
| nuclease-free Tris Cl (1M), pH 7.4 | Lonza | 51237 | Stock solution |
| Phase Lock Gel Heavy tubes (2.0 ml) | 5Prime | 2302830 | Use in step 1.3.4. |
| Polypropylene 15 ml centrifuge tubes | Greiner Bio-One | 188271 | Or equivalent; they have to tolerate up to 15,000 × g |
| QIAzol Lysis Reagent (200 ml) | Qiagen | 79306 | Use this or equivalent TRI reagent for RNA isolation, WARNING – CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol) |
| Qubit RNA HS assay kit | Life Technologies | Q32852 | Use this kit for quantifying RNA quantity in step 1.4.11 |
| RNeasy MinElute Kit | Qiagen | 74204 | Optional; includes Buffer RLT |
| Sodium citrat | Sigma Aldrich | C8532 | Prepare 1.6 M stock solution using nuclease-free water |
| Tween 20 | Sigma Aldrich | P1379 | |
| µMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store at 4°C, includes µMacs columns used in step 1.4.6. (store at RT) |
| Cell viability and stress assay | |||
| PE Annexin V Apoptosis Detection Kit I | BD Biosciences | 559763 | Optional |
| Thapsigargin | Sigma-Aldrich | T9033 | Optional |
| p53 | abcam | ab26 | Optional |
| p-EIF2a (Ser51) | Cell Signaling | 9721 | Optional |
| BH3I-1 | Sigma-Aldrich | B 8809 | Optional |
| Buffers | |||
| 4sU Washing Buffer | store at RT | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O | |
| Biotinylation Buffer (10x) | store at 4 °C | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water; make aliquots of 1 ml; store at 4°C | |
| RNA precipitation buffer | store at RT | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | |
| RNA 6000 Nano Kit | Agilent | 5067-1511 | Use this kit to verify RNA integrity in step 1.3.10 |
| RNA 6000 Pico Kit | Agilent | 5067-1513 | Optional |
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use in step 1.2.2/3.1.8/3.3.3/3.4.3 |
| High-speed centrifuge | Thermo Scientific | Heraeus Multifuge X3R | Or equivalent equipment capable of reaching 13,000 × g |
| High-speed rotor | Thermo Scientific | Fiberlite F15-6 x 100y | |
| Adaptors for 15 ml tubes | |||
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer C | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 µMacs columns. |
| Ultra-fine scale | Mettler Toledo | ML204T | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
| DynaMag-2 Magnet-1 each | Life Technologies | 12321D | |
| RNaseZap | Sigma | R2020 | Optional |
| TruSeq stranded total RNA library prep kit | Illumina | RS-122-2201 | Or equivalent. For T cells we used 400 ng 4sU and Total RNA with 11 cycles for PCR amplification. rRNA depletion is included in this kit |
| Nanodrop | Thermo Scientific | use a Nanodrop or equivalent instrument to measure RNA concentration | |
| Ribosome Profiling | |||
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPYSC12116 (Yeast) | |
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPHMR12126 (Mammalian) | |
| Illustra MicroSpin S-400 HR Columns | GE Healthcare | 27-5140-01 | |
| RNA Clean & Concentrator-25 kit | Zymo Research | R1017 | |
| RNA Clean & Concentrator-5 kit | Zymo Research | R1015 | |
| Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) | Illumina | MRZG126 or MRZG12324 | |
| (High Sensitivity DNA Kit) | Agilent Technologies | 5067-4626 | Already needed for 4sU-seq |
| All other consumables and equipment are listed in the User guide | !!! | Carefully read the user guide and order required consumables in advance (consider a long delivery time for some consumables e.g. gels) | |
| ChIP | |||
| 10 mM Tris-HCl (pH 8.0) | gereral lab supplier | ||
| 100 bp Plus Marker | Thermo Fisher | SM0323 | |
| 16 % Formaldehyde | Thermo Fisher | 28908 | Add to a final concentration of 1 % |
| 70% EtOH | gereral lab supplier | Always prepare fresh | |
| Agarose | gereral lab supplier | ||
| Agencourt RNAClean XP beads | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| ChIP library preparation kit | KapaBiosystems | KK8504 | Or use the kit of your choice |
| DNA low bind microcentrifuge tubes | Eppendorf | Z666548-250EA | or equivalent |
| Dynabeads Protein G | Invitrogen | 10004D | Use these superparamagnetic beads coupled to protein G in step 4.3.1.; Bring to RT before use |
| Glycine | gereral lab supplier | Prepare a 2M stock solution | |
| Glycogen | Roche | 10-901-393-001 | |
| MinElute PCR Purification Kit | Qiagen | 28004 | Use this kit (or equivalent) to purify chromatin in step 4.2.4. |
| Phosphatase Inhibitor (PhosStop) | Roche | 4906837001 | Add freshly to the buffer and keep on ice |
| Power SYBRgreen Master mix | Thermo Fisher | 4367659 | |
| Protease Inhibitor (cOmplete, EDTA-free) | Roche | 11873580001 | Add freshly to the buffer and keep on ice |
| Proteinase K | Invitrogen | 25530049 | |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32851 | Use this kit for quantifying DNA quantity in step 4.6.4. on a Qubit Fluorometer |
| Rnase, DNase free | Roche | 11-119915001 | |
| Salmon sperm (sonicated to around 100bp) | Sigma | D1626 | |
| TE pH 8.0 | gereral lab supplier | ||
| Antibodies (ChIP grade if possible) | |||
| anti-RNA Pol II [8WG16] | abcam | ab817 | |
| anti-Histon H3K36me3 | abcam | ab9050 | |
| or antibody of interest | |||
| Buffers | |||
| Binding/Blocking buffer | Store at RT | PBS with 0.5 % BSA and 0.5 % Tween 20 | |
| Cell-Lysis buffer | Store at RT | 5 mM Pipes [pH 8.0], 85 mM KCl, and 0.5 % NP40 | |
| ChIP IP buffer | Store at RT | 0.01 % SDS; 1. 1% Triton X-100;1.2 mM EDTA; 16.7 mM Tris-HCl, pH 8.1; 16.7 mM NaCl | |
| Elution buffer | Store at RT up to 6 months | 10 mM Tris-HCl (pH 8.0), 5 mM EDTA (pH 8.0), 300 mM NaCl and 0.5 % SDS | |
| Nuclei-Lysis buffer | Store at RT | 50 mM Tris [pH 8.0], 10 mM EDTA, and 1 % SDS | |
| Wash buffer I | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20mM Tris-HCL pH 8.1; 150 mM NaCl | |
| Wash buffer II | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20 mM Tris-HCL pH 8.1; 500 mM NaCl | |
| Wash buffer III | Store at RT | 0.25 M LiCl; 1% NP-40; 1 mM EDTA; 10 mM Tris-HCl, pH 8.1 | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | use this instrument for electrophoretical analysis |
| Nanodrop | Thermo Scientific | ||
| Bioruptor TBX microtubes 1.5 ml | Diagenode | C30010010 | |
| or tubes special for your sonication device | |||
| Bioruptor sonication device or sonication device of your choice | Sonication of T cells with Bioruptor: 20 - 25 cycles (30 s on, 30 s off at high in two 1.5 ml bioruptor microtubes with 500 µl each tube) | ||
| Magnetic stand for tubes | |||
| Thermomixer | |||
| Agarose gel electrophoresis | |||
| Qubit Fluorometer | Thermo Scientific | Use this Fluorometer for quantifying low amounts of RNA/DNA |
Referencias
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 5 (7), 621-628 (2008).
- Chavez, S., Garcia-Martinez, J., Delgado-Ramos, L., Perez-Ortin, J. E. The importance of controlling mRNA turnover during cell proliferation. Curr Genet. 62 (4), 701-710 (2016).
- Rutkowski, A. J., et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun. 6, 7126 (2015).
- Ozsolak, F., Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 12 (2), 87-98 (2011).
- Carninci, P., et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 38 (6), 626-635 (2006).
- Churchman, L. S., Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 469 (7330), 368-373 (2011).
- Core, L. J., Waterfall, J. J., Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 322 (5909), 1845-1848 (2008).
- Hah, N., et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 145 (4), 622-634 (2011).
- Dolken, L., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14 (9), 1959-1972 (2008).
- Paulsen, M. T., et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67 (1), 45-54 (2014).
- Davari, K., et al. Rapid Genome-wide Recruitment of RNA Polymerase II Drives Transcription, Splicing, and Translation Events during T Cell Responses. Cell Rep. 19 (3), 643-654 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 22 (10), 2031-2042 (2012).
- Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 10 (10), 669-680 (2009).
- Blecher-Gonen, R., Barnett-Itzhaki, Z., Jaitin, D., Amann-Zalcenstein, D., Lara-Astiaso, D., Amit, I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc. 8 (3), 539-554 (2013).
- Schwanhausser, B., et al. Global quantification of mammalian gene expression control. Nature. 473 (7347), 337-342 (2011).
- Larsson, O., Tian, B., Sonenberg, N. Toward a genome-wide landscape of translational control. Cold Spring Harb Perspect Biol. 5 (1), a012302 (2013).
- Brar, G. A., Weissman, J. S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 16 (11), 651-664 (2015).
- Hafner, M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Radle, B., Rutkowski, A. J., Ruzsics, Z., Friedel, C. C., Koszinowski, U. H., Dolken, L. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. (78), (2013).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10-12 (2011).
- Bonfert, T., Kirner, E., Csaba, G., Zimmer, R., Friedel, C. C. ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics. 16, 122 (2015).
- Li, H., Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26 (5), 589-595 (2010).
- Liao, Y., Smyth, G. K., Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30 (7), 923-930 (2014).
- Zhang, Y., et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9 (9), R137 (2008).
- Guo, Y., Mahony, S., Gifford, D. K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 8 (8), e1002638 (2012).
- Team, R. D. C. . R: A Language and Environment for Statistical Computing. , (2016).
- Huber, W., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nature Methods. 12 (2), 115-121 (2015).
- Eisenberg, E., Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29 (10), 569-574 (2013).
- Friedel, C. C., Dolken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol Biosyst. 5 (11), 1271-1278 (2009).
- Burger, K., et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 10 (10), 1623-1630 (2013).
- Duffy, E. E., Rutenberg-Schoenberg, M., Stark, C. D., Kitchen, R. R., Gerstein, M. B., Simon, M. D. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 59 (5), 858-866 (2015).
- Parekh, S., Ziegenhain, C., Vieth, B., Enard, W., Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 6, 25533 (2016).
- Baudrimont, A., et al. Multiplexed gene control reveals rapid mRNA turnover. Sci Adv. 3 (7), e1700006 (2017).
- Rabani, M., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 29 (5), 436-442 (2011).
- Schlackow, M., Nojima, T., Gomes, T., Dhir, A., Carmo-Fonseca, M., Proudfoot, N. J. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol Cell. 65 (1), 25-38 (2017).
- Mukherjee, N., Calviello, L., Hirsekorn, A., de Pretis, S., Pelizzola, M., Ohler, U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. 24 (1), 86-96 (2017).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados