
Method Article
Echtzeit-Analyse des Transkriptionsfaktors verbindlich, Transkription, Übersetzung und Umsatz, globale Ereignisse während der zellulären Aktivierung anzuzeigen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die kombinatorische Verwendung von ChIP-Seq, 4sU-Seq, total RNA-Seq und Ribosom profiling für Zelllinien und Primärzellen. Nachverfolgen von Änderungen im Transkriptionsfaktor verbindlich, de Novo Transkription, RNS-Verarbeitung, Umsatz und Übersetzung im Laufe der Zeit und zeigt den gesamten Verlauf der Ereignisse in aktivierten und/oder sich rasch verändernden Zellen ermöglicht.
Zusammenfassung
Nach der Aktivierung Zellen rasch ändern ihre funktionale Programme und damit ihre Genexpressionsprofil. Massive Veränderungen in der Genexpression auftreten, beispielsweise bei der Zelldifferenzierung, Morphogenese und funktionelle Stimulation (z. B. die Aktivierung von Immunzellen), oder nach der Exposition gegenüber Drogen und andere Faktoren aus der lokalen Umgebung. Je nach Reiz und Zelle treten diese Änderungen schnell und auf jeder Ebene der Genregulation. Anzeigen aller molekularen Prozesse einer Zelle reagiert auf eine bestimmte Art von Reiz/Medikamenten ist eine der schwierigsten Aufgaben im Bereich der Molekularbiologie. Hier beschreiben wir eine Protokoll, die die simultane Analyse von mehreren Schichten der Genregulation ermöglicht. Wir vergleichen, insbesondere Transkriptionsfaktor verbindlich (Chromatin Immunopräzipitation-Sequenzierung (ChIP-Seq)), de-Novo -Transkription (4-Thiouridine-Sequenzierung (4sU-Seq)), mRNA-Verarbeitung, sowie Umsatz und Übersetzung (Ribosom Profiling). Durch die Kombination dieser Methods, ist es möglich, eine detaillierte und genomweite Vorgehensweise angezeigt.
Neu transkribierten RNA Sequenzierung empfiehlt sich besonders bei der Analyse von schnell anzupassen oder Wechselsysteme, da dies die transkriptionelle Aktivität aller Gene während der Zeit der 4sU Exposition zeigt (unabhängig davon, ob sie up- oder herunterreguliert). Die kombinatorische Verwendung von total RNA-Seq und Ribosom Profilierung ermöglicht darüber hinaus die Berechnung der RNA Umsatz und Übersetzung raten. Bioinformatische Analyse von Hochdurchsatz-Sequenzierung Ergebnisse ermöglicht viele Mittel für die Analyse und Interpretation der Daten. Die generierten Daten können auch verfolgen, co-transcriptional und alternative Spleißen, um nur einige Möglichkeiten zu nennen.
Der hier beschriebene kombinierten Ansatz kann für verschiedenen Modellorganismen oder Zelltypen, einschließlich Primärzellen angewendet werden. Wir bieten detaillierte Protokolle für jede Methode verwendet, einschließlich Qualitätskontrollen, und besprechen Sie mögliche Probleme und Fallstricke.
Einleitung
In den letzten Jahren geworden RNA-Sequenzierung (RNA-Seq) das Standardwerkzeug, alle geäußerten RNAs innerhalb einer Zelle oder eines Organismus1zu analysieren. Um den gesamten Prozess der Zellen als Reaktion auf einen bestimmten Reiz/Medikament Anpassung zu verstehen, ist es jedoch notwendig, vollständig alle zugrunde liegenden Prozesse, Bearbeitung, Umsatz und Übersetzung von mRNA-Transkription bis fest. Kurzfristige Änderungen im RNA-Transkription können kaum von insgesamt RNAseq gemessen werden, da Änderungen von Gesamt-RNS abhängig von Faktoren wie z.B. RNA Halbwertszeiten und transkriptionelle Aktivität sind eine schlechte Vorlage zu reflektieren die Anpassung der Zellen zu Auswirkungen auf die Umwelt2,3. In der Tat eine Vielzahl von neuen Sequenzierung Techniken wurden entwickelt, die eine Analyse der verschiedenen Schritte im Prozess der gen Verordnung4 Kombination in der richtigen Weise zu ermöglichen. Dieses Protokoll beschreibt, wie einige ziemlich leicht anwendbare Sequenzierung Techniken kombinieren, die Verfolgung der Verordnung der wesentlichen Schichten der mRNA in vergleichender Weise zu ermöglichen. Für die Analyse von transkriptionelle Aktivität, eine Vielzahl von Methoden beschrieben worden, wie z.B. GAP-Analyse von gen Ausdruck (CAGE)5, native Dehnung Transkript Sequenzierung (NET-Seq)6und genomweite nuklearen Nachlauf (GRO-Seq)7 , 8, sowie Bromouridine-Sequenzierung (Bru-Seq) und 4-Thiouridine-Sequenzierung (4sU-FF), die Metaboliten zu verwenden, die in neu eingebaut sind transkribierte RNA9,10, um nur um einige wenige zu nennen. Während CAGE die genaue Transkription Startsite identifiziert, NET-Seq und GRO-Seq genauere Informationen liefern über das Lesen von Richtungen und 4sU-Seq (das ist die hier beschriebene Methode) nur neu erkennt transkribiert RNA. Jedoch 4sU-Seq ist sehr empfindlich und kann angewendet werden, in verschiedenen Zeitrahmen zu messen quantitative transkriptionelle Aktivität aktiv zu ändern, Zellen sowie quantitative Veränderungen in der mRNA-Verarbeitung (was innerhalb von Minuten)9, 11,12. Darüber hinaus ist 4sU-Seq ideal kombinieren mit RNA-Seq RNA Umsatzraten für Gene9zu berechnen. Die Transkription der mRNA durch RNA-Polymerase II (RNAPII) erfolgt, die wiederum durch eine Vielzahl von Faktoren beeinflusst wird, wie z. B. Transkriptionsfaktoren, Histon-Modifikationen und allgemeine Aktivatoren/Repressoren, die können sogar die transkriptionelle gehören komplexe. Um zu testen wie viele gen/Veranstalter/Enhancer Regionen von einem Faktor gebunden sind, hat ChIP-Seq entwickelt, das ist jetzt die standard-Methode für diesen Zweck, aufgrund der vielen im Handel erhältlichen Antikörper13. Jedoch obwohl ChIPseq klaren Informationen gibt über wo Bind regulieren Faktoren, spiegelt es nicht wenn es in der Tat zu Veränderungen der Transkription14 führt. Durchführung von ChIP-Seq mit 4sU-Seq ist daher, die ideale Kombination für solche biologischen Fragen. Regulation der Genexpression kann auch zu einem späteren Zeitpunkt auftreten, da mRNA und Protein Ebenen nicht unbedingt15,16 korreliert, Angabe möglicherweise erhebliche Verordnung über die translational oder post-translationalen Ebene, je nach Kontext. Im Jahr 2011 Ribosom Profilierung hatte zunächst mit RNA-Seq kombiniert worden und ist jetzt die Methode der Wahl, um Änderungen zu quantifizieren, die schnell an Eiweiß, auftreten, da gibt es noch einige Sensitivitätslimite mit Massenspektrometrie17. In der Tat Umrechnungskurse von solchen Methoden erhalten haben gezeigt, dass bieten eine relativ gute Einschätzung der Änderungen im Protein-Ebene (zumindest für langfristige Veränderungen gemessen) und ermöglichen eine noch detailliertere Sicht auf den Prozess der Übersetzung, z. B. die Bestimmung der Start-Seite und alternative Übersetzung Rahmen17. Die kombinatorische Verwendung aller vier Methoden im Steady-State, zwischen verschiedenen Zelltypen, verwendet werden oder in die Zeit-Serie von einer sich rasch verändernden Zelle11zu experimentieren. Dieser Einsatz bietet eine genomweite Übersicht über Änderungen in Transkription Faktor Bindung RNA-Transkription, Bearbeitung und Übersetzung zu beeinflussen.
Protokoll
Alle Methoden sind entsprechend und Einhaltung der Helmholtz Zentrum München institutionelle, bundesstaatlichen und staatlichen Richtlinien.
1. Vorbereitung
- Machen einen detaillierten Plan des experimentellen Aufbaus auch einen Zeitplan, wann das Zellkulturmedium 4sU hinzu, und wenn die Zellen für jede Methode zu ernten. Abhängig von der biologischen Frage abwägen Sie sorgfältig Zeitpunkte für Beispielzeichnung, 4sU Kennzeichnung Zeit und Konzentration (Tabelle 1).
Hinweis: Überprüfen Sie die Auswirkungen von 4sU im Zellviabilität und betonen Sie die Antwort im Voraus (siehe "Verify 4sU Kennzeichnung Optimalbedingungen" Vertreter und Abbildung 1). Es wird empfohlen, eine Vorprüfung der einzelnen Methoden mit mindestens eine Probe durchführen. Überprüfen Sie, ob die Qualität und die Menge an RNA/DNA ist ausreichend für tiefe Sequenzierung (siehe gewidmet Teile des Protokolls), und schnell aber schonende Behandlung der Zellen während des Experiments. - Berechnen Sie die Anzahl der Zellen, die für jeden Zeitpunkt der einzelnen Methoden benötigt (siehe Tabelle 2 für eine grobe Abschätzung bei primären T-Zellen). Berücksichtigen Sie auch die Sequenzierung weniger Proben von Gesamt-RNS als nur das 4sU RNA (z. B.nur zum Zeitpunkt Übersetzung Preise oder RNA-Umsatz, die Preise berechnet werden soll). Erstellen Sie zur Bestätigung und Überprüfung der Signifikanz der Ergebnisse (empfohlen), mindestens eine biologische replizieren.
Hinweis: Wichtig ist, müssen für alle Methoden und aufeinander folgenden Zeitpunkten Proben aus dem gleichen Ausgangspunkt Pool der Zellen sein. Mindestens eine engagierte Forscher für jede Methode wird empfohlen. - Bereiten Sie alles notwendige im Voraus (z.B., regelmÄÑig 4sU Cycloheximide, Mammalian Lysis Puffer und 1 % Formaldehyd). Vermeiden Sie nach Zugabe von 4sU die Zellen auf helles Licht, da dies zur Vernetzung mit der Bezeichnung von 4sU RNA auf zelluläre Proteine18führen kann.
- Bündeln Sie alle Zellen von Interesse in einem Kolben kurz vor Behandlung (Abbildung 2). Zählen der Zellen (z.B.mit einem Hemocytometer) und verwenden Sie die erforderliche Menge für die unbehandelten Kontrolle der einzelnen Methoden (vergessen Sie nicht, auch die unbehandelte Kontrolle für 4sU-Seq mit 4sU kennzeichnen). Verbleibende Zellen zu behandeln und unverzüglich die erforderliche Anzahl von Zellen für jeden Zeitpunkt und Methode aufgeteilt. Behandeln Sie Zellen so schnell wie möglich Stress aufgrund von Änderungen in der Temperatur oder CO2 -Konzentration zu minimieren.
Hinweis: zum Beispiel werden Proben entnommen, 1 h, 2 h, 3 h nach der Behandlung, dann ein Viertel der Zellen dient zur unbehandelten Kontrolle und drei Viertel sind für die behandelten Steuerung verwendet.
2. 4sU-Kennzeichnung
Hinweis: Dieses Protokoll ist geändert von Rädle, Et Al. 19 beziehen sich auf deren Protokoll für weitere Informationen zu metabolischen Etikettierung mit 4sU. Für alle Methoden und aufeinander folgenden Zeitpunkten müssen Proben aus dem gleichen Ausgangspunkt Pool der Zellen stammen.
-
Beginn der Kennzeichnung
- RegelmÄÑig 4sU kurz vor Gebrauch auftauen. Fügen Sie 4sU zu jedem Zeitpunkt direkt zu Zellen von Interesse-haltigem Medium (siehe Tabelle 2 für empfohlene T-Zell-Zahlen, mindestens 60 µg RNA pro Zeitpunkt), mischen, und setzen Sie wieder in den Inkubator. Entsorgen Sie verbleibende 4sU (nicht wieder einfrieren).
- Am Ende der Beschriftung sammeln Sie Zellen (z.B. Zelle Schaber) und Zentrifuge bei 330 X g für 5 min bei 4 ° C in Polypropylenröhrchen (die hohen g-Kräften zu widerstehen). Aspirieren Medium und fügen Reagenz zur RNA-Isolierung (≥1 mL pro 3 x 106 Zellen, siehe Materialien für Empfehlung welche zu benutzen) um jedes Rohr. Voll Aufschwemmen Sie Pellet (≥1 mL pro 3 x 106 Zellen), 5 min bei Raumtemperatur (RT) inkubieren Sie und frieren Sie der Proben bei-20 ° C ein. Proben können für mindestens 1 Monat bei-20 ° C gelagert werden.
Vorsicht: Die Reagenzien zur RNA-Isolierung sind extrem gefährlich, wenn immer in Kontakt mit Haut oder Augen. Behandeln Sie diese pfleglich zu und beachten Sie die Sicherheitshinweise.
-
RNA Vorbereitung mit RNA Isolierung Protokoll geändert
- 0,2 mL Chloroform pro 1 mL Reagenz zur RNA-Isolierung, und gründlich mischen durch Schütteln für 15 S. fortfahren, wie bereits in den metabolischen Kennzeichnung Protokoll (Schritt 1-12, 2. RNA Vorbereitung Nutzung veränderter Trizol Protokoll) von Rädle Et Al. 19
- RNA-Konzentration zu messen (siehe Tabelle der Materialien), gemäß den Anweisungen des Herstellers. Verwenden Sie diese RNA für total RNA-Seq (siehe Schritt 3. Total RNA-Seq) oder bei-80 ° C für mindestens 1 Monat aufbewahren.
-
Thiol-spezifische Biotinylierungen neu transkribierten RNA
- Beginnen Sie mit 30-80 µg der gesamten zellulären RNA. 60 µg RNA sollten ausreichende Mengen an neu transkribierten RNA ergeben.
- Kennzeichnung Reaktion vorzubereiten. Pipette in der folgenden Reihenfolge (pro µg RNA): 1 µL 10 x Biotinylierungen Puffer, 7 µL RNA (mit 1 µg RNA verdünnt in Nuklease-freie H2O), und 2 µL Biotin-HPDP (1 mg/mL). Biotin-HPDP letzten und mischen Sie sofort durch pipettieren. Wickeln Sie Rohre mit Alu-Folie, um helles Licht zu vermeiden. Siehe die Diskussion nach einer Alternative zu Biotin-HPDP. 1,5 h mit Rotation bei RT inkubieren.
- Zentrifuge entsprechende 2 mL Röhrchen (siehe Tabelle der Materialien) bei 15.000 x g für 2 min. Pipette aller biotinylierte RNA in der Pre-gesponnen 2 mL Tube, fügen Sie ein gleiches Volumen von Chloroform und mischen Sie kräftig. Inkubieren Sie für 2-3 min bis die Phasen anfangen zu trennen und Luftblasen anfangen zu verschwinden.
- Zentrifuge bei 15.000 x g für 15 min bei 4 ° C. Übertragen Sie die oberen wässrige Phase sorgfältig in einen neuen Schlauch.
- Wiederholen Sie die Schritte 2.3.3. und 2.3.4. einmal. Fügen Sie 10 % des Volumens von NaCl (5 M) und ein gleiches Volumen von Isopropanol in der wässrigen Phase. Zentrifuge bei 20.000 x g für 20 min bei 4 ° C. Verwerfen Sie überstand.
- Fügen Sie ein gleiches Volumen 75 % Ethanol frisch zubereitet. Zentrifugieren bei 20.000 x g. verwerfen der Überstand, kurz drehen und entfernen die restlichen Ethanol. 30-100 µL H2O (Nutzung 1 µL H2O pro 1 µg Eingabe RNA aus Schritt 2.3.1) durch das Mischen von Pipette voll aufzuwirbeln.
- Integrität der RNA durch elektrophoretische Analyse zu überprüfen oder eine aliquote und später bestätigen.
-
Trennung von neu transkribiert (beschriftet) und bereits vorhandenen (unbeschriftete) RNA
- Entfernen Sie paramagnetischen Beads (siehe Tabelle der Materialien) aus 4 ° C Speicher und lassen Sie es für mindestens 30 Minuten stehen, um sie auf Raumtemperatur zu bringen. Wärme 4sU waschen Puffer (3 mL pro Probe) bis 65 ° C.
- 100 mM Dithiothreitol (DTT) Lösung ansetzen. Wiegen Sie DVB-t auf eine ultra-feine Waage und fügen Sie die erforderliche Menge an Nuklease-freies Wasser. Immer frisch zubereiten. Verwenden Sie 200 µL pro Probe.
- Erwärmen Sie biotinylierte RNA-Proben (1 µg/µL) auf 65 ° C für 10 min zu denaturieren und sofort auf Eis. Biotinylierte RNA 100 µL Streptavidin Perlen hinzufügen und 15 min. mit Rotation bei RT inkubieren.
- Eine entsprechende Spalte zu platzieren (siehe Tabelle der Materialien für Empfehlungen) für jede Probe in die Magnetstativ und Pre-equilibrate jede Spalte mit 1 mL Raumtemperatur 4sU waschen Puffer.
Hinweis: Dies dauert etwa 5-10 Minuten. Wenn eine der Spalten die Entleerung nach 5 min nicht starten, drücken Sie vorsichtig auf die Stütze mit einem behandschuhten Finger. - Eine RNA/Perlen-Mischung in die Mitte jeder Spalte anwenden. Flow-through zu verwerfen, es sei denn, unbeschriftete RNA wiederhergestellt werden muss. Wenn ja, sammeln Sie die Durchströmung und mindestens der ersten Wäsche. Führen Sie die Wiederherstellung der RNA wie Rädle Et Al. beschrieben (Schritt 1-7, 7. Wiederherstellung der unbeschrifteten, ungebundene RNA)19.
- Waschen Sie dreimal mit 0,9 mL 4sU waschen Puffer (bis 65 ° C vorgewärmt aus Schritt 2.4.2.) und 0,9 mL RT 4sU waschen Puffer, beziehungsweise.
- Verwenden Sie paramagnetische Perlen neu transkribierten RNA zu erholen. Pipette 400 µL gut dispergierte RT paramagnetischen Beads in einem Röhrchen pro Sample und Ort unter jeder Spalte. Eluieren Sie neu transkribierten RNA mit 100 µL 100 mM DTT. 3 min. warten Sie, und führen Sie eine zweite Elution mit 100 µL 100 mM DTT. (Optional: Elutions- und Wiederherstellung durchführen, wie beschrieben durch Rädle Et Al.) 19
- Mix neu transkribiert RNA/Perlen gründlich mit Pipette 10 Mischzeiten und gehen Sie nach den Hersteller-Richtlinie. Eluieren RNA in 11 µL Nuklease-freie H2O.Quantify RNA mit einem geeigneten Fluorometer (siehe Tabelle der Materialien). RNA kann mindestens 1 Monat bei-80 ° C aufbewahrt werden.
Hinweis: Neu transkribiert RNA kann verwendet werden, um die cDNA-Bibliotheken für Next Generation Sequencing vorzubereiten (siehe Tabelle der Materialien für einen Vorschlag, welche Kit zu verwenden) oder weitere nachgelagerte Analysen. 100 - 500 ng RNA sind ausreichend für die meisten Bibliothek Vorbereitung Kits (siehe Diskussion).
3. total RNA-Seq
- Nehmen Sie direkt RNA von 4sU beschriftet RNA nach RNA-Vorbereitung mit der modifizierten Reagenz für RNA Isolierung Protokoll für total RNA-Seq (siehe Punkt 2.2.2.)
- Vorbereitung der Bibliothek verdünnen einer RNA Aliquoten, eine Endkonzentration von 50-100 ng / µL. verwenden Sie das gleiche Kit Bibliothek Vorbereitung des neu transkribierten RNA. 100 - 500 ng RNA sind ausreichend für die meisten Bibliothek Vorbereitung Kits.
(4) Ribosom Profilierung
Hinweis: Für alle Methoden und aufeinander folgenden Zeitpunkten, müssen Proben aus dem gleichen Ausgangspunkt Pool der Zellen sein. Für Empfehlungen, welche Kit verwenden, beziehen sich auf die Tabelle der Materialien.
- Vorbereitung und Isolierung von Ribosom geschützt Fragmente (RPFs):
- Verwenden Sie entsprechende Mengen an Zellen für jeden Zeitpunkt (siehe Tabelle 2 für empfohlene T-Zell-Zahlen). Adhärente Zellen mit Cycloheximide zu behandeln, wie im Protokoll des Herstellers beschrieben.
Achtung: Cycloheximide ist hochgiftig und Mutationen verursachen kann. Vermeiden Sie Hautkontakt und Inhalation. - Sammeln und Pool nicht oder semi - adherent Zellen von jedem Punkt in einem Polypropylen-Rohr und passen Sie die Endkonzentration, 1 x 106 Zellen pro mL zellspezifische Medium (z. B.ergänzt RPMI für T-Zellen). Cycloheximide mit einer Endkonzentration von 0,1 mg/mL, mischen durch das Polypropylen Röhrchen umdrehen, und 1 min. Zentrifuge Zellen für 5 min bei 330 X g bei 4 ° c inkubieren Aspirieren Sie Medium und waschen Sie Zellen mit mindestens 10 mL PBS ergänzt mit Cycloheximide (Endkonzentration von 0,1 mg/mL).
- Zentrifuge Zellen für 5 min bei 330 X g bei 4 ° C. Aspirieren Medium und 100 µL Säugetier-Zelle Lysis Puffer pro 10 x 106 Zellen. Mischen von pipettieren und durch eine sterile 22 25-Gauge-Nadel zur lyse der Zellen vollständig zu vertreiben.
- Übertragen Sie die Zelle lysate auf eine vorgekühlte 1,5 mL-Tube. 10 min auf Eis mit periodischen Inversionen inkubieren. Zentrifuge für 10 min bei 20.000 x g bei 4 ° C, die lysate zu klären. Übertragen Sie den Überstand auf eine vorgekühlte 1,5 mL-Tube.
- Bereiten Sie eine 01:10 Verdünnung der lysate mit Nuklease-freies Wasser und Datensatz A260Lesung mit einem Spektralphotometer. Nuklease-freies Wasser zu verwenden, als eine leere und eine 01:10 Verdünnung der Säugetier-Zelle Lysis Puffer als Standard. Berechnen Sie die A260/mL Konzentration von lysate nach folgender Gleichung:
(Eine260 Zelle lysate - ein260 Säugetier-Lysis Puffer) x 10 Verdünnungsfaktor = ein260-/mL - Erstellen von 200 µL-Aliquots von der lysate auf Eis und mit Nuklease Behandlung fortfahren.
Hinweis: Optional, total RNA einer 100 µL aliquoten vorbereiten, 10 µL 10 % SDS und verrühren. Bei 4 ° C lagern und mit 4.3.2 fortfahren. Gesamt-RNS von 4sU beschriftet RNA wird empfohlen (siehe Schritt 3. Total RNA-Seq).
- Verwenden Sie entsprechende Mengen an Zellen für jeden Zeitpunkt (siehe Tabelle 2 für empfohlene T-Zell-Zahlen). Adhärente Zellen mit Cycloheximide zu behandeln, wie im Protokoll des Herstellers beschrieben.
- Ribosom-Fußabdruck
- Durchführen Sie Nuklease Behandlung sofort ohne zu frieren die lysate. Fügen Sie 7,5 Einheiten der Nuklease (enthalten im empfohlenen Kit) für jedes A260 von lysate. Zum Beispiel: 80 A260/mL lysate x 0,2 mL x 7,5 U/A260Nuklease lysate = 120 U Nuklease.
Hinweis: Optional, Titrieren Sie Nuclease für die Verdauung, wie vom Hersteller beschrieben. - Inkubieren Sie die Nuklease-Reaktion für 45 min bei RT mit schonendes Mischen. Eingefroren Sie 200 µL-Aliquots der lysate mit flüssigem Stickstoff und bei-80 ° C lagern Sie oder stoppen Sie Nuklease Reaktion zu, indem Sie 15 µL RNase-Inhibitor zu je 200 µL Aliquoten, und fahren Sie mit Schritt 4.3.2.
- Durchführen Sie Nuklease Behandlung sofort ohne zu frieren die lysate. Fügen Sie 7,5 Einheiten der Nuklease (enthalten im empfohlenen Kit) für jedes A260 von lysate. Zum Beispiel: 80 A260/mL lysate x 0,2 mL x 7,5 U/A260Nuklease lysate = 120 U Nuklease.
- Reinigung von RPFs
- Eine Probe der Nuklease verdaut RPFs aufzutauen und 15 µL RNase-Inhibitor hinzufügen. Halten Sie die Proben auf Eis.
- Reinigen der RPFs laut Protokoll des Herstellers (Spalte Reinigung wird empfohlen) und RNA-Konzentration auf ein Spektralphotometer messen.
- rRNA Erschöpfung
- Verwenden Sie 5 µg des gereinigten RPFs für rRNA Erschöpfung.
- Folgen des Herstellers Protokoll (Stufe 1-2, primäre rRNA Erschöpfung) für rRNA Erschöpfung. Maßnahme-RNA-Konzentration von rRNA erschöpft RPFs auf einem Spektrophotometer.
- Seite Reinigung des RPFs
- Einsatz 500 ng der rRNA erschöpft RPFs zur Seite Reinigung.
- Seite Reinigung RNA Kontrolle, Samples und Leiter vorbereiten. Mix 5 µL, die RNA-Kontrolle und 5 µL Denaturierung gel laden Farbstoff in einem 0,5 mL Microcentrifuge Schlauch. Mischung 10 µL jeder RPF mit 10 µL Gel geladen bzw. Denaturierung. Bereiten Sie eine Leiter aliquoten (4 µL 20/100 Leiter, 1 µL Nuklease-freies Wasser und 5 µL Denaturierung gel laden Farbstoff). Legen Sie es zwischen den einzelnen Probe und Kontrolle um Kreuzkontamination zu verhindern.
- Denaturieren Sie Proben und Leiter durch Inkubation bei 95 ° C für 5 min zu und sofort auf Eis. Laden Sie 20 µL jeder Probe (optional laden 10 µL und Einfrieren, die restlichen Proben bei 20 ° C) durch 10 µL vorbereiteten Leiter auf eine 12 % oder 15 % Harnstoff-Polyacrylamid-Gel getrennt. 10 µL RNA-Steuerelement zu laden. Das Gel laufen, bis die Bromophenol blue Band den Boden des Gels (180 V, ~ 70 min) erreicht (Abbildung 3).
- Fleck das Gel nach Protokoll des Herstellers bei 4 ° C. Verwenden Sie ein Dark-Feld Transilluminator, das blaues Licht zur Visualisierung der RNS emittiert. Verbrauchssteuern Gel Scheiben für jede Probe entsprechend ~ 28 und 30 nt in der Länge. Kontrollieren Sie RNA als Referenz und Verbrauchsteuern Sie es.
Hinweis: RPFs sind kaum sichtbar. Verbrauchssteuern Sie Scheiben in der Größe von der RNA-Steuerung - haltigen zwei Oligos von 28 und 30 nt Länge - angegeben, auch wenn Proben nicht sichtbar sind. - Durchstechen Sie ein Loch im Boden des Mikrozentrifugenröhrchen 0,5 mL mit einer sterilen 20-Gauge-Nadel. Übertragen Sie jede Gel-Scheibe in ein separates Rohr und Ort begrenzt Röhren in einem 1,5 mL-Tube. Zentrifuge für 2 min bei 12.000 x g. Wiederholung Zentrifugation wenn Gel Scheiben nicht vollständig in der 1,5 mL Tube vernichtet sind.
- Eluieren Sie die RNA aus gestörten Gel Scheiben mit 400 µL Nuklease-freies Wasser, 40 µL Ammoniumacetat (5 M) und 2 µL SDS (10 %) pro Nacht bei 4 ° c
- Übertragen Sie die Gülle auf 1,5 mL Filterschläuche (versehen mit dem empfohlenen Kit) mit 1 mL Pipettenspitze (Wide-Bohrung Tipp oder selbstgebauten 1 mL Spitze mit abgeschnittenen Ende). Zentrifuge für 3 min bei 2.000 x g trennen eluiert RNA aus Gel Scheiben. Sanft pipette wässrigen Lösung in einem 1,5 mL-Tube. Fügen Sie 2 µL Glykogen (versehen mit dem empfohlenen Kit) und 700 µL 100 % Isopropanol und Speicher bei-20 ° C für mindestens 1 h.
- Zentrifugieren Sie bei 4 ° C für 20 min bei 13.000 x g. überstand verwerfen. Das Pellet mit vorgekühlt frisch zubereitete 80 % Ethanol bei 4 ° C für 10 min bei 13.000 x g. verwerfen überstand zu waschen und trocknen. Jede Probe in 20 µL und die RNA-Kontrolle in 8 µL Nuklease-freies Wasser Aufschwemmen. Aufbewahren Sie bei-20 ° C, bei Bedarf.
- Fragmentierung, Ende Reparatur, 3' Adapter Ligation, Reverse Transkription
- Führen Sie das Verfahren, wie durch den Hersteller-Protokoll (Fragmentierung und Ende Reparatur, 3' Adapter Ligatur und reversen Transkription) beschrieben.
- Seite Reinigung des cDNA
- Bereiten Sie Proben, RNA-Kontrolle und Leiter zur Seite Reinigung: Mix 10 µL jeder Probe und RNA-Kontrolle mit 10 µL Denaturierung Gel Farbstoff, bzw. laden. Bereiten Sie eine Leiter aliquoten (4 µL 20/100 ladder, 1 µL Nuklease-freies Wasser, 5 µL Denaturierung Gel laden Farbstoff). Legen Sie es zwischen den einzelnen Probe und Kontrolle um Kreuzkontamination zu verhindern.
- Denaturieren Sie Proben und Leiter durch Inkubation bei 95 ° C für 5 min zu und sofort auf Eis. Laden Sie 20 µL jeder Probe (optional laden 10 µL und Einfrieren, die restlichen Proben bei 20 ° C) durch 10 µL vorbereiteten Leiter auf eine 10 % Polyacrylamid/7 - 8 M Harnstoff/FSME Gel getrennt. 10 µL RNA-Steuerelement zu laden. Führen Sie das Gel, bis die Bromophenol blue komplett aus dem Gel (180 V, ~ 60 min) wandert.
- Fleck das Gel nach Protokoll des Herstellers bei 4 ° C. Verwenden Sie ein Dark-Feld Transilluminator, das blaues Licht zur Visualisierung der RNS und Verbrauchsteuern die Gel-Scheiben für jede Probe entspricht ~ 70-80 nt emittiert.
- Gehen Sie wie in Schritt 4.5.5 - 4.5.8 beschrieben und Aufschwemmen Sie jede Probe in 10 µL Nuklease-freies Wasser.
- cDNA Circularization
- Bereiten Sie genügend Circularization master-Mix für alle Reaktionen durch die Kombination der folgenden Reagenzien für jede Probe auf Eis: 4.0 µL Circularization Reaktion Mix, 2.0 µL ATP und 2.0 µL MnCl22.0 µL Ligase.
- Jede Probe 10 µL des master-Mix hinzufügen. Vorsichtig mischen und kurz zentrifugieren. Inkubieren Sie Proben bei 60 ° C für 2 h sofort Platz auf dem Eis Proben.
- PCR-Amplifikation
- Folgen des Herstellers Protokoll (Schritt 1-3, PCR-Amplifikation) für PCR Verstärkung. Verwenden Sie 4 µL circularized cDNA für Verstärkung mit 9 PCR-Zyklen für primäre T-Zellen, um beste Ergebnisse zu erzielen.
- Reinigen Sie Bibliotheken zu und überprüfen Sie ihre Größenverteilung, laut Protokoll des Herstellers (Schritt 4-8, PCR-Amplifikation). Die erwartete Größe der verstärkten Bibliothek ist 140-160 bp (siehe Abbildung 4).
- Sequenzierung Bibliotheken finden Sie unter Protokoll des Herstellers und die Sequenzierung Anlage für weitere Hinweise.
(5) ChIP-Seq
Hinweis: Dieses Protokoll ist geändert von Blecher-Gonen, Et Al. 14 beziehen sich auf deren Protokoll für weitere Informationen zu ChIP-seq. Für alle Methoden und aufeinander folgenden Zeitpunkten müssen Proben aus dem gleichen Ausgangspunkt Pool der Zellen sein.
- Vernetzung und Ernte der Zellen
- Crosslink entsprechende Anzahl von Zellen (siehe Tabelle 2 für empfohlene T-Zell-Zahlen) für jeden Zeitpunkt mit einer Endkonzentration von 1 % Formaldehyd in einem Cellspecific Medium (z. B.ergänzt RPMI für T-Zellen) für 10 min bei Raumtemperatur mit sanften Schaukeln. Stoppen Sie die Vernetzungsreaktion durch die Zugabe von Glycin, eine Endkonzentration von 0,125 M.
- Zentrifuge Zellen bei 330 X g für 5 min bei 4 ° C. Den Überstand verwerfen und die Zellen in eiskaltem PBS waschen. Wiederholen Sie Schritt 5.1.2 zweimal und frieren Sie Zelle Pellets bei-80 ° C ein. Gefrorene Pellets können für mindestens 6 Monate gelagert werden.
- Lyse der Zelle und Beschallung
Hinweis: Bei allen Zellen lyse und Beschallung Arbeitsschritten, Proben auf Eis oder bei 4 ° C, Crosslink Umkehr und Protein Degradation zu minimieren aufzubewahren.- Aufschwemmen Sie Zelle Pellets in 1 mL eiskalte Zelle Lysis Puffer mit frisch hinzugefügten Protease-Inhibitoren, die Kerne zu isolieren (fügen Sie optional Phosphatase-Inhibitoren). 10 min auf Eis und Zentrifuge bei 2.600 x g für 5 min bei 4 ° c inkubieren
- Aspirieren überstand und Aufschwemmen Kerne Pellet in 1 mL eiskaltes Kerne Lyse Puffer mit frisch hinzugefügten Protease-Inhibitoren (optional: Fügen Sie Phosphatase-Inhibitoren). Inkubieren Sie für 10 min auf Eis. Beschallen Sie die Zellen, um eine mittlere DNA Größe Bruchteil von 0,2 - 1,0 kb zu generieren (siehe Abbildung 5).
Hinweis: Beschallung Bedingungen müssen optimiert werden, je nach Zelltyp und weitere Bedingungen (z.B., Zellzahl, Volumen und Puffer). Für primäre T-Zellen empfiehlt Beschallung für 20-25 Zyklen (siehe Materialien für detaillierte Beschreibung). - Nehmen Sie eine 20-50 µL aliquoten geschert Chromatin und Wärme für 10 min bei 95 ° C und 1.000 u/min schütteln um schneller Rücklauf Crosslink und Chromatin Größe überprüfen. Fügen Sie 2 bis 5 µL Proteinase K und 20 min bei 56 ° C und 1.000 u/min schütteln inkubieren. Durchführen Sie Hitzeinaktivierung für 10 min bei 95 ° C und 1.000 u/min schütteln. Chromatin mit einem geeigneten Kit zu reinigen (siehe Tabelle der Materialien). Überprüfen Sie Chromatin Größe auf einem 1 % Agarose-Gel und verwenden Sie 100 bp Plus Marker zu.
- Zentrifugieren Sie geschert Chromatin mit einem Mittelwert DNA Größe Bruchteil von 0,2 - 1,0 kb für 10 min bei 20.000 x g und 4 ° C um unlösliche Chromatin und Schutt pellet. Übertragen Sie den Überstand auf einen neuen Schlauch und halten Sie auf dem Eis.
- Halten Sie ca. 5-10 % der beschallten Chromatin als Eingabe. Frieren Sie bei-20 ° C (in Schritt 5.5.2 verwendet ein).
- Paar Antikörper zu Perlen
- Paar 10 µg Antikörper (z. B.Anti-RNA Pol II; Anti-Histon-H3K36me3) in 220 µL PBS (mit 0,5 % BSA und 0,5 % Tween 20) zu 80 µL superparamagnetischen Perlen verbunden zu G Protein (siehe Tabelle der Materialien) für mindestens 1 h bei Raumtemperatur mit Rotation.
- Legen Sie die Rohre auf einen Magneten. Warten Sie, bis alle Perlen sind, der Magnet verpflichtet und den Überstand zu entfernen. Weitere Block mit 6 µL sonorisiert Lachse Samenzellen DNA mit PBS-Puffer (mit 0,5 % BSA und 0,5 % Tween 20) für 30 min bei Raumtemperatur mit Rotation.
- Legen Sie die Rohre auf einen Magneten. Warten Sie, bis alle Perlen sind, der Magnet verpflichtet und den klare überstand zu entfernen. Waschen Sie Perlen mit ChIP IP-Puffer dreimal.
- Chromatin Immunopräzipitation
- Verdünnen Sie Chromatin zu 1 mL Gesamtvolumen in Kernen Lyse Puffer mit frisch hinzugefügten Protease-Inhibitoren (optional hinzufügen Phosphatase-Inhibitoren). ChIP-IP-Puffer mit frisch hinzugefügten Protease-Inhibitoren (optional hinzufügen Phosphatase-Hemmer) zu einem Endvolumen von 3 mL. Halten Sie auf dem Eis oder bei 4 ° C, während Antikörper an die Perlen gekoppelt ist.
- Fügen Sie verdünnte Chromatin an den Antikörper gekoppelt Perlen aus Schritt 5.3.3 und Inkubation über Nacht bei 4 ° C mit sanften Drehung.
- Waschen mit der folgenden Puffer (jeweils 1 mL siehe Tabelle of Materials) bei Raumtemperatur für 5 min mit Rotation, Rohre zurück auf den Magneten legen und entfernen Sie überstand: Wash Buffer I, waschen Puffer II, waschen Puffer III und 2 x TE pH 8.0 bzw..
- Überstand verwerfen und für ~ 5 min an der Luft trocknen.
- Umgekehrte Vernetzung
- Proben von den Magneten zu entfernen. 50 µL Elution Buffer und Mischen von pipettieren, um Protein-DNA-komplexen aus Perlen eluieren.
- Gehören Sie Eingabe Proben aus diesen Schritt nach vorn. Elution Puffer zur Eingabe Probe(n) für ein Endvolumen von 50 µL (um die Puffer Zusammensetzung ähnlich wie die ChIP-Proben zu halten) und zusammen mit dem ChIP-Proben.
- Mix 3 µL Elution Buffer und 2 µL RNase (DNase frei). Jede Probe 5 µL der Mischung hinzu und 30 min bei 37 ° c inkubieren
- Mix 2,5 µL Proteinase K, 1 µL Glykogen und 1,5 µL Elution Puffer pro Probe. Jede Probe (1 U Proteinase K und 20 µg Glykogen pro Probe) 5 µL der Mischung hinzu und 2 h bei 37 ° c inkubieren
- Inkubieren Sie die Proben bei 65 ° C über Nacht (mindestens 4 h) mit schütteln um umgekehrte Vernetzung durchzuführen.
- Legen Sie die Rohre auf den Magneten für mindestens 30 s und Übertragung der Überstand zu einem neuen Schlauch. Proben können bis zu 12 Monate bei-20 ° C eingefroren werden.
- DNA-Reinigung
- 140 µL gut dispergierte paramagnetischen Beads zu 60 µL der Probe (2.3:1 Verhältnis) hinzufügen. Pipette vorsichtig nach oben und unten 25 mal gründlich mischen. Stellen Sie sicher, dass die Flüssigkeit in jedem Röhrchen homogen ist. Inkubation bei Raumtemperatur für 2 min. Platz die Rohre auf den Magneten für 4 Minuten, oder bis alle Perlen sind verpflichtet, der Magnet und den Überstand verwerfen.
- Lassen Sie die Rohre auf den Magneten und fügen Sie 200 µL von frisch zubereiteten 70 % Ethanol. Inkubieren Sie die Rohre für 30 s ohne die Perlen zu stören. Verwerfen Sie den Überstand und wiederholen Sie diesen Schritt noch einmal. Aspirieren Ethanol komplett und lassen Sie die paramagnetischen Perlen für 4 Minuten an der Luft trocknen.
Hinweis: Unvollständige Ethanol Entfernung kann ernsthaft reduzieren DNA Erholung und Ertrag. Trocknen Sie das Pellet, nur, bis es trocken ist. Trocknung über das Pellet kann DNA Erholung und Ertrag. - Entfernen Sie die Rohre vom Magneten zu, und fügen Sie 20 µL 10 mM Tris-HCl (pH 8,0). Pipette vorsichtig das gesamte Volumen nach oben und unten 25 mal gründlich mischen. 2 min bei Raumtemperatur inkubieren. Setzen Sie die Rohre wieder auf der Magnet für 4 min und übertragen Sie den Überstand auf eine andere Röhre.
- Messen Sie die Menge der DNA mit einem geeigneten Fluorometer (siehe Tabelle der Materialien).
- Stellen Sie sicher, dass der ChIP von qPCR (verdünnter 1 µL in 100 µl H2O und Nutzung 2 bis 5 µL für qPCR) erfolgreich war. Verwenden Sie spezifische Primer für eine Positive (bekannte Bindungsstelle des Proteins des Interesses) und negativ-Kontrolle (z.B. ein Gen, das Stillen und/oder kein Ziel des Proteins des Interesses ist).
Hinweis: Bibliothek Vorbereitung kann durchgeführt werden, mit 2 ng DNA-ChIP-je nach Kit (siehe Tabelle der Materialien für einen Vorschlag, welche Kit zu verwenden).
Ergebnisse
4 sU Kennzeichnung: Überprüfen Sie optimale 4 sU-Kennzeichnung Bedingungen (zytoplasmatischen Stress, Apoptose, nukleare Stress), Zeit und Konzentration: Hohes Maß an 4sU können hemmen die Produktion und Verarbeitung von rRNA und zytoplasmatischen sowie nukleare Stress30zu induzieren. Daher sollte die Zellen von Interesse für 4sU-induzierten Stress sowie Apoptose getestet werden. Western-Blot Analyse empfiehlt sich zur Visualisierung von p53-Akkumulation, die nukleare Belastung angibt, Phospho-EIF2a Anhebung der zytoplasmatischen Stress und Fluoreszenz-aktivierte Zelle sortieren (FACS) Analyse für die Apoptose angezeigt. Hohe und lange Exposition gegenüber 4sU oder Medikamente wie Thapsigargin oder Arsenat können zur zellulären Stress auslösen. Um Apoptose oder Zelltod induzieren, Zellen mit BH3I-1 behandelt wurden (500 ng/µL) oder inkubierten für 5 min bei 95 ° C (Hitzeschock). Annexin V/7-AAD Färbung wurde zur Apoptose (Annexin V) und Toten (7-AAD) Zellen zu bestimmen. Kennzeichnung von in-Vitro erzeugte primäre Th1 Zellen für 0,5 h mit 500 µM 4sU (Endkonzentration) oder 1 h mit 200 µM 4sU weder Anzeichen von zellulären Stress induziert oder Apoptose (Abbildung 1) aber zu ausreichend 4sU Aufnahme führen.
RNA Kennzeichnung Zeit verkürzt werden (≤5 min) das führt zu einem Anstieg der kurzlebigen intronischen Sequenzen im Vergleich zu mehr Kennzeichnung Zeiten. Um co-transcriptional Spleißen Preise sichtbar zu machen, sollte 4sU Kennzeichnung mal 30 min nicht überschreiten. Weitere Einzelheiten zum 4sUlabeling finden Sie unter Rädle Et Al. 19
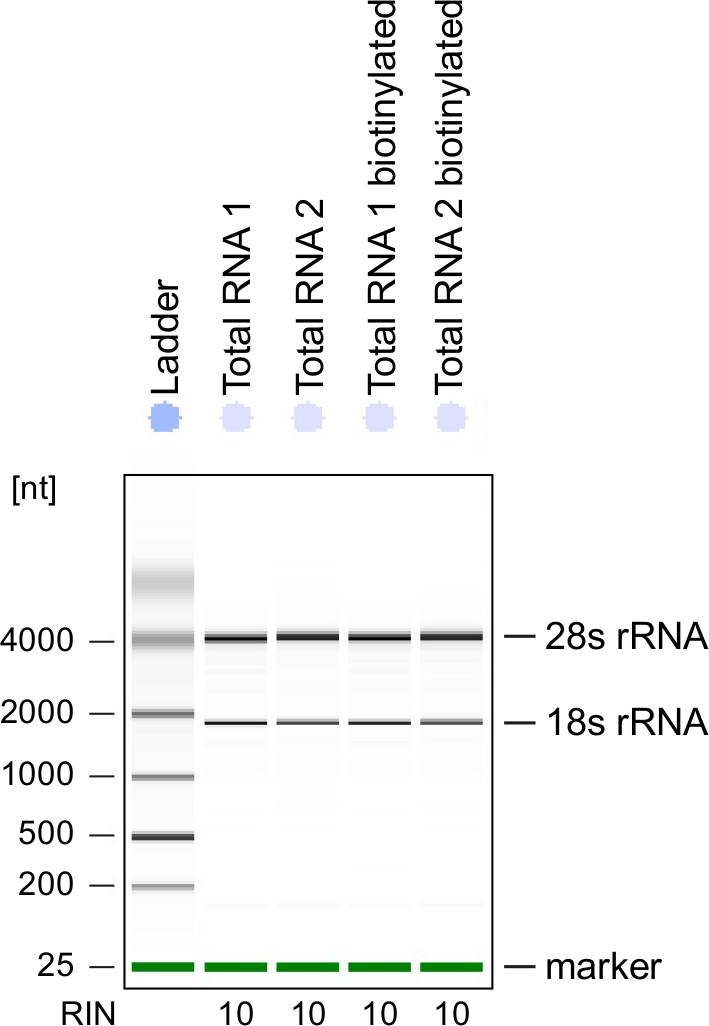
Qualitätskontrolle: Integrität der RNA ist von großer Bedeutung bei der Verarbeitung von RNA. Es ist am bequemsten, die RNA mit der Bezeichnung von 4sU RNA nach Biotinylierungen durch eine electrophoretical Analyse Qualitätsprüfung (siehe Tabelle der Materialien). Betrachten Sie isolierten RNA aus Schritt 2.2.2, zu überprüfen, vor allem, wenn es für die Sequenzierung von Gesamt-RNS. RNA-Integrität-Anzahl (RIN) sollte ≥8, Integrität der RNA zur Weiterverarbeitung (Abbildung 3).
Electrophoretical Analyse kann auch verwendet werden, um neu transkribierten RNA zu überprüfen. Beachten Sie, dass neu transkribierten RNA enthält deutlich weniger Reifen rRNAs im Vergleich zum Gesamt-RNS mit den typischen rRNA-Bands viel weniger prominenten.
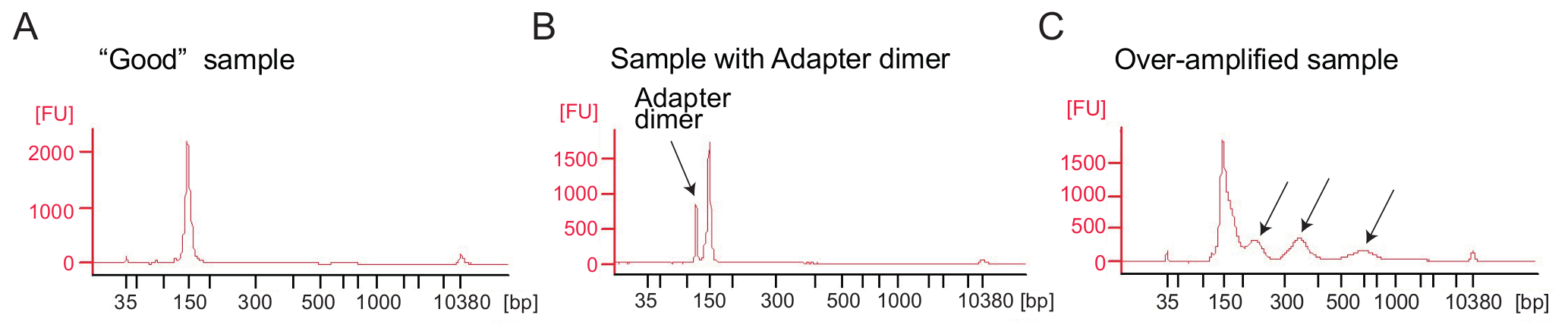
Ribosom Profilerstellung: PCR-Amplifikation der cDNA-Bibliothek: cDNA Amplification (Schritt 4.9.1) ist ein wichtiger Schritt um gute Sequenzierung Ergebnisse zu gewährleisten. Analysieren Sie verstärkte Bibliotheken durch eine electrophoretical Analyse. Ein gutes Beispiel für verstärkte Bibliotheken zeigt einen Höhepunkt etwa 140-160 bp (Abb. 4A). Übermäßige Mengen an Adapter, die Dimere sollten vermieden werden (Abbildung 4 b) und die Proben sollten weiter gereinigt werden, mit der Seite Reinigungsprozedur nach der Hersteller-Protokoll (Seite Reinigung der PCR-Produkte). Zuviel Vorlage oder zuviele PCR-Zyklen führen Überverstärkung gekennzeichnet durch das Auftreten von höher als erwartet Molekulargewicht Bands, verschmierten PCR-Produkte und Adapter Dimer Produkte (Abbildung 4). Für die meisten Proben ergibt 1 bis 5 µL circularized cDNA und 9 PCR-Zyklen für Verstärkung in der Regel ausreichende Mengen an das richtige PCR-Produkt.
ChIP: Chromatin Scheren: Schur optimale Bedingungen für jede Art von Zelle angepasst werden müssen. Bestimmen Sie Schere Bedingungen (z.B. Anzahl der Zyklen, hohe oder niedrige Leistung) im Voraus. Verwenden Sie die gleiche Anzahl von Zellen und das gleiche Volumen zu Testzwecken, da ein geringer Zelldichte die Schur Effizienzsteigerungen. Versuchen Sie, über - oder unter-Scheren das Chromatin zu vermeiden. Großen Chromatin Fragmente ChIP Ergebnisse durch verstopfen dramatisch beeinflussen können, und über Scheren Epitope auf das Protein des Interesses, was zu einen niedrigeren Wirkungsgrad der Bindung durch die Antikörper zerstören kann. In diesem Experiment wurden die besten Ergebnisse erzielt, wenn die Hauptfraktion des Chromatins geschert rund 1.000 wurde bp oder etwas niedriger (Abb. 5A).
Überprüfung der ChIP von qPCR: Vor Beginn des Chips ist es ratsam zu testen, ob die verwendeten Antikörper für ChIP eignet (wenn möglich, verwenden Sie ChIP Grade Antikörper) durch ChIP-qPCR. Überprüfen den ChIP für die Sequenzierung von qPCR vor Beginn der Bibliothek-Vorbereitung (siehe Schritt 5.6.5). Design-Primer, die an eine bekannte Zielstandort des Proteins des Interesses zu binden. Wenn die genaue Zielsite innerhalb eines Gens unbekannt ist, können mehrere Primer-Paaren, die Gene und damit verbundenen regulatorischen Elemente scannen verwendet werden. Für RNAPII-ChIP von Th1 Ifng Zellen, die ist transcriptionally hochreguliert nach Stimulation und Aktin Primer können als Positivkontrolle verwendet werden. Sox9 und Insulin dienen als Negativkontrolle, da diese Gene nicht in Th1 Zellen (Abb. 5 b) zum Ausdruck kommen. Vergesst nicht, Exon-spanning Primer zu verwenden, die normalerweise für qPCR mRNA verwendet werden. Ein IgG-Steuerelement kann auch verwendet werden, Spezifität der verwendeten Antikörper nachzuweisen. Immunoprecipitated DNA kann mit einem geeigneten Fluorometer gemessen werden (siehe Tabelle der Materialien). Mengen an nonspecifically gebundenen DNA durch das IgG-Steuerelement sollte deutlich niedriger, die DNA-Menge durch die Antikörper von Interesse gebunden verglichen werden.
Repliziert: Nachweis der biologischen Bedeutung: Es wird dringend empfohlen, führen die kinetische Experiment für alle Methoden, die ausgehend von den gleichen Pool von Zellen um sicherzustellen, dass Zellen haben die gleiche Identität für alle unbehandelten und behandelten Proben (Abbildung 2). Dennoch empfiehlt es sich, nehmen kleine aliquoten Hauptzeit Punkte für jede Methode, Proben, um eine biologische Replikation (z. B.durch qPCR, FACS-Analyse) zu vergleichen. Dies ermöglicht für eine grobe Abschätzung, ob die Behandlung für beide Replikate reproduzierbar war und Sie konnte mit der Sequenzierung fortfahren. Validierung von den Wiederholungen sollten mittels Laborprotokollen Analyse durchgeführt werden. Reproduzierbarkeit der Ergebnisse in Bezug auf die Korrelation zwischen FPKM Werten auf Wiederholungen beurteilt werden kann und mit Streudiagrammen (Abbildung 6) visualisiert.

Abbildung 1: Überprüfung der optimalen 4sU Kennzeichnung Bedingungen ohne störend Zellphysiologie (Figur aus Davari Et Al. 11)
(A) Erkennung der Zellapoptose durch FACS Analyse: In Vitro erzeugt bzw. Th0 Zellen wurden mit verschiedenen Konzentrationen von 4sU (in Klammern angegeben) für 0,5 h, 1 h und 2 h behandelt. BH3I-1 Behandlung diente induzieren Apoptose durch Annexin V bestimmt, während Hitzeschock (5 min bei 95 ° C) verwendet wurde, um Zelltod durch 7-AAD bestimmt zu induzieren. (B) Western-Blot Analyse für p53 4sU behandelt und T-Zellen aktiviert: Proben wurden mit 200 µM 4sU für die angegebene Zeit der Aktivierung, mit Ausnahme der 0,5 h-Zeit-Punkt, der mit 500 µM 4sU beschriftet wurde beschriftet. (C) Western-Blot Analyse des Phospho-EIF2a und insgesamt EIF2a im aktivierten Th1 Zellen mit der gleichen Kennzeichnung wie in (B). Thapsigargin wurde als Positivkontrolle verwendet. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: Schematische Übersicht einer kinetischen Installation genomweite Änderungen nachverfolgen
Dieses Schema zeigt das Setup für die Kombination von 4sU-Seq, total RNA-Seq Ribosom Profiling und ChIP-Seq, genomweite Veränderungen bei der Behandlung zu studieren. Zellen zu bündeln und die erforderliche Anzahl von Zellen für unbehandelten Kontrolle beiseite stellen. Verbleibende Zellen zu behandeln und für jede Methode und Zeit Punkt geteilt. Beschriften Sie unbehandelt/behandelten Zellen für 4sU-Seq mit 4sU wie beschrieben. Zeitpunkte und Proben für jede Methode hängt die spezifischen biologischen Frage geprüft wird. Proben für jeden Zeitpunkt und Methode, und folgen Sie den speziellen Teil des Protokolls. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Qualitätskontrolle mit der Bezeichnung von 4sU RNA
Gesamt-RNA und biotinylierte RNA aus aktivierten Th1 Zellen gewonnen wurden auf einem Bioanalyzer analysiert. 18 s rRNA und 28 s rRNA gezeigt und RNA Integrität Anzahl (RIN) ergibt sich aus dem Instrument Integrität der RNA zu bestimmen. RIN sollte ≥8, RNA-Integrität zu gewährleisten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: Bioanalyzer Profile der Ribosom Profilerstellung Bibliotheken
(A) A gute Bibliothek: das Beispiel zeigt einen Spitzenwert bei den erwarteten Größenbereich (140-160 bp) und keine weitere Reinigung erforderlich ist. (B) dieses Beispiel zeigt übermäßige Adapter Dimer verstärkten Produkt (120 bp) bezogen auf das gewünschte Produkt (140-160 bp). Diese Bibliothek erfordert weitere Reinigung. (C) eine übermäßig verstärkten Probe: Molekulargewicht Gipfel höher als erwartet und verschmiert PCR Amplifikate sind sichtbar (durch Pfeile angedeutet). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: Optimale Chromatin Größe nach Scheren und Überprüfung der ChIP durch qPCR
(A) Agarose Gel Bild zeigt die optimale Fragmentgröße geschert Chromatin von drei Proben, die für 25 Zyklen auf einem Sonikator geschoren und gereinigt wie zuvor im Protokoll beschrieben wurden. (B) Q-PCR-Ergebnisse eines gesamten RNAPII Chips (Anti-RNA Pol II, 8WG16, ab817) wird als ein Prozentsatz des Eingangs dargestellt. IFNG und Aktin Primern dienten als positiv, während Sox9 und Insulin Negativkontrollen (beide Gene sind nicht in aktivierten Th1-Zellen exprimiert). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 6: Vergleich von biologischen repliziert (Abbildung von Davari Et al. ( 11)
Repräsentative Streudiagramm Vergleich Ausdruckswerte (FPKM) zwischen repliziert neu transkribierten (4sU) RNA-4 h nach der Stimulation der aktivierten Th1 Zellen. Die grüne Linie zeigt gleich FPKM Werte und Rangkorrelation wird in jeder Parzelle.
| Dauer der Beschriftung (min) | Empfohlene 4sU Konzentration (µM) |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| < 10 | 500 - 2000 |
Tabelle 1: Empfohlene 4sU Konzentrationen (von Rädle, Et Al. 19)
Die Palette der empfohlenen 4sU Konzentrationen ist für unterschiedliche Tageszeiten Kennzeichnung angegeben.
| Methode | Anzahl von Zellen | RNA-Menge |
| 4sU-Beschriftung | ≥2 x 107 | ≥60µg |
| Ribosom Profilierung | ≥2 X107 | |
| ChIP-seq | ≥2 x 107 - 3 x 107 |
Tabelle 2: Erforderliche Menge an primären T-Zellen
Mindestbetrag der erforderlichen primären T-Zellen für jede Methode. Mengen können kleiner sein, wenn andere Zelltypen zu verwenden.
Diskussion
Analyse des gesamten Prozesses der Genregulation ist notwendig, zelluläre Anpassungen als Reaktion auf einen bestimmten Reiz oder Behandlung voll und ganz verstehen. Total RNA-Seq kombinierend führt 4sU-Seq, Ribosom, Profilierung und ChIP-Seq zu unterschiedlichen Zeitpunkten zu einer umfassenden Analyse der wichtigsten Prozesse der Genregulation im Laufe der Zeit. Ein tiefes Verständnis der biologischen Prozessen ist erforderlich, um den Versuchsaufbau sowie optimale Zeitpunkte definieren.
Da Methoden zur Genregulation studieren rasch verbessern, können diese Protokolle zu raschen Veränderungen angepasst werden. Dennoch bieten sie die wichtigsten Methoden für grundlegende gen Regulationsmechanismen in jeder Art von Zelle zu studieren. Hier diskutieren wir einige der Fallstricke und Fakten muss man berücksichtigen, wenn Sie diese Methoden verwenden.
Zellen: Zellen müssen höchst lebensfähig sein und wenn primäre isolierte Zellen verwenden, Reinheit des Zell-Populationen gewährleistet sein muss (z. B.FACS Analyse für primäre T-Zellen). Sogar leicht gestresste Zellen können Einfluss auf die Ergebnisse von diesen sehr sensiblen Sequenziermethoden und senken die Menge von neu transkribierten oder übersetzten RNA und führen zu unerwünschten Auslesen der Stress-Reaktion in der Sequenzierung Ergebnisse. Die Zentrifugation Geschwindigkeit erwähnt in diesem Protokoll zu pellet-Zellen ist für primäre T-Zellen optimiert. So passen Sie die Geschwindigkeit je nach Zelltyp.
4 sU-Auswirkungen auf die Zellphysiologie: Zusätzlich zu den oben genannten Optionen für minimale Störung für Zellphysiologie auf 4sU Zugabe überprüfen kann weitere bzw. zusätzliche Analyse durchgeführt werden, vor allem wenn Zellzahlen begrenzt sind. Auswirkungen auf die Zellproliferation können durch die Überprüfung der Verdopplungszeit der Zellen durch einfach zählen beschriftete und unbeschriftete Zellen getestet werden. Nukleolus Stress Induktion könnte auch geprüft werden, durch die Analyse der Zellmorphologie über Immunfluoreszenz-Färbung des Nucleolin und Kerne. Um die Auswirkungen der 4sU weiter zu überprüfen, konnte veränderten globalen Genexpression gemessen werden, indem Sie korrelieren, Grafen von beschrifteten Gesamt-RNS nach unbeschriftete Gesamt-RNS gelesen.
Handy-Nummern: Für in-Vitro generierten T-Zellen empfiehlt es sich, beginnend mit mindestens die Höhe der Zellen in Tabelle 2angegeben. Wählen Sie angemessene zahlen pro Verfahren nach der Art der Zellen. Da T-Zellen weniger Zytoplasma und RNA im Vergleich zu anderen Zellen haben, werden am ehesten geringere Mengen an andere Zellen ausreichend sein. Für ChIP-Seq hängen Zellzahlen hoch der Antikörper verwendet und die Expression des Proteins des Interesses innerhalb der Zellen. Für Histon oder RNAPII ChIP können liegt Zellen verwendet werden, während Zellzahlen müssen erhöht werden, wenn Transkriptionsfaktoren verwendet werden, vor allem, wenn sie auf einem niedrigen Niveau zum Ausdruck kommen.
4 sU-labeling und RNA-Biotinylierungen: Bei der Verwendung von adhärenten Zellen kann 4sU Kennzeichnung geleitet werden, wie durch Rädle Et al.beschrieben. 19 seit Zellen sehr schnell integrieren 4sU, kann es direkt auf das Medium der Aussetzung, Anhänger oder semi-adhärente Zellen hinzugefügt werden.
Es wird empfohlen, die Biotinylierungen mit 60-80 µg RNA zu beginnen. Jedoch geringere Mengen an RNA können verwendet werden, obwohl wir nicht testen für weniger als 30 µg. Hinzufügen einer Coprecipitant (z.B.GlycoBlue) als RNA Ausfällen, wenn das Pellet schwer zu erkennen ist. Duffy Et Al. haben gezeigt, dass diese Methylthiosulfonate aktiviert Biotin (MTS-Biotin) effizienter mit 4sU beschriftet RNA als HPDP-Biotin31reagiert. Daher kann es sinnvoll unter Berücksichtigung der Umstellung auf MTS-Biotin, insbesondere für die Verwertung von kleinen RNAs, die neigen, weniger Uridin Rückstände haben (beziehen sich auf das Biotinylierungen-Protokoll von Duffy Et Al.; siehe Reinigung von 4sU beschriftet RNA genannt, sein Experimentelle Verfahren).
Für die Verwertung von neu transkribierten RNA ist es möglich, paramagnetischen Beads oder RNA Cleanup Perlen Ihrer Wahl verwenden. Berücksichtigen Sie immer, die diese Kits können oder können nicht für bestimmte RNAs reinigen. Wenn Sie MiRNAs interessiert sind, betrachten Sie z. B. mit speziellen Kits für MiRNA Abscheidung und Sequenzierung.
Quantifizierung der neu transkribierten RNA: Um neu transkribierten RNA genau zu quantifizieren, Messung durch eine geeignete Fluorometer durchgeführt werden (siehe Tabelle der Materialien). Innerhalb 1 h nach 4sU Exposition stellt neu transkribierten RNA ca. 1-4 % der Gesamt-RNS. Neu transkribierten RNA von 1 h gekennzeichnet, aktivierte T-Zellen besteht aus ca. 90-94 % der rRNA11.
Ribosom Profilerstellung: Bei der Festlegung der Methode wird herausgestellt, dass mit 1,5 Mal die Menge der Nuklease als das ursprüngliche Protokoll vorgeschlagen richtige Verdauung garantiert. Auch wurden keine negativen Auswirkungen für erhöhte Mengen an Nuklease berichtet. Denn es ziemlich schwierig ist, den RPFs overdigest während sie Bestandteil der RNA durch ribosomale Proteine gebunden sind, können Sie den Betrag ein, um optimale Nuklease Verdauung titrieren noch leicht erhöhen.
Wenn weniger als 500 ng RPF RNA wurden geborgen, in Schritt 4.4.2, wiederholen Sie die rRNA Erschöpfung und Pool gereinigt RPFs mit RNA Clean & Konzentrator-5 Spalten. Alternativ laden Sie zwei identische Proben nebeneinander auf das Gel (Schritt 4.5.3) und Pool Gel Scheiben während RNA Elution aus dem Gel (Schritt 4.5.6).
Es wird empfohlen, schneiden die RPFs auf einem Gel so dicht wie möglich an die 28 und 30 nt-Bands. Dies hilft bei der Beseitigung von unerwünschten Fragmente von rRNAs und tRNAs, die später Bestandteil Ihrer Bibliothek und Sequenzierung liest für Ihre RPFs reduzieren.
Es wird auch empfohlen, UV-Licht während Gel Reinigung zu vermeiden. Dies kann Nicks erstellen, in der RNA-Fragmente sowie Pyrimidine Dimere, die am Ende der Bibliothek Vorbereitung und Sequenzierung Ergebnisse erheblich beeinträchtigen können.
Bibliothek Vorbereitung und Sequenzierung von Daten: Ribosom Profilerstellung Protokoll ermöglicht eine cDNA-Bibliothek für Sequenzierung zu generieren. Proben von 4sU Beschriftung erzeugt können direkt für die Vorbereitung der Bibliothek mit einer entsprechenden RNA Sequenzierung Kit verwendet werden. Seit neu transkribierten RNA vor allem, wenn mit kurzen Zeiten, Kennzeichnung noch nicht Polyadenylated, ist sollte keine Poly-A-Auswahl durchgeführt werden. Stattdessen empfehlen wir rRNA Erschöpfung zu verhindern, dass Verringerung der Sequenzierung Tiefe für die eigentliche Probe. Mit T-Zellen, wir begannen mit 400 ng neu transkribiert und total RNA (abhängig von dem Kit, siehe Materialien), durchgeführt rRNA Erschöpfung und reduziert Zyklen für PCR-Amplifikation, PCR Verzerrungen zu minimieren. Vorbereiten der Bibliothek kann mit weniger Ausgangsmaterial durchgeführt werden. Um Bibliothek Komplexität vieler PCR-Zyklen berücksichtigen sollte optimiert werden.
Für ChIP-Seq gibt es auch viele Kits für Bibliothek Vorbereitung zur Verfügung. In unseren Händen-Bibliothek Vorbereitung arbeitete auch beginnend mit 2 ng DNA-ChIP (siehe Materialien für einen Vorschlag, welche Kit zu verwenden). Achten Sie darauf, die Indizes für Farb-Balance während der Sequenzierung zu überprüfen. Wir empfehlen eine Sequenzierung Tiefe von ≥40 x 106 liest jedes 4sU-Seq, total RNA-Seq und ChIP-Seq-Proben und ≥80 x 106 liest für Ribosom Profilerstellung Proben. Die Sequenzierung Tiefe richtet sich nach der Probe und der nachgeschalteten Bioinformatik-Analyse und sollte gut überlegt sein. Um zu analysieren, intronischen liest für cotranscriptional Spleißen, 100 muss bp gepaart-End Sequenzierung gewählt werden.
Bias-Sequenzierung: Sequenzierung ist der Goldstandard geworden, bei der Festlegung globale Veränderungen in Transkription, Übersetzung und Transkription Faktor Bindung. In den letzten Jahren bestehende Methoden waren an ihre Grenzen gestoßen oder neue Techniken wurden entwickelt, um immer kleinere Startbeträge RNA-Sequenzierung. Dies erfordert eine Verstärkung der cDNA, die Rauschen oder Verzerrungen führt. Vor kurzem wurden einzigartige molekulare Bezeichner (UMIs) entwickelt, um Duplikate von PCR eingeführt experimentell zu identifizieren. Vor kurzem zeigte sich, dass UMIs nur milde Sequenzierung macht und false Discovery Rate für differenzielle gen Ausdruck32verbessern. Dennoch erwägen Sie, einzigartige molekulare Bezeichner (UMIs) für alle Bibliotheken der Sequenzierung zu steuern für Bibliothek Komplexität, vor allem beim Starten mit geringen Mengen an RNA und wenn viele PCR-Zyklen benötigt werden.
Puffer und Stammlösungen: Alle Puffer für 4sU-Seq und Ribosom Profilerstellung müssen unter strengen RNase-freie mit Nuklease-freies Wasser vorbereitet werden. Es empfiehlt sich, vorgefertigte Nuklease-freie NaCl, Tris-HCl, EDTA, Natriumcitrat und Wasser kaufen. Um Nuklease-freien Bedingungen zu gewährleisten, kann eine RNase dekontaminierende Lösung verwendet werden, um Pipetten oder Oberflächen zu säubern. Alle Puffer für ChIP-Seq müssen mindestens DNase-frei und kann bei Zimmertemperatur gelagert werden. Immer fügen Sie Protease-Inhibitoren und optional Phosphatase-Hemmer direkt vor Gebrauch hinzu und auf Eis.
Bioinformatik: Analyse aller Sequencing-Daten (d. h., ChIP-Seq, RNA-Seq und Ribsosome profiling) beinhaltet Qualitätskontrolle (z.B.mit FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), Adapter trimmen (z. B.mit Cutadapt20) gefolgt von Mapping auf das Referenz-Genom für die Zellen untersucht. Für RNA-Seq-Daten (insgesamt und 4sU-Seq) sowie Ribosom Profilerstellungsdaten ist eine gespleißte RNA-Seq-Mapper erforderlich, z. B. ContextMap 221. Für ChIP-Seq Daten unspliced Achsen reicht mit BWA-MEM-22 . Genexpression kann berechnet werden, mit dem RPKM Modell (gelesen pro Kilobase Exon pro Million Fragmente abgebildet)1, nach der Bestimmung Zählungen pro gen mit einem Programm, z. B. FeatureCounts23zu lesen. Für den Gipfel Aufruf von ChIP-Seq-Daten, gibt es eine Reihe von Programmen, z.B., MACS24 oder GEM25. Weiter können nachgeschaltete Analysen in R26, insbesondere unter Verwendung von Bioconductor Projekt27bereitgestellten Tools durchgeführt werden.
Hier ist eine große Herausforderung bei der Integration von 4sU und Gesamt-RNA-Ebene und translationale Aktivität vom Ribosom Profilerstellung Normalisierung. Ein klassischer Ansatz zur Lösung dieses Problems ist, auf ein Niveau von Housekeeping Gene zu normalisieren. Durch zufällige Schwankungen für einzelne Housekeeping Gene reduzieren, es empfiehlt sich nicht nur ein paar Housekeeping Gene Median Ebenen für eine größere Gruppe, z. B. zu verwenden, aber die > 3.000 Housekeeping Gene zusammengestellt von Eisenberg und Levanon28 . Für die Berechnung der RNA Umsatzraten von Übersetzungen von 4sU-Gesamt-RNS, Normalisierung auf mittlere Umsatz basiert Preise (z.B., vorausgesetzt eine RNA-Halbwertszeit von 5 h)29. Da dies keine allgemeinen Änderungen für Housekeeping Gene wird davon ausgegangen, empfehlen wir jedoch mit Analyse-Ansätze unabhängig von Normalisierung, z.B.Korrelation basierende Cluster einer Zeitreihe der verschiedenen Datentypen um Gruppen von Genen identifizieren mit unterschiedlichen Verhalten in Transkription und Übersetzung während der Aktivierung. Für eine detaillierte Beschreibung über Laborprotokollen Integration der verschiedenen Datentypen verweisen wir auf die ursprüngliche Publikation11.
Analyse von Umsatz Preise und Datenintegration: Einer kürzlich veröffentlichten Papier33 Halbwertszeiten bestimmt durch eine gebündelte gen (MGC) im Vergleich zu globale Methoden konnten zeigen, dass die Halbwertszeiten korreliert am besten mit denen, die durch metabolische Kennzeichnung Methoden im Vergleich zu anderen Methoden (z.B., Allgemeine Hemmung der Transkription durch Medikamente). Allerdings sei darauf hingewiesen, dass Unterschiede zwischen Halbwertszeit Berechnungen entstehen können und beschriebenen 15,34gewesen. Wir machen den größten Teil der Probleme und Differenzen, die durch die Stress-Reaktion aufgrund der längeren 4sU Exposition eingeführt werden. Daher ist es unerlässlich, die Stress-Reaktion von 4sU-Kennzeichnung eingeführt auszuschließen. Um weitere Umsatzraten zu überprüfen, empfehlen wir die Verwendung von MGCs.
Darüber hinaus könnte ein Dataset als hier erzeugt auch ein integrativer Daten Analyse (z.B. Regelung der lange nicht-kodierende RNAs)35,36verwendet werden.
Offenlegungen
Die Autoren erklären, dass sie keine finanziellen Interessenkonflikte.
Danksagungen
Wir danken Lars Dölken für Beratung, 4sU Kennzeichnung für primäre T-Zellen zu etablieren; Elisabeth Graf und Thomas Schwarzmayr für kritische Hilfe bei Bibliothek Generationen und Sequenzierung; Dirk Eick und Andrew Flatley für die Bereitstellung von RNAPII und T-Zell-Antikörper; N. Henriette Uhlenhaut und Franziska Greulich für Hilfe bei der Vorbereitung der Bibliothek für ChIP-Seq; Caroline C. Friedel wurde durch Zuschüsse FR2938/7-1 und CRC 1123 (Z2) von der Deutschen Forschungsgemeinschaft (DFG) unterstützt. Elke Glasmacher wurde durch den Zuschuss GL 870/1-1 von der Deutschen Forschungsgemeinschaft (DFG) und dem Deutschen Zentrum für Diabetes Research (DZD), Helmholtz Zentrum München unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| 4sU-labeling | |||
| 4-thiouridine (100 mg) | Carbosynth | 13957-31-8 | Prepare 50 mM stock in sterile H2O/PBS; store at –20°C in aliquots of 50-500 µl; do not refreeze. |
| 1.5 ml safe-lock tubes | Eppendorf | 30121589 | Optional |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72692005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72694005 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| Agencourt RNAClean XP | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| Chloroform | Sigma Aldirch | 372978 | WARNING – HAZARDOUS TO HEALTH |
| Dimethylformamide | Sigma Aldrich | D4551 | |
| Dithiothreitol (DTT) | Roth | 6908.1 | Prepare as 100 mM DTT in nuclease-free H2O; always prepare fresh |
| Ethanol | Merck | 1.00983.1000 | |
| EZ-Link Biotin-HPDP (50 mg) | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg Biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4°C in aliquots of 1 ml. DMF dissolves some plastic materials. We recommend to use glass pipettes to transfer DMF from ist stock glass bottle to 50 ml Falcon tubes. |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| Isopropanol | Merck | 1.09634.1011 | |
| NaCl (5M) | Sigma Aldrich | 71386 | Stock solution |
| nuclease-free EDTA (500 mM ), pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| Nuclease-free H2O | Sigma Aldrich | W4502 | Stock solution |
| nuclease-free Tris Cl (1M), pH 7.4 | Lonza | 51237 | Stock solution |
| Phase Lock Gel Heavy tubes (2.0 ml) | 5Prime | 2302830 | Use in step 1.3.4. |
| Polypropylene 15 ml centrifuge tubes | Greiner Bio-One | 188271 | Or equivalent; they have to tolerate up to 15,000 × g |
| QIAzol Lysis Reagent (200 ml) | Qiagen | 79306 | Use this or equivalent TRI reagent for RNA isolation, WARNING – CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol) |
| Qubit RNA HS assay kit | Life Technologies | Q32852 | Use this kit for quantifying RNA quantity in step 1.4.11 |
| RNeasy MinElute Kit | Qiagen | 74204 | Optional; includes Buffer RLT |
| Sodium citrat | Sigma Aldrich | C8532 | Prepare 1.6 M stock solution using nuclease-free water |
| Tween 20 | Sigma Aldrich | P1379 | |
| µMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store at 4°C, includes µMacs columns used in step 1.4.6. (store at RT) |
| Cell viability and stress assay | |||
| PE Annexin V Apoptosis Detection Kit I | BD Biosciences | 559763 | Optional |
| Thapsigargin | Sigma-Aldrich | T9033 | Optional |
| p53 | abcam | ab26 | Optional |
| p-EIF2a (Ser51) | Cell Signaling | 9721 | Optional |
| BH3I-1 | Sigma-Aldrich | B 8809 | Optional |
| Buffers | |||
| 4sU Washing Buffer | store at RT | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O | |
| Biotinylation Buffer (10x) | store at 4 °C | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water; make aliquots of 1 ml; store at 4°C | |
| RNA precipitation buffer | store at RT | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | |
| RNA 6000 Nano Kit | Agilent | 5067-1511 | Use this kit to verify RNA integrity in step 1.3.10 |
| RNA 6000 Pico Kit | Agilent | 5067-1513 | Optional |
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use in step 1.2.2/3.1.8/3.3.3/3.4.3 |
| High-speed centrifuge | Thermo Scientific | Heraeus Multifuge X3R | Or equivalent equipment capable of reaching 13,000 × g |
| High-speed rotor | Thermo Scientific | Fiberlite F15-6 x 100y | |
| Adaptors for 15 ml tubes | |||
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer C | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 µMacs columns. |
| Ultra-fine scale | Mettler Toledo | ML204T | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
| DynaMag-2 Magnet-1 each | Life Technologies | 12321D | |
| RNaseZap | Sigma | R2020 | Optional |
| TruSeq stranded total RNA library prep kit | Illumina | RS-122-2201 | Or equivalent. For T cells we used 400 ng 4sU and Total RNA with 11 cycles for PCR amplification. rRNA depletion is included in this kit |
| Nanodrop | Thermo Scientific | use a Nanodrop or equivalent instrument to measure RNA concentration | |
| Ribosome Profiling | |||
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPYSC12116 (Yeast) | |
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPHMR12126 (Mammalian) | |
| Illustra MicroSpin S-400 HR Columns | GE Healthcare | 27-5140-01 | |
| RNA Clean & Concentrator-25 kit | Zymo Research | R1017 | |
| RNA Clean & Concentrator-5 kit | Zymo Research | R1015 | |
| Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) | Illumina | MRZG126 or MRZG12324 | |
| (High Sensitivity DNA Kit) | Agilent Technologies | 5067-4626 | Already needed for 4sU-seq |
| All other consumables and equipment are listed in the User guide | !!! | Carefully read the user guide and order required consumables in advance (consider a long delivery time for some consumables e.g. gels) | |
| ChIP | |||
| 10 mM Tris-HCl (pH 8.0) | gereral lab supplier | ||
| 100 bp Plus Marker | Thermo Fisher | SM0323 | |
| 16 % Formaldehyde | Thermo Fisher | 28908 | Add to a final concentration of 1 % |
| 70% EtOH | gereral lab supplier | Always prepare fresh | |
| Agarose | gereral lab supplier | ||
| Agencourt RNAClean XP beads | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| ChIP library preparation kit | KapaBiosystems | KK8504 | Or use the kit of your choice |
| DNA low bind microcentrifuge tubes | Eppendorf | Z666548-250EA | or equivalent |
| Dynabeads Protein G | Invitrogen | 10004D | Use these superparamagnetic beads coupled to protein G in step 4.3.1.; Bring to RT before use |
| Glycine | gereral lab supplier | Prepare a 2M stock solution | |
| Glycogen | Roche | 10-901-393-001 | |
| MinElute PCR Purification Kit | Qiagen | 28004 | Use this kit (or equivalent) to purify chromatin in step 4.2.4. |
| Phosphatase Inhibitor (PhosStop) | Roche | 4906837001 | Add freshly to the buffer and keep on ice |
| Power SYBRgreen Master mix | Thermo Fisher | 4367659 | |
| Protease Inhibitor (cOmplete, EDTA-free) | Roche | 11873580001 | Add freshly to the buffer and keep on ice |
| Proteinase K | Invitrogen | 25530049 | |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32851 | Use this kit for quantifying DNA quantity in step 4.6.4. on a Qubit Fluorometer |
| Rnase, DNase free | Roche | 11-119915001 | |
| Salmon sperm (sonicated to around 100bp) | Sigma | D1626 | |
| TE pH 8.0 | gereral lab supplier | ||
| Antibodies (ChIP grade if possible) | |||
| anti-RNA Pol II [8WG16] | abcam | ab817 | |
| anti-Histon H3K36me3 | abcam | ab9050 | |
| or antibody of interest | |||
| Buffers | |||
| Binding/Blocking buffer | Store at RT | PBS with 0.5 % BSA and 0.5 % Tween 20 | |
| Cell-Lysis buffer | Store at RT | 5 mM Pipes [pH 8.0], 85 mM KCl, and 0.5 % NP40 | |
| ChIP IP buffer | Store at RT | 0.01 % SDS; 1. 1% Triton X-100;1.2 mM EDTA; 16.7 mM Tris-HCl, pH 8.1; 16.7 mM NaCl | |
| Elution buffer | Store at RT up to 6 months | 10 mM Tris-HCl (pH 8.0), 5 mM EDTA (pH 8.0), 300 mM NaCl and 0.5 % SDS | |
| Nuclei-Lysis buffer | Store at RT | 50 mM Tris [pH 8.0], 10 mM EDTA, and 1 % SDS | |
| Wash buffer I | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20mM Tris-HCL pH 8.1; 150 mM NaCl | |
| Wash buffer II | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20 mM Tris-HCL pH 8.1; 500 mM NaCl | |
| Wash buffer III | Store at RT | 0.25 M LiCl; 1% NP-40; 1 mM EDTA; 10 mM Tris-HCl, pH 8.1 | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | use this instrument for electrophoretical analysis |
| Nanodrop | Thermo Scientific | ||
| Bioruptor TBX microtubes 1.5 ml | Diagenode | C30010010 | |
| or tubes special for your sonication device | |||
| Bioruptor sonication device or sonication device of your choice | Sonication of T cells with Bioruptor: 20 - 25 cycles (30 s on, 30 s off at high in two 1.5 ml bioruptor microtubes with 500 µl each tube) | ||
| Magnetic stand for tubes | |||
| Thermomixer | |||
| Agarose gel electrophoresis | |||
| Qubit Fluorometer | Thermo Scientific | Use this Fluorometer for quantifying low amounts of RNA/DNA |
Referenzen
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 5 (7), 621-628 (2008).
- Chavez, S., Garcia-Martinez, J., Delgado-Ramos, L., Perez-Ortin, J. E. The importance of controlling mRNA turnover during cell proliferation. Curr Genet. 62 (4), 701-710 (2016).
- Rutkowski, A. J., et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun. 6, 7126 (2015).
- Ozsolak, F., Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 12 (2), 87-98 (2011).
- Carninci, P., et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 38 (6), 626-635 (2006).
- Churchman, L. S., Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 469 (7330), 368-373 (2011).
- Core, L. J., Waterfall, J. J., Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 322 (5909), 1845-1848 (2008).
- Hah, N., et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 145 (4), 622-634 (2011).
- Dolken, L., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14 (9), 1959-1972 (2008).
- Paulsen, M. T., et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67 (1), 45-54 (2014).
- Davari, K., et al. Rapid Genome-wide Recruitment of RNA Polymerase II Drives Transcription, Splicing, and Translation Events during T Cell Responses. Cell Rep. 19 (3), 643-654 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 22 (10), 2031-2042 (2012).
- Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 10 (10), 669-680 (2009).
- Blecher-Gonen, R., Barnett-Itzhaki, Z., Jaitin, D., Amann-Zalcenstein, D., Lara-Astiaso, D., Amit, I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc. 8 (3), 539-554 (2013).
- Schwanhausser, B., et al. Global quantification of mammalian gene expression control. Nature. 473 (7347), 337-342 (2011).
- Larsson, O., Tian, B., Sonenberg, N. Toward a genome-wide landscape of translational control. Cold Spring Harb Perspect Biol. 5 (1), a012302 (2013).
- Brar, G. A., Weissman, J. S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 16 (11), 651-664 (2015).
- Hafner, M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Radle, B., Rutkowski, A. J., Ruzsics, Z., Friedel, C. C., Koszinowski, U. H., Dolken, L. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. (78), (2013).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10-12 (2011).
- Bonfert, T., Kirner, E., Csaba, G., Zimmer, R., Friedel, C. C. ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics. 16, 122 (2015).
- Li, H., Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26 (5), 589-595 (2010).
- Liao, Y., Smyth, G. K., Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30 (7), 923-930 (2014).
- Zhang, Y., et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9 (9), R137 (2008).
- Guo, Y., Mahony, S., Gifford, D. K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 8 (8), e1002638 (2012).
- Team, R. D. C. . R: A Language and Environment for Statistical Computing. , (2016).
- Huber, W., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nature Methods. 12 (2), 115-121 (2015).
- Eisenberg, E., Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29 (10), 569-574 (2013).
- Friedel, C. C., Dolken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol Biosyst. 5 (11), 1271-1278 (2009).
- Burger, K., et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 10 (10), 1623-1630 (2013).
- Duffy, E. E., Rutenberg-Schoenberg, M., Stark, C. D., Kitchen, R. R., Gerstein, M. B., Simon, M. D. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 59 (5), 858-866 (2015).
- Parekh, S., Ziegenhain, C., Vieth, B., Enard, W., Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 6, 25533 (2016).
- Baudrimont, A., et al. Multiplexed gene control reveals rapid mRNA turnover. Sci Adv. 3 (7), e1700006 (2017).
- Rabani, M., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 29 (5), 436-442 (2011).
- Schlackow, M., Nojima, T., Gomes, T., Dhir, A., Carmo-Fonseca, M., Proudfoot, N. J. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol Cell. 65 (1), 25-38 (2017).
- Mukherjee, N., Calviello, L., Hirsekorn, A., de Pretis, S., Pelizzola, M., Ohler, U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. 24 (1), 86-96 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten