
Method Article
Análise em tempo real do fator de transcrição, ligação, transcrição, tradução e volume de negócios para exibir eventos globais durante a ativação celular
* Estes autores contribuíram igualmente
Neste Artigo
Resumo
Este protocolo descreve a utilização combinatória de ChIP-seq, seq-4sU, RNA total-seq e Ribossoma profiling para linhas de células e as células primárias. Permite rastreamento de alterações em fator de transcrição vinculativas, novo de transcrição, processamento de RNA, volume de negócios e tradução ao longo do tempo e exibindo o curso geral dos acontecimentos nas células ativadas e/ou rápida mudança.
Resumo
Após a ativação, células alterar rapidamente os seus programas funcionais e, desse modo, seu perfil de expressão do gene. Grandes mudanças na expressão gênica ocorrem, por exemplo, durante a diferenciação celular, morfogênese e estimulação funcional (como a ativação de células do sistema imunológico), ou após a exposição a drogas e outros fatores do ambiente local. Dependendo do tipo de estímulo e célula, essas mudanças ocorrem rapidamente e em qualquer nível possível de regulação gênica. Exibir todos os processos moleculares de uma célula de resposta para um determinado tipo de estímulo/droga é uma das tarefas mais difíceis em biologia molecular. Aqui, descrevemos um protocolo que permite a análise simultânea de múltiplas camadas de regulação gênica. Podemos comparar, em particular, fator de transcrição vinculação (cromatina-imunoprecipitação-sequenciamento (ChIP-seq)), transcrição de novo (4-thiouridine-sequenciamento (seq-4sU)), processamento de mRNA e volume de negócios, bem como tradução (Ribossoma criação de perfil). Combinando estes métodos, é possível exibir um curso detalhado e todo o genoma de ação.
Sequenciamento do RNA recém transcrito é especialmente recomendado quando analisando rapidamente adaptar ou alterar sistemas, desde que este retrata a atividade transcricional de genes de todos durante o tempo de exposição 4sU (independentemente se eles são acima - ou ativador). A utilização combinatória de RNA total-seq e perfilamento Ribossoma, além disso, permite o cálculo das taxas de rotatividade e tradução de RNA. Análise de Bioinformatic dos resultados do sequenciamento do elevado-throughput permite muitos meios para análise e interpretação dos dados. Os dados gerados também permite rastreamento co-transcriptional e alternativas emenda, só para mencionar alguns resultados possíveis.
A abordagem combinada descrita aqui pode ser aplicada para organismos modelo diferentes ou tipos de células, incluindo as células primárias. Além disso, podemos fornecer protocolos detalhados para cada método utilizado, incluindo controles de qualidade e discutir potenciais problemas e armadilhas.
Introdução
Nos últimos anos, sequenciamento de RNA (RNA-seq) tornou-se a ferramenta padrão para analisar tudo expressas RNAs dentro de uma célula ou um organismo1. No entanto, para compreender todo o processo de adaptação em resposta a uma estímulo/droga específica de células, é necessário determinar completamente todos os processos subjacentes, variando de transcrição de mRNA para processamento, faturamento e tradução. A curto prazo alterações na transcrição do RNA dificilmente podem ser medidas por RNAseq total, desde que as alterações do RNA total dependem de fatores por exemplo, RNA meias-vidas e atividade transcricional, que são um modelo pobre para refletindo a adaptação de células para efeitos ambientais2,3. Com efeito, uma variedade de novas técnicas de sequenciamento foram desenvolvidos que permitem uma análise das diferentes etapas no processo de gene Regulamento4 quando combinados da maneira correta. Este protocolo descreve como combinar algumas técnicas de sequenciamento bastante facilmente aplicável que permitem o acompanhamento do Regulamento das camadas essenciais do mRNA de forma comparativa. Para analisar a atividade transcricional, uma variedade de métodos têm sido descritos, como Cap-análise do gene expressão (CAGE)5, alongamento nativo transcrição sequenciamento (NET-seq)6e todo o genoma nuclear pperigoso (GRO-seq)7 , 8, bem como bromouridine-sequenciamento (Bru-seq) e 4-thiouridine-sequenciamento (seq-4sU), que usam metabólitos que são incorporados no recém transcrito de RNA9,10, só para citar alguns. Enquanto gaiola identifica o local de início de transcrição exata, NET-seq e GRO-seq fornecem informações mais precisas sobre as direções de leitura e 4sU-seq (que é o método descrito aqui) detecta apenas recentemente transcrito de RNA. No entanto, 4sU-seq é altamente sensível e pode ser aplicado em diferentes períodos de tempo para medir a atividade transcricional quantitativa em ativamente mudando células, bem como alterações quantitativas no processamento de mRNA (que acontece em poucos minutos)9, 11,12. Além disso, 4sU-seq é ideal para combinar com o RNA-seq para calcular taxas de rotatividade de RNA para genes9. A transcrição do mRNA é realizada pela RNA polimerase II (RNAPII), que por sua vez é influenciado por uma multiplicidade de fatores, tais como fatores de transcrição, modificações do histone e ativadores/repressores gerais que pode até mesmo ser parte do transcriptional complexo. Para testar quantas regiões do gene/promotor/realçador obrigam-se em um fator, ChIP-seq desenvolveu-se, que agora é o método padrão para essa finalidade, devido a muitos anticorpos comercialmente disponíveis13. No entanto, apesar de ChIPseq dá informações claras sobre onde regulamenta fatores vincular, não reflete se ele realmente leva a alterações na transcrição14. Portanto, realizar ChIP-seq com 4sU-seq é a combinação ideal para tais questões biológicas. Regulação da expressão genética também pode ocorrer numa fase posterior, desde que os níveis de mRNA e proteína não se correlacionam necessariamente15,16, indicando Regulamento potencialmente significativo na translação ou borne-translational nível, dependendo do contexto. No ano de 2011, o ribossoma profiling tinha foram combinado pela primeira vez com RNA-seq e é agora o método de escolha para quantificar as mudanças que ocorrem rapidamente em proteínas, uma vez que ainda existem alguns limites de sensibilidade com espectrometria de massa17. Com efeito, taxas de conversão obtidas de tais métodos foram mostradas para fornecer uma estimativa relativamente bom de alterações nos níveis de proteína (medidas pelo menos para mudanças de longo prazo) e permitem uma visão mais detalhada sobre o processo de tradução, por exemplo, o determinação da tradução alternativa e iniciar-lado quadros17. A utilização combinatória de todos os quatro métodos podem ser usados em estado estacionário, entre vários tipos de células, ou em uma série de tempo experimentar de uma rápida mudança de cela11. Este uso fornece uma visão geral de todo o genoma de alterações na ligação do fator de transcrição influenciando RNA transcrição, processamento e tradução.
Protocolo
Todos os métodos estão em conformidade e consonância com as diretrizes de Helmholtz Zentrum München institucionais, estaduais e federais.
1. preparação
- Fazer um plano detalhado da instalação da experimental, incluindo um calendário, quando adicionar 4sU para o meio de cultura celular e quando a colheita de células para cada método. Dependendo a questão biológica, considere cuidadosamente os pontos de tempo para o desenho da amostra, tempo 4sU-rotulagem e concentração (tabela 1).
Nota: Verificar o impacto da 4sU na viabilidade celular e stress resposta com antecedência (consulte "Verificar 4sU-rotulagem condições ideais" nos resultados de representante e Figura 1). É recomendável realizar um teste preliminar de cada método pelo menos uma amostra. Verificar se a qualidade e a quantidade de RNA/DNA é suficiente para sequenciamento profundo (ver partes do protocolo) e prática rápida mas suave manipulação das células durante o experimento. - Calcular o número de células necessárias para cada ponto de tempo de cada método (ver tabela 2 para uma estimativa grosseira quando usando células-T primárias). Além disso, considere o sequenciamento menos amostras de RNA total, só que do RNA 4sU (por exemplo, apenas para taxas de conversão de ponto de tempo ou volume de negócios de RNA taxas devem ser calculadas). Para confirmar e verificar o significado dos resultados (recomendado), crie replicar biológica pelo menos um.
Nota: Importante, para todos os métodos e pontos de vez consecutiva, as amostras devem ser do mesmo pool inicial de células. Recomenda-se pelo menos um pesquisador dedicado para cada método. - Prepare todo o necessário com antecedência (por exemplo, 4sU aliquotadas, cicloheximida, mamíferos do lysis e 1% de formaldeído). Após a adição de 4sU, Evite expor as células à luz brilhante, como isso pode levar a reticulação do RNA 4sU-rotulado de proteínas celulares18.
- Piscina de todas as células de interesse em um frasco só antes do tratamento (Figura 2). Contar as células (por exemplo, com um hemocytometer) e usar a quantidade necessária para o controle não tratado de cada método (não se esqueça de também rotular o controle não tratado para seq-4sU com 4sU). Tratar as células restantes e imediatamente dividir o número necessário de células para cada ponto de tempo e método. Manipular as células mais rapidamente possível para minimizar o estresse devido às mudanças de temperatura ou CO2 níveis.
Nota: por exemplo, amostras 1h, 2h, 3h após o tratamento, então um quarto das células é usado para controle não tratado e três quartos são usados para controle de tratados.
2. 4sU-rotulagem
Nota: Este protocolo é modificado de Rädle, et al. 19 se referem ao seu protocolo para mais informações referentes à rotulagem metabólicos com 4sU. Para todos os métodos e pontos de vez consecutiva, as amostras devem ser provenientes de partida mesmo pool de células.
-
Início da rotulagem
- Degelo aliquotadas 4sU imediatamente antes da utilização. Adicionar 4sU em cada ponto de tempo diretamente para a mídia que contém células de interesse (consulte a tabela 2 para números recomendados de célula T, menos de 60 µ g RNA por ponto de tempo), misture delicadamente e coloque de volta para a incubadora. Dispose restante 4sU (não congelar).
- No final da rotulagem, colete células (por exemplo, raspador de célula) e centrifugar 330 x g por 5 min a 4 ° C em tubos de polipropileno (que resistem às forças de alto g). Aspire médio e adicionar o reagente para isolamento de RNA (≥ 1 mL por 3 x 106 células, consulte materiais para recomendação para usar) para cada tubo. Resuspenda o pellet (≥ 1 mL por 3 x 106 células) totalmente, incubar durante 5 min à temperatura ambiente (RT) e congelar amostras a-20 ° C. As amostras podem ser armazenadas a-20 ° C durante pelo menos 1 mês.
Atenção: Os reagentes utilizados para isolamento de RNA são extremamente perigosos quando entrar em contato com a pele ou olhos. Lidar com isso com cuidado e considere as instruções de segurança.
-
Preparação de RNA usando modificado protocolo de isolamento de RNA
- Adicionar 0,2 mL de clorofórmio por 1 mL de reagente para isolamento de RNA e homogeneizar por agitação por 15 s. Proceda como mencionado no protocolo de rotulagem metabólico (etapa 1-12, 2. Preparação de RNA usando modificado protocolo) de Rädle et al. 19
- Medir a concentração de RNA (ver Tabela de materiais), de acordo com as instruções do fabricante. Use este RNA para RNA total-seq também (consulte a etapa 3. Total do RNA-seq) ou armazenar a-80 ° C durante pelo menos 1 mês.
-
Thiol-Specific Biotinylation do RNA recém transcrito
- Comece com 30-80 µ g de RNA celular total. 60 µ g de RNA devem produzir quantidades suficientes de RNA recém transcrito.
- Prepare a reação de rotulagem. Pipeta na seguinte ordem (por µ g do RNA): 1 µ l 10 x Biotinylation Buffer, 7 µ l de RNA (contendo 1 µ g de RNA diluído em livre de nuclease H2O) e 2 µ l biotina-HPDP (1 mg/mL). Adicionar biotina-HPDP última e misture imediatamente por pipetagem. Envolva os tubos com folha de alumínio para evitar a exposição à luz brilhante. Consulte a discussão de uma alternativa à biotina-HPDP. Incube a RT por 1,5 h com rotação.
- Apropriado mL 2 tubos de centrífuga de (ver Tabela de materiais) a 15.000 x g por 2 min. Pipetar todos o biotinilado RNA para o tubo previamente fiado 2 mL, adicionar um volume igual de clorofórmio e misturam vigorosamente. Incubar durante 2-3 min até as fases começam a separar e as bolhas começam a desaparecer.
- Centrifugar a 15.000 x g por 15 min a 4 ° C. Transferi com cuidado a fase aquosa superior para um tubo novo.
- Repita os passos de 2.3.3. e 2.3.4. uma vez. Adicione 10% do volume de NaCl (5m) e um volume igual de isopropanol para a fase aquosa. Centrifugar a 20.000 x g por 20 min a 4 ° C. Descarte o sobrenadante.
- Adicione que um volume igual preparadas de 75% de etanol. Centrifugar a 20.000 x g. descartar o sobrenadante, girar brevemente e remover o restante do etanol. Totalmente Resuspenda em 30-100 µ l de H2O (uso 1 µ l H2O por 1 µ g de RNA entrada da etapa 2.3.1) pela mistura de pipeta.
- Verificar a integridade do RNA por análise eletroforética, ou tomar uma alíquota e verificar mais tarde.
-
Separação de recentemente transcrito (rotulado) e preexistente do RNA (sem rótulo)
- Remover contas paramagnéticos (ver Tabela de materiais) de armazenamento de 4 ° C e deixe pelo menos 30 min para trazê-los à temperatura ambiente. Tampão de lavagem de 4sU calor (3 mL por amostra) a 65 ° C.
- Prepare a solução de ditiotreitol (DTT) de 100 mM. Pesa TDT numa escala ultra-fina e adicionar a quantidade necessária de água livre de nuclease. Sempre prepare fresco. Use 200 µ l por amostra.
- Aqueça biotinilado amostras do RNA (1 µ g / µ l) a 65 ° C por 10 min desnaturar e coloque imediatamente no gelo. Adicionar contas de estreptavidina 100 µ l a biotinilado RNA e incubar a RT por 15 min com rotação.
- Coloque uma coluna apropriada (consulte a Tabela de materiais para recomendações) para cada amostra no carrinho magnético e pre-equilibrar cada coluna com 1 mL temperatura 4sU lavagem.
Nota: Isto vai demorar cerca de 5-10 min. Se qualquer uma das colunas não iniciar drenagem após 5 min, pressione suavemente em cima da coluna com um dedo com luva. - Aplica uma mistura de RNA/grânulos para o meio de cada coluna. Descarte o escoamento, a menos que o RNA sem rótulo precisa ser recuperado. Em caso afirmativo, colete o escoamento e pelo menos a primeira lavagem. Realizar a recuperação de RNA, como descrito por Rädle et al . (etapa 1-7, 7. Recuperação do RNA sem rótulo, não vinculado)19.
- Lave três vezes com tampão de lavagem 4sU 0,9 mL (pré-aquecido a 65 ° C da etapa 2.4.2.) e 0,9 mL RT 4sU de tampão de lavagem, respectivamente.
- Uso de grânulos paramagnéticos recuperar recém transcrito de RNA. Distribuir 400 µ l de grânulos de paramagnético RT bem dispersos em um tubo por amostra e lugar debaixo de cada coluna. Eluir recém transcrito de RNA com 100 µ l 100 mM DTT. Esperar 3 min e realizar uma segunda eluição com 100 µ l 100 mM DTT. (Opcional: executar a eluição e recuperação conforme descrito por Rädle et al) 19
- Mix recentemente transcrito RNA/grânulos completamente por pipeta 10 vezes de mistura e proceder de acordo com a orientação do fabricante. Eluir RNA em 11 µ l livre de nuclease H2O.Quantify do RNA usando um apropriado dados (ver Tabela de materiais). RNA pode ser armazenado a-80 ° C durante pelo menos 1 mês.
Nota: Recentemente transcrito RNA pode ser usado para preparar as bibliotecas de cDNA para sequenciamento de nova geração (consulte Tabela de materiais para uma sugestão na qual kit de usar) ou mais a jusante análises. 100 - 500 ng RNA são suficientes para a maioria dos kits de preparação de biblioteca (ver discussão).
3. total do RNA-Seq
- Tomar diretamente do RNA de 4sU rotulado RNA após a preparação do RNA usando o reagente modificado para protocolo de isolamento de RNA para RNA total-seq (consulte a etapa 2.2.2).
- Para a preparação de biblioteca, diluir uma alíquota do RNA para uma concentração final de 50-100 ng / µ l. Use o mesmo kit quanto à preparação de biblioteca do RNA recém transcrito. 100 - 500 ng RNA são suficientes para a maioria dos kits de preparação de biblioteca.
4. Ribossoma perfilamento
Nota: Para todos os métodos e pontos de vez consecutiva, as amostras devem ser do mesmo pool inicial de células. Para recomendações sobre qual kit para usar, consulte a Tabela de materiais.
- Preparação e isolamento do Ribossoma protegido fragmentos (RPFs):
- Use quantidades adequadas de células para cada ponto do tempo (consulte a tabela 2 para celulares T recomendada). Trate as células aderentes com cicloheximida, conforme descrito no protocolo do fabricante.
Cuidado: Cicloheximida é altamente tóxica e pode causar mutações. Evite a inalação e contacto com a pele. - Coletar e piscina não - ou semi - adherent células de cada vez que apontam para um tubo de polipropileno e ajustam a concentração final de 1 x 106 células por mL de meio de célula específica (por exemplo, RPMI complementado por células T). Adicionar cicloheximida com uma concentração final de 0,1 mg/mL, misturar por inversão do tubo de polipropileno e incubar durante 1 min. células Centrifugue por 5 min a 330 x g a 4 ° C. Aspire médio e lavam-se células pelo menos 10 mL de PBS suplementado com cicloheximida (concentração final de 0,1 mg/mL).
- Células de centrifugar durante 5 min à 330 x g a 4 ° C. Aspire médio e adicionar 100 µ l pilha mamífera lise por 10 x 106 células. Misture por pipetagem e expulsar através de uma agulha estéril de 25 gauge 22 para lisar as células completamente.
- Transfira o lisado celular para um tubo de pré-resfriado 1,5 mL. Incube durante 10 min no gelo com inversões periódicas. Centrifugação por 10min a 20.000 x g a 4 ° C, para esclarecer o lisado. Transferi o sobrenadante para um tubo pré-resfriado 1,5 mL.
- Preparar um 01:10 diluição do lisado com água livre de nuclease e leitura de registro um A260usando um espectrofotômetro. Usar água nuclease-livre como um espaço em branco e um 01:10 diluição de tampão de lise de células de mamíferos como o padrão. Calcule a concentração de /mL de260A do lisado de acordo com a seguinte equação:
(Um260 lisado celular -260 mamíferos do lysis) x fator de diluição 10 = um260/mL - Criar 200 alíquotas µ l do lisado no gelo e prosseguir com o tratamento de nuclease.
Nota: Opcionalmente, preparar uma alíquota 100 µ l de RNA total, adicionar 10 µ l de SDS 10% e misture. Armazenar a 4 ° C e prosseguir com o 4.3.2. É recomendado usar o RNA total do RNA 4sU-rotulado (consulte a etapa 3. RNA total-seq).
- Use quantidades adequadas de células para cada ponto do tempo (consulte a tabela 2 para celulares T recomendada). Trate as células aderentes com cicloheximida, conforme descrito no protocolo do fabricante.
- Footprinting do Ribossoma
- Realizar o tratamento de nuclease imediatamente sem congelar o lisado. Adicione 7,5 unidades de nuclease (incluído no kit recomendado) para cada A260 do lisado. Por exemplo: 80 A260/mL lisado x 0,2 mL de lisado x 7,5 U/A260nuclease = 120 nuclease U.
Nota: Opcionalmente, titule da nuclease para digestão conforme descrito pelo fabricante. - Incube a reação de nuclease por 45 min em RT com uma mistura suave. Congelar 200 alíquotas µ l do lisado com nitrogênio líquido e armazenar a-80 ° C, ou parar a reação de nuclease adicionando inibidor de RNase 15 µ l de cada alíquota 200 µ l e continue com o passo 4.3.2.
- Realizar o tratamento de nuclease imediatamente sem congelar o lisado. Adicione 7,5 unidades de nuclease (incluído no kit recomendado) para cada A260 do lisado. Por exemplo: 80 A260/mL lisado x 0,2 mL de lisado x 7,5 U/A260nuclease = 120 nuclease U.
- Purificação de RPFs
- Descongelar uma amostra de nuclease digerida RPFs e adicionar inibidor de RNase 15 µ l. Mantenha as amostras no gelo.
- Purificar os RPFs de acordo com o protocolo do fabricante (purificação da coluna é recomendada) e medir a concentração de RNA em um espectrofotômetro.
- rRNA depleção
- Use 5 µ g de RPFs purificados para esgotamento de rRNA.
- Siga o protocolo do fabricante (etapa 1-2, depleção primário rRNA) para esgotamento de rRNA. Concentração de RNA medida de rRNA empobrecido RPFs em um espectrofotômetro.
- PÁGINA de purificação dos RPFs
- Uso 500 ng de rRNA empobrecido RPFs para purificação de página.
- Prepare o RNA controle, amostras e escada para a purificação de página. Mix 5 µ l do RNA controle e 5 µ l de desnaturação do gel carregando o corante em um tubo de microcentrifugadora de 0,5 mL. Mix 10 µ l de cada RPF com 10 µ l de desnaturação gel de carregamento, respectivamente. Preparar uma escada alíquota (4 µ l 20/100 escada, água livre de nuclease 1 µ l e 5 µ l desnaturação gel corante carregamento). Carregá-lo entre cada amostra e controle para evitar contaminação cruzada.
- Desnature amostras e escada incubando a 95 ° C por 5 min e coloque imediatamente no gelo. Carregar a 20 µ l de cada amostra (opcionalmente, carregar 10 µ l e freeze restantes amostras a 20 ° C) separados por 10 µ l de escada preparada em um gel de poliacrilamida-ureia 12% ou 15%. Carrega 10 µ l de RNA controle. Funcione o gel até a banda de azul de bromofenol atinge a parte inferior do gel (180 V, ~ 70 min) (Figura 3).
- Manchar o gel de acordo com o protocolo do fabricante a 4 ° C. Use um transiluminador de campo escuro que emite luz azul para visualizar o RNA. Gel de fatias para cada amostra correspondente a ~ 28 e 30 de excisar nt em comprimento. Assumir o controle do RNA como referência e impostos especiais de consumo.
Nota: RPFs são dificilmente visíveis. Impostos especiais de consumo fatias para o tamanho indicado pelo controle do RNA - contendo dois oligos do nt 28 e 30 de comprimento - mesmo se as amostras não são visíveis. - Perfure um buraco no fundo dos tubos de 0,5 mL microcentrifuge com uma agulha estéril de calibre 20. Transferi cada fatia de gel em um tubo separado e tubos de lugar tampado em um tubo de 1,5 mL. Centrifugue por 2 min em 12.000 x centrifugação de repetição de g. se gel de fatias não são completamente destruir no tubo de 1,5 mL.
- Eluir o RNA das fatias do gel de interrompidos com 400 µ l livre de nuclease água, acetato de amônio 40 µ l (5 M) e 2 µ l de SDS (10%) a cada noite a 4 ° C.
- Transferi o chorume para tubos de filtro de 1,5 mL (fornecidos com o kit recomendado) com uma ponta de pipeta de 1 mL (largo-furo ponta ou ponta de self-made 1ml com corte final). Centrífuga para 3 min a 2.000 x g para separar eluída RNA da gel de fatias. Suavemente, pipete solução aquosa em um tubo de 1,5 mL. Adicionar 2 glicogênio de µ l (fornecido com o kit recomendado) e 700 µ l 100% isopropanol e loja a-20 ° C durante pelo menos 1 h.
- Centrifugar a 4 ° C por 20 min em 13.000 x g. Discard sobrenadante. Lave a pelota com etanol a 80% recentemente preparada previamente refrigerados a 4 ° C durante 10 minutos a 13.000 x sobrenadante de descarte de g.... e deixe secar naturalmente. Resuspenda cada amostra em 20 µ l e o controle do RNA em água de nuclease livre 8 µ l. Armazenar a-20 ° C, se necessário.
- Fragmentação, reparação final, 3' adaptador ligadura, transcrição reversa
- Execute o procedimento conforme descrito pelo protocolo do fabricante (fragmentação e reparação final 3' ligadura de adaptador e transcrição reversa).
- PÁGINA de purificação do cDNA
- Preparar amostras, controle de RNA e escada para a purificação de página: Mix 10 µ l de cada amostra e controle de RNA com 10 µ l de desnaturação gel carregando tintura, respectivamente. Preparar uma escada alíquota (4 µ l 20/100 da escada, 1 µ l da nuclease-livre de água, 5 µ l de desnaturação gel de tintura de carregamento). Carregá-lo entre cada amostra e controle para evitar contaminação cruzada.
- Desnature amostras e escada incubando a 95 ° C por 5 min e coloque imediatamente no gelo. Carregar a 20 µ l de cada amostra (opcionalmente, carregar 10 µ l e freeze restantes amostras a 20 ° C) separados por 10 µ l de escada preparada para uma poliacrilamida 10% / 7 - 8 M gel de ureia/TBE. Carrega 10 µ l de RNA controle. Funcione o gel até o azul de bromofenol migra completamente fora do gel (180 V, ~ 60 min).
- Manchar o gel de acordo com o protocolo do fabricante a 4 ° C. Use um transiluminador de campo escuro que emite luz azul para visualizar o RNA e as fatias de gel para cada amostra correspondente a nt ~ 70-80 impostos especiais de consumo.
- Prossiga conforme descrito no passo 4.5.5 - 4.5.8 e ressuspender cada amostra em água de nuclease-livre 10 µ l.
- cDNA circularização
- Prepare-se bastante mistura de circularização mestre para todas as reações, combinando os seguintes reagentes para cada amostra em gelo: 4,0 µ l circularização reação Mix, 2,0 µ l ATP, 2,0 µ l MnCl2e 2,0 µ l Ligase.
- Adicione 10 µ l da mistura mestre para cada amostra. Misture delicadamente e centrifugar brevemente. Incube as amostras a 60 ° C por 2 h. imediatamente lugar amostras no gelo.
- Amplificação por PCR
- Siga o protocolo do fabricante (etapa 1-3, amplificação por PCR) para amplificação por PCR. Use 4 µ l do cDNA circular para amplificação com 9 ciclos PCR para células T primárias para alcançar melhores resultados.
- Purificar as bibliotecas e verificar sua distribuição de tamanho, de acordo com o protocolo do fabricante (passo 4-8, amplificação por PCR). O tamanho esperado da biblioteca amplificado é bp 140-160 (ver Figura 4).
- Para bibliotecas de sequenciamento, consulte protocolo do fabricante e o mecanismo de sequenciamento para orientações adicionais.
5. chIP-Seq
Nota: Este protocolo é modificado de Blecher-Gonen, et al. 14 se referem ao seu protocolo para mais informações sobre o ChIP-Seq Para todos os métodos e pontos de vez consecutiva, as amostras devem ser do mesmo pool inicial de células.
- Reticulação e colheita das células
- Crosslink número adequado de células (consulte a tabela 2 para celulares T recomendado) para cada ponto de tempo, com uma concentração final de 1% de formaldeído em um meio de cellspecific (por exemplo, RPMI complementado por células T) por 10 min à temperatura ambiente com um balanço suave. Pare a reação de reticulação pela adição de glicina a uma concentração final de 0,125 M.
- Células de centrífuga a 330 x g por 5 min a 4 ° C. Desprezar o sobrenadante e lavar as células em PBS gelado. Repita a etapa 5.1.2 duas vezes e congelar pelotas de célula a-80 ° C. Pelotas congeladas podem ser armazenadas pelo menos 6 meses.
- Lise celular e Sonication
Nota: Durante todas as etapas célula Lise e sonication, as amostras devem ser mantidas no gelo ou a 4 ° C para minimizar a degradação de reversão e proteína de ligação cruzada.- Resuspenda pelotas de célula em tampão de lise celular gelada 1 mL com inibidores de protease recentemente adicionado para isolar os núcleos (adicionar inibidores da fosfatase opcionalmente). Incubar durante 10 min no gelo e centrifugar 2.600 x g por 5 min a 4 ° C.
- Aspire o sobrenadante e ressuspender núcleos em tampão de lise de núcleos gelada de 1ml com inibidores de protease recentemente adicionado (opcionalmente: Adicionar inibidores da fosfatase). Incube durante 10 min no gelo. Proceda à sonicação das células para gerar uma fração média de tamanho de DNA de 0,2 - 1,0 kb (ver Figura 5).
Nota: Sonication condições precisam ser otimizadas de acordo com o tipo de célula e outras condições (por exemplo, número de telemóvel, o volume e reserva). Para células T primárias sonication para ciclos de 20-25 é recomendado (ver materiais para descrição detalhada). - Pegue um 20-50 µ l alíquota da cromatina cortada e calor durante 10 minutos a 95 ° C e 1.000 rpm agitação para realizar um rápido reverso crosslink e verificar o tamanho da cromatina. Adicione 2-5 μL de Proteinase K e incube por 20 min a 56 ° C e 1.000 rpm a tremer. Execute a inactivação de calor durante 10 minutos a 95 ° C e 1.000 rpm a tremer. Purificar a cromatina com um kit apropriado (consulte a Tabela de materiais). Verifique o tamanho da cromatina em um gel de agarose 1% e use 100 bp Plus marcador.
- Centrifugue a cromatina cortada com uma média de fração de tamanho de DNA de 0,2 - 1,0 kb para 10 min a 20.000 x g e 4 ° C para detritos e cromatina insolúvel de Pelotas. Transferir o sobrenadante para um tubo novo e manter no gelo.
- Manter 5-10% da cromatina lisada como entrada. Congele a-20 ° C (usado na etapa 5.5.2).
- Casal de anticorpo para grânulos
- Anticorpo de casal 10 µ g (por exemplo, anti-RNA Pol II; H3K36me3 anti-Histon) em 220 µ l PBS (com BSA de 0,5% e 0,5% Tween 20) 80 talões superparamagnético µ l acoplado à proteína G (ver Tabela de materiais) pelo menos 1 h à temperatura ambiente com rotação.
- Coloque os tubos em um ímã. Espere até que todos os grânulos estão vinculados ao ímã e remover o sobrenadante. Lote suplementar com 6 µ l lisados esperma de salmão DNA em PBS (com 0,5% de BSA e 0.5% Tween 20) por 30 min à temperatura ambiente com rotação.
- Coloque os tubos em um ímã. Espere até que todos os grânulos estão vinculados ao ímã e remover o sobrenadante claro. Lave grânulos com buffer de ChIP IP três vezes.
- Imunoprecipitação da cromatina
- Diluir a cromatina ao volume total de 1 mL de tampão de Lise núcleos com inibidores de protease recentemente adicionado (opcionalmente, adicionar inibidores da fosfatase). Adicionar o buffer de ChIP IP com inibidores de protease recentemente adicionado (opcionalmente, adicionar inibidores da fosfatase) até um volume final de 3 mL. Manter no gelo ou a 4 ° C, enquanto o anticorpo é acoplado aos talões.
- Adicione a cromatina diluída ao anticorpo acoplado grânulos da etapa 5.3.3 e incubar durante a noite a 4 ° C, com rotação suave.
- Lavar com os seguintes buffers (1 mL cada, consulte Tabela de materiais) à temperatura ambiente por 5 min com rotação, coloque o ímã tubos e remover o sobrenadante: Wash Buffer eu lavar Buffer II, III de tampão de lavagem e 2 x pH TE 8.0 respectivamente.
- Descartar o sobrenadante e deixe secar naturalmente para ~ 5 min.
- Reverter a reticulação
- Retire amostras do ímã. Juntar 50 µ l de tampão de eluição e misture pipetando para eluir complexos proteína-ADN de grânulos.
- Inclui entrada amostra deste passo em frente. Adicionar o tampão de eluição para amostras para um volume final de 50 µ l (para manter a composição de reserva semelhante para as amostras de ChIP) de entrada e de processo, juntamente com as amostras de ChIP.
- Mix 3 µ l de tampão de eluição e 2 µ l de RNase (DNase livre). Adicionar 5 µ l da mistura para cada amostra e incube por 30 min a 37 ° C.
- Da protease µ l de mistura 2.5 K, 1 µ l glicogénio e tampão de eluição 1,5 µ l por amostra. Adicionar 5 µ l da mistura para cada amostra (1 protease U K e 20 µ g glicogênio, por exemplo) e incube por 2 h a 37 ° C.
- Incube as amostras a 65 ° C durante a noite (pelo menos 4 h) agitando para executar reticulação reversa.
- Coloque os tubos sobre o ímã pelo menos 30 s e transferir o sobrenadante para um tubo novo. As amostras podem ser congeladas a-20 ° C por até 12 meses.
- Purificação de DNA
- Adicione 140 µ l de grânulos paramagnéticos bem dispersos para 60 µ l de amostra (2.3:1 ratio). Cuidadosamente, pipete acima e abaixo de 25 vezes para homogeneizar. Certifique-se de que o líquido em cada tubo é homogêneo. Incube a temperatura ambiente por 2 min. lugar dos tubos sobre o ímã para 4 min, ou até todos os grânulos são vinculados ao ímã e descartar o sobrenadante.
- Deixe os tubos sobre o ímã e adicione 200 µ l de etanol a 70% preparados na hora. Incubar os tubos por 30 s sem perturbar os grânulos. Desprezar o sobrenadante e repita essa etapa mais uma vez. Etanol, Aspire completamente e deixe o paramagnético grânulos secar ao ar durante 4 minutos.
Nota: Remoção incompleta do etanol a sério pode reduzir a recuperação do DNA e rendimento. Seque a pelota apenas até que esteja seco. Ressecamento da pelota pode reduzir a recuperação do DNA e rendimento. - Retirar os tubos do ímã e adicionar 20 µ l 10 mM Tris-HCl (pH 8.0). Pipete suavemente todo o volume acima e abaixo de 25 vezes para homogeneizar. Incube durante 2 min à temperatura ambiente. Coloque os tubos volta sobre o ímã para 4 min e transferir o sobrenadante para outro tubo.
- Medir a quantidade de DNA com um apropriado dados (ver Tabela de materiais).
- Verifique se que o ChIP foi bem-sucedida por qPCR (diluído 1 µ l em 100 µ l de H2O e uso 2-5 µ l para qPCR). Use primers específicos para um positivo (sítio de ligação conhecida da proteína de interesse) e o controlo negativo (por exemplo, um gene que é silencioso e/ou não um alvo da proteína de interesse).
Nota: A preparação da biblioteca pode ser executada com 2 ng de DNA ChIP dependendo do kit (consulte Tabela de materiais para uma sugestão na qual kit de usar).
Resultados
4 sU rotulagem: verificar ideal 4 sU-rotulagem condições (apoptose, nuclear stress, stress citoplasmática), tempo e concentração: Altos níveis de 4sU podem inibir a produção e processamento de RNA ribossomal e induzir stress citoplasmática, bem como nuclear30. Portanto, as células de interesse devem ser testadas para 4sU induzida por estresse, bem como a apoptose. Análise ocidental do Borrão é recomendado para a visualização de acumulação de p53, que indica a tensão nuclear, aumentando os níveis de fosfo-EIF2a que apresentam estresse citoplasmática e fluorescência-ativado da pilha classificação análise (FACS) por apoptose. Níveis elevados e longa exposição ao 4sU ou drogas como thapsigargin ou Arsenito podem ser usados para induzir o estresse celular. Para induzir apoptose morte celular, as células foram tratadas com BH3I-1 (500 ng / µ l) ou incubadas por 5 min a 95 ° C (choque térmico). Anexina V/7-AAD coloração foi usada para determinar a apoptose (anexina V) e células de mortos (7-AAD). Rotulagem de in vitro gerado primárias células Th1 para 0,5 h com 500 µM 4sU (concentração final) ou 1 h com 200 µM 4sU induz nem sinais de estresse celular nem apoptose (Figura 1), mas leva a incorporação de 4sU suficientes.
RNA rotulagem tempo também pode ser encurtado (≤ 5 min) que leva ao aumento de curta duração intrônicas sequências vezes em comparação com mais de rotulagem. Para visualizar taxas emenda co-transcriptional, vezes 4sU-rotulagem não devem ultrapassar 30 min. Para obter mais detalhes sobre 4sUlabeling, por favor consulte Rädle et al. 19
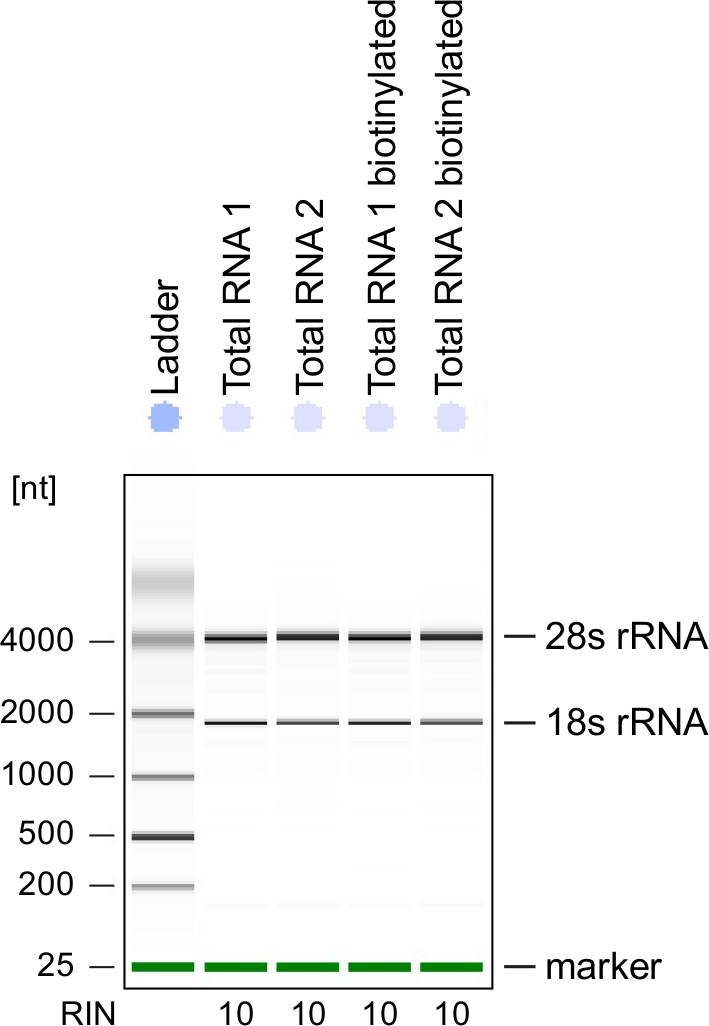
Controle de qualidade: Integridade do RNA é de grande importância quando o processamento do RNA. É mais conveniente verificar a qualidade do RNA do RNA 4sU-rotulado depois biotinylation pela análise electrophoretical (ver Tabela de materiais). Considere a possibilidade de verificar RNA isolado da etapa 2.2.2, especialmente quando usá-lo para sequenciamento do RNA total. Número de integridade do RNA (RIN) deve ser ≥ 8 para garantir a integridade do RNA para tratamento posterior (Figura 3).
Electrophoretical análise também pode ser usado para verificar recentemente transcrito de RNA. Esteja ciente de que o RNA recém transcrito contém significativamente menos maduro rRNAs comparado ao RNA total com as bandas rRNA típico, sendo muito menos proeminente.
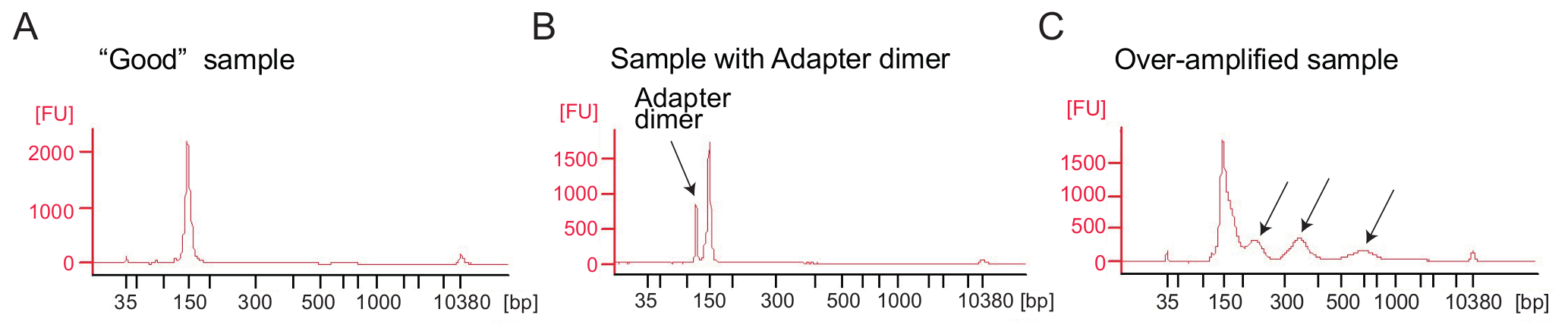
De perfil Ribossoma: amplificação por PCR da biblioteca de cDNA: amplificação do cDNA (etapa 4.9.1) é um passo fundamental para garantir resultados bom sequenciamento. Analise bibliotecas amplificadas pela análise electrophoretical. Uma boa amostra de bibliotecas amplificadas mostra um pico em torno de 140-160 bp (Figura 4A). Quantidades excessivas de adaptador dímeros devem ser evitado (Figura 4B) e as amostras devem ser purificadas ainda mais usando o procedimento de purificação de página de acordo com o protocolo do fabricante (purificação da página de produtos do PCR). Muito modelo ou muitos ciclos PCR podem resultar em amplificação excessiva, caracterizada pelo aparecimento de bandas de peso molecular maior do que o esperado, produtos PCR borrados e produtos de dímero de adaptador (Figura 4). Para a maioria das amostras 1-5 µ l do cDNA circular e 9 ciclos PCR para amplificação tipicamente produzirá quantidades suficientes do produto correto do PCR.
ChIP: cromatina de corte: Óptimas condições de corte precisam ser ajustado para cada tipo de célula. Determine as condições de corte (por exemplo, o número de ciclos, alta ou baixa potência) com antecedência. Use o mesmo número de células e o mesmo volume para testes, uma vez que uma menor densidade celular aumenta a eficiência de corte. Tente evitar sobre ou sob-corte a cromatina. Fragmentos de cromatina grande podem afetar drasticamente os resultados de ChIP por entupimento, e excesso de corte pode destruir resumos sobre a proteína de interesse, levando a uma menor eficiência de ligação pelo anticorpo. Neste experimento, os melhores resultados foram alcançados quando a fração principal da cromatina cortada era cerca de 1.000 bp ou ligeiramente inferior (Figura 5A).
Verificar o ChIP por qPCR: Antes de iniciar o ChIP, é aconselhável testar se o anticorpo utilizado é apropriado para ChIP (se possível, usar os anticorpos da classe de ChIP) por ChIP-qPCR. Verifique se o ChIP para sequenciamento por qPCR antes de iniciar a preparação de biblioteca (consulte a etapa 5.6.5). Projetar primers que se ligam a um site conhecido alvo da proteína de interesse. Se o site de destino exato dentro de um gene é desconhecido, vários pares de primeira demão podem ser usados para escanear o gene e elementos normativos associados. Para RNAPII ChIP de Th1 células Ifng, que transcricionalmente é upregulated após estimulação e actina primeiras demão podem ser usadas como controle positivo. Sox9 e insulina servem como um controle negativo, uma vez que esses genes não são expressos em células Th1 (Figura 5B). Lembra de não usar exon-abrangendo as primeiras demão, que são normalmente utilizadas para qPCR de mRNA. Um controle de IgG também pode ser usado para provar a especificidade do anticorpo usado. Immunoprecipitated DNA pode ser medido com um apropriado dados (ver Tabela de materiais). Quantidades de DNA nonspecifically acoplado pelo controle IgG devem ser significativamente mais baixo comparadas com a quantidade de DNA vinculada o anticorpo de interesse.
Replica: prova de importância biológica: É altamente recomendável realizar o experimento cinético para todos os métodos a partir do mesmo pool de células para garantir que as células têm a mesma identidade para todas as amostras não tratadas e tratadas (Figura 2). No entanto, é aconselhável levar pequenas alíquotas de tempo principais pontos para cada método comparar amostras biológica idêntico (por exemplo, por qPCR, análise de FACS). Isto permite uma estimativa grosseira se o tratamento para ambas as repetições foi reprodutível e você pode prosseguir com o sequenciamento. Validação das repetições deve ser executada por meio da análise de bioinformatical. Reprodutibilidade dos resultados pode ser avaliada em termos de correlação entre os valores FPKM entre repetições e visualizadas usando gráficos de dispersão (Figura 6).

Figura 1: Verificação de condições óptimas 4sU-rotulagem sem distorçer fisiologia celular (figura de Davari et al. 11)
(A) deteção de apoptosis da pilha por análise de FACS: In vitro gerado Th0 células foram tratadas com diferentes concentrações de 4sU (entre parênteses) para 0,5 h, 1 h e 2 h, respectivamente. BH3I-1 tratamento foi usado para induzir apoptose determinado pela anexina V, Considerando que o choque térmico (5 min a 95 ° C) foi usado para induzir a morte celular, determinada pelo 7-AAD. (B) análise ocidental do borrão para p53 de 4sU tratados e ativadas as células T: amostras foram rotuladas com 200 µM 4sU para a hora indicada de ativação, exceto o ponto de tempo de 0,5 h que foi rotulado com 500 µM 4sU. (C) análise ocidental do borrão de fosfo-EIF2a e EIF2a total em células Th1 ativadas com as mesmas condições de rotulagem como em (B). Thapsigargin foi usado como controle positivo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Visão geral esquemática de uma instalação cinética para controlar as alterações de todo o genoma
Este esquema ilustra a configuração para a combinação de 4sU-seq, total RNA-seq, Ribossoma Profiling e ChIP-seq para estudar todo o genoma alterações após tratamento com células. Células do pool e reserve o número necessário de células para controle não tratado. Tratar as células restantes e dividir para cada ponto de tempo e método. Rótulo não tratada/tratados células para seq-4sU com 4sU como descrito. Pontos de tempo e amostras para cada método dependem a questão biológica específica a ser examinada. Colher amostras para cada ponto de tempo e método e siga a parte dedicada do protocolo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: controle de qualidade de RNA 4sU-etiquetado
Total do RNA e biotinilado RNA obtido de células Th1 ativadas foram analisados em um Bioanalyzer. RRNA 18s e rRNA 28S são mostrados e número de integridade do RNA (RIN) é dado pelo instrumento para determinar a integridade do RNA. RIN deve ser ≥ 8 para garantir a integridade do RNA. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Bioanalyzer perfis de Ribossoma perfis de bibliotecas
(A), A boa biblioteca: A amostra mostra um pico na faixa de tamanho esperado (bp 140-160) e sem depuração adicional é necessária. (B), este exemplo mostra o produto amplificado de dímero de adaptador excessiva (120 bp) em relação ao produto desejado (bp 140-160). Esta biblioteca requer mais purificação. (C) uma amostra de excesso amplificada: picos de peso molecular maior do que o esperado e manchado PCR amplicons são visíveis (indicado pelas setas). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Tamanho de cromatina ideal depois da tosquia e verificação do ChIP por qPCR
(A) Agarose gel de imagens mostra o tamanho do fragmento ideal da cromatina cortado de três amostras que foram cortados para 25 ciclos em um sonicador e purificado conforme descrito antes no protocolo. (B) resultados Q-PCR de um ChIP de RNAPII total (anti-RNA Pol II, 8WG16, ab817) é representado como uma porcentagem de entrada. As primeiras demão IFNG e actina foram usadas como um positivo enquanto Sox9 e insulina são controles negativos (ambos os genes não são expressos em células Th1 ativadas). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Comparação de biológico Replica (figura de Davari et al 11.)
Representante de dispersão comparando valores de expressão (FPKM) entre Replica de recentemente transcrito (4sU) RNA 4 h após a estimulação de células Th1 ativadas. A linha verde indica valores iguais FPKM e correlação é indicada em cada parcela.
| Duração de rotulagem (min) | Concentração recomendada 4sU (µM) |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| < 10 | 500 - 2000 |
Tabela 1: Recomendado 4sU concentrações (de Rädle, et al. 19)
A gama de concentrações 4sU recomendada é indicada para diferentes tempos de rotulagem.
| Método | Número do celular | Quantidade de RNA |
| 4sU-rotulagem | ≥2 x 107 | ≥60µg |
| Ribossoma perfilamento | ≥2 x107 | |
| ChIP-seq | ≥2 x 107 - 3 x 107 |
Tabela 2: Quantidade necessária de células primárias de T
Quantidade mínima de pilhas de T primárias necessárias para cada método. Quantidades podem ser menor quando usando outros tipos de células.
Discussão
Analisar todo o processo de regulação gênica é necessário compreender plenamente as adaptações celulares em resposta a um estímulo ou tratamento. Combinando o RNA total-seq, seq-4sU, Ribossoma perfilamento e ChIP-seq em pontos diferentes do tempo leva a uma análise abrangente dos principais processos de regulação dos genes ao longo do tempo. Uma compreensão profunda dos processos biológicos é necessária para definir a configuração experimental, bem como os pontos de tempo ideal.
Desde métodos para estudar a regulação gênica melhorar rapidamente, estes protocolos podem ser adaptados para mudanças rápidas. No entanto, eles fornecem os métodos mais importantes para estudar os mecanismos de regulação genética básica em qualquer tipo de célula. Aqui, vamos discutir alguns dos fatos, deve-se considerar quando usando estes métodos e armadilhas.
Células: As células devem ser altamente viáveis e, se usando células isoladas primárias, pureza de populações de células deve ser garantida (por exemplo, análise de FACS para primárias células T). Células até mesmo um pouco estressadas podem influenciar os resultados desses métodos de sequenciamento muito sensível e diminuir a quantidade de RNA recém transcrito ou traduzido e levar a leituras indesejadas da resposta stress nos resultados do sequenciamento. A velocidade de centrifugação mencionada neste protocolo para células de Pelotas é otimizada para primárias células T. Assim, ajuste a velocidade de acordo com o tipo de célula.
4 sU efeitos na fisiologia celular: Além das opções acima para verificar a mínima perturbação de fisiologia celular sobre adição 4sU, análise adicional e/ou adicional pode ser executada, especialmente quando os números de celular são limitados. Efeitos sobre a proliferação celular podem ser testados por verificar o tempo de duplicação das células simplesmente contando células etiquetadas e sem rótulo. Indução de estresse nucleolar também poderia ser testada através da análise de morfologia celular através de mancha de nucleolin e núcleos da imunofluorescência. Para ainda mais verificar o impacto de 4sU, expressão gênica global alterado poderia ser medido pela correlação ler contagens de RNA total rotulado de RNA total sem rótulo.
Números de celular: Para em vitro gerado as células T, recomendamos começar pelo menos a quantidade de células indicado na tabela 2. Escolha números adequados por método de acordo com o tipo de células. Uma vez que as células T têm menos citoplasma e RNA em comparação com outras células, provavelmente menor quantidade de outras células será adequada. Para ChIP-seq, número de células altamente depende o anticorpo usado e o nível de expressão da proteína de interesse dentro das células. Para histona ou ChIP RNAPII, células fewe podem ser usadas, Considerando que o número de células precisa ser aumentada quando são utilizados fatores de transcrição, especialmente se eles são expressos em níveis baixos.
4 sU-rotulagem e RNA biotinylation: Ao usar células aderentes, 4sU rotulagem pode ser direcionado conforme descrito por Rädle et al. 19 desde 4sU muito rapidamente incorporar células, ele pode ser adicionado diretamente ao meio de suspensão, aderente ou células semi aderentes.
Recomenda-se iniciar o biotinylation com 60-80 µ g de RNA. No entanto, a menor quantidade de RNA pode ser usada, embora não testámos para menos de 30 µ g. adicionar uma coprecipitant (por exemplo, GlycoBlue) quando precipitação do RNA se o sedimento é difícil de ver. Duffy et al . demonstraram também que biotina methylthiosulfonate ativado (MTS-biotina) mais eficientemente reage com RNA 4sU-rotulado HPDP-biotina31. Por isso, valeria a pena pensando em mudar para MTS-biotina, em particular, para a recuperação de pequenos RNAs, que tendem a ter menos resíduos de uridina (referem-se ao protocolo biotinylation mencionado pela Sé de Duffy et al; purificação do RNA 4sU-rotulados, Procedimentos experimentais).
Para a recuperação do RNA recém transcrito, é possível usar grânulos paramagnéticos ou grânulos de limpeza de RNA de sua escolha. Sempre leve em conta que estes kits podem ou não podem purificar para RNAs específicos. Por exemplo, se você está interessado em miRNAs, considere o uso de jogos específicos para sequenciamento e captura de miRNA.
Quantificação do RNA recém transcrito: Para quantificar com precisão recentemente transcrito de RNA, medição deve ser realizada por um dados adequados (ver Tabela de materiais). Dentro de 1 h de exposição 4sU, recentemente transcrito de RNA representa cerca de 1-4% do RNA total. Recentemente transcrito de RNA de 1h rotulado, células T ativadas consiste em ~ 90-94% de rRNA11.
De perfil Ribossoma: Ao estabelecer o método, determinamos que usando 1,5 x a quantidade de nuclease do sugerido no protocolo original garante a boa digestão. Além disso, sem efeitos adversos têm sido relatados para quantidades elevadas de nuclease. Desde que é muito difícil para overdigest os RPFs enquanto eles são parte do RNA vinculado por proteínas ribossomais, ainda ligeiramente pode aumentar a quantidade para dosear a digestão de nuclease ideal.
Se inferior a 500 ng do RPF RNA foram recuperadas na etapa 4.4.2, repita a depleção de rRNA e piscina purificado RPFs com RNA Clean & concentrador-5 colunas. Em alternativa, carrega duas amostras idênticas ao lado uns aos outros sobre o gel (etapa 4.5.3) e fatias de gel de piscina durante a eluição de RNA do gel (etapa 4.5.6).
Recomendamos que os RPFs de corte em um gel tão firmemente quanto possível, para as bandas de nt 28 e 30. Isso ajuda na eliminação de fragmentos indesejados de rRNAs e tRNAs, que mais tarde tornar-se parte de sua biblioteca e reduzirá a leituras de sequenciamento para seus RPFs.
Também é aconselhável evitar a luz durante a purificação de gel UV. Isto pode criar nicks em fragmentos de RNA, os dímeros de pirimidina, que no final podem afetar severamente a biblioteca preparação e resultados de sequenciamento.
Preparação de biblioteca e sequenciamento de dados: Ribossoma protocolo de criação de perfil permite que para gerar uma biblioteca de cDNA apropriada para sequenciamento. Gerado pelo 4sU-rotulagem de amostras podem ser usadas diretamente para a preparação de biblioteca com qualquer kit de sequenciamento de RNA apropriado. Desde recentemente transcrito RNA, especialmente quando usando curto rotulagem vezes, pode ainda não estar polyadenylated, nenhuma seleção de poli-A deve ser realizada. Em vez disso, recomendamos a depleção de ARNr para evitar reduzir a profundidade de sequenciamento para a amostra real. Usando células-T, começamos com depleção de rRNA 400 ng de RNA total e recentemente transcrito (dependendo do kit, veja materiais), executada e reduzidos ciclos de amplificação por PCR minimizar o viés PCR. Preparação de biblioteca pode ser executada com menos matéria-prima. Conta para números de complexidade biblioteca de ciclos PCR deve ser otimizada.
Para ChIP-seq há também muitos kits disponíveis para preparação de biblioteca. Em nossa biblioteca de mãos preparação trabalhou bem, começando com 2 ng de DNA de ChIP (ver materiais para uma sugestão na qual kit de usar). Não se esqueça de verificar os índices para o equilíbrio de cor durante o sequenciamento. Recomendamos uma profundidade de sequenciamento de ≥ 40 x 106 lê cada para 4sU-seq, total RNA-seq e amostras de ChIP-seq e ≥ 80 x 106 leituras para Ribossoma amostras de criação de perfil. A profundidade de sequenciamento depende da amostra e a análise de Bioinformática a jusante e deve ser considerada cuidadosamente. Para analisar intrônicas leituras para a emenda cotranscriptional, 100 sequenciamento de bp emparelhado-final precisa ser escolhido.
Viés de sequenciamento: Sequenciamento tornou-se o padrão de ouro ao determinar mudanças globais na ligação do fator de transcrição, tradução ou transcrição. Em anos recentes, os métodos existentes foram empurrados para os seus limites ou novas técnicas foram desenvolvidas para cada vez mais pequenos montantes iniciais do RNA de sequenciamento. Isto requer amplificação do cDNA, que introduz ruído ou preconceito. Recentemente, identificadores exclusivos moleculares (UMIs) foram desenvolvidos para identificar experimentalmente duplicatas introduzidas pelo PCR. Recentemente, foi demonstrado que UMIs apenas ligeiramente melhorar o poder de sequenciamento e taxa de falsa descoberta de expressão diferencial do gene32. No entanto, considere usando identificadores exclusivos moleculares (UMIs) para todas as bibliotecas de sequenciamento de controle para a complexidade da biblioteca, especialmente quando começar com pequenas quantidades de RNA e quando são necessários muitos ciclos de PCR.
Buffer e soluções estoque: Todos os buffers para 4sU-seq e perfilamento Ribossoma devem ser preparados em condições rigorosas, RNase-livre usando água livre de nuclease. É recomendável comprar pre-feita livre de nuclease NaCl, Tris-HCl, EDTA, citrato de sódio e água. Para garantir condições de nuclease-livre, uma solução de RNase descontaminação pode ser usada para limpar superfícies ou pipetas. Todos os buffers para ChIP-seq precisam ser pelo menos livre de DNase e podem ser armazenados à temperatura ambiente. Sempre adicionar inibidores de protease e, opcionalmente, inibidores da fosfatase bem antes de usar e manter no gelo.
Bioinformática: Análise de todos os dados de sequenciamento (i.e., ChIP-seq, RNA-seq e criação de perfil ribsosome) envolve o controle de qualidade (por exemplo, usando FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), aparamento de adaptador (por exemplo, com cutadapt20) seguido de mapeamento para o genoma de referência para as células em estudo. Para dados de RNA-seq (total e seq-4sU), bem como dados de perfil de Ribossoma, um mapeador de RNA-seq emendado é necessário, tais como 2 ContextMap21. Para alinhamentos de dados unspliced ChIP-seq, usando BWA-MEM22 é suficiente. Expressão do gene pode ser calculada usando o modelo (leituras por quilobase do Exon por milhões de fragmentos mapeados) RPKM1, depois de determinar a ler contagens por gene usando um programa, por exemplo, featureCounts23. Para chamada de pico de dados ChIP-seq, uma série de programas está disponível, por exemplo, MACS24 ou GEM25. Mais análises a jusante podem ser executadas em R26, em particular, usando ferramentas fornecidas pelo projeto Bioconductor27.
Aqui, um grande desafio em integrar os níveis de RNA - 4sU e total e atividade translacional de perfilamento do ribossoma é a normalização. É uma abordagem clássica para resolver este problema para normalizar níveis de genes de manutenção da casa. Para reduzir o ruído devido a flutuações aleatórias de genes individuais de manutenção da casa, é recomendável usar não apenas alguns genes domésticas mas níveis medianos para um conjunto maior, por exemplo, a > 3.000 genes domésticas, compilados por Eisenberg e Levanon28 . Para o cálculo das taxas de rotatividade de RNA de rácios de 4sU-para que RNA total, normalização é baseado no volume de negócios médio de taxas (por exemplo, supondo que uma meia-vida do RNA de 5h)29. No entanto, já que isto supõe nenhuma alteração global para genes de manutenção da casa, recomendamos a utilização de abordagens de análise independente de normalização, por exemplo, de uma série de tempo dos tipos de dados diferentes para identificar grupos de genes de clustering baseado em correlação com comportamento distinto em transcrição e tradução durante a ativação. Para uma descrição detalhada sobre a integração de bioinformatical dos tipos de dados diferentes, consultar a publicação original11.
Análise de integração de dados e taxas de volume de negócios: Um artigo recentemente publicado33 comparando a meia-vida determinada por um controle de gene multiplexado (MGC) com métodos globais poderia mostrar que meias-vidas correlacionaram melhor com aqueles obtidos por métodos de rotulagem metabólicos em comparação com outros métodos (por exemplo, inibição geral da transcrição por drogas). No entanto, deve ser mencionado que as diferenças entre os cálculos de Half-Life podem surgir e tem sido descrito 15,34. Nós representam a maioria dos problemas e diferenças introduzidas pela resposta de estresse devido à exposição prolongada 4sU. Portanto, é indispensável para excluir a resposta ao stress introduzida por 4sU-rotulagem. Para validar ainda mais taxas de rotatividade, recomendamos o uso de MGCs.
Além disso, um conjunto de dados, conforme gerado aqui também poderia ser usado para um mais Integrativa dados análise (por exemplo, regulamento de tempo não-codificantes RNAs)35,36.
Divulgações
Os autores declaram que eles têm não tem interesses financeiro concorrente.
Agradecimentos
Agradecemos a Lars Dölken para o Conselho para estabelecer 4sU rotulagem para primárias células T; Elisabeth Graf e Thomas Schwarzmayr pela crítica ajuda nas gerações de biblioteca e sequenciamento; Dirk Eick e Andrew Flatley para fornecimento de anticorpos RNAPII e células T; S. Henriette Uhlenhaut e Franziska Greulich para ajudar na preparação da biblioteca para ChIP-seq; Caroline C. Friedel foi apoiado por subsídios FR2938/7-1 e CRC 1123 (Z2) da Deutsche Forschungsgemeinschaft (DFG); Elke Glasmacher foi apoiado pela subvenção GL 870/1-1 da Deutsche Forschungsgemeinschaft (DFG) e do centro alemão de pesquisa para Diabetes (DZD), Helmholtz Zentrum München.
Materiais
| Name | Company | Catalog Number | Comments |
| 4sU-labeling | |||
| 4-thiouridine (100 mg) | Carbosynth | 13957-31-8 | Prepare 50 mM stock in sterile H2O/PBS; store at –20°C in aliquots of 50-500 µl; do not refreeze. |
| 1.5 ml safe-lock tubes | Eppendorf | 30121589 | Optional |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72692005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72694005 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| Agencourt RNAClean XP | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| Chloroform | Sigma Aldirch | 372978 | WARNING – HAZARDOUS TO HEALTH |
| Dimethylformamide | Sigma Aldrich | D4551 | |
| Dithiothreitol (DTT) | Roth | 6908.1 | Prepare as 100 mM DTT in nuclease-free H2O; always prepare fresh |
| Ethanol | Merck | 1.00983.1000 | |
| EZ-Link Biotin-HPDP (50 mg) | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg Biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4°C in aliquots of 1 ml. DMF dissolves some plastic materials. We recommend to use glass pipettes to transfer DMF from ist stock glass bottle to 50 ml Falcon tubes. |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| Isopropanol | Merck | 1.09634.1011 | |
| NaCl (5M) | Sigma Aldrich | 71386 | Stock solution |
| nuclease-free EDTA (500 mM ), pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| Nuclease-free H2O | Sigma Aldrich | W4502 | Stock solution |
| nuclease-free Tris Cl (1M), pH 7.4 | Lonza | 51237 | Stock solution |
| Phase Lock Gel Heavy tubes (2.0 ml) | 5Prime | 2302830 | Use in step 1.3.4. |
| Polypropylene 15 ml centrifuge tubes | Greiner Bio-One | 188271 | Or equivalent; they have to tolerate up to 15,000 × g |
| QIAzol Lysis Reagent (200 ml) | Qiagen | 79306 | Use this or equivalent TRI reagent for RNA isolation, WARNING – CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol) |
| Qubit RNA HS assay kit | Life Technologies | Q32852 | Use this kit for quantifying RNA quantity in step 1.4.11 |
| RNeasy MinElute Kit | Qiagen | 74204 | Optional; includes Buffer RLT |
| Sodium citrat | Sigma Aldrich | C8532 | Prepare 1.6 M stock solution using nuclease-free water |
| Tween 20 | Sigma Aldrich | P1379 | |
| µMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store at 4°C, includes µMacs columns used in step 1.4.6. (store at RT) |
| Cell viability and stress assay | |||
| PE Annexin V Apoptosis Detection Kit I | BD Biosciences | 559763 | Optional |
| Thapsigargin | Sigma-Aldrich | T9033 | Optional |
| p53 | abcam | ab26 | Optional |
| p-EIF2a (Ser51) | Cell Signaling | 9721 | Optional |
| BH3I-1 | Sigma-Aldrich | B 8809 | Optional |
| Buffers | |||
| 4sU Washing Buffer | store at RT | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O | |
| Biotinylation Buffer (10x) | store at 4 °C | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water; make aliquots of 1 ml; store at 4°C | |
| RNA precipitation buffer | store at RT | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | |
| RNA 6000 Nano Kit | Agilent | 5067-1511 | Use this kit to verify RNA integrity in step 1.3.10 |
| RNA 6000 Pico Kit | Agilent | 5067-1513 | Optional |
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use in step 1.2.2/3.1.8/3.3.3/3.4.3 |
| High-speed centrifuge | Thermo Scientific | Heraeus Multifuge X3R | Or equivalent equipment capable of reaching 13,000 × g |
| High-speed rotor | Thermo Scientific | Fiberlite F15-6 x 100y | |
| Adaptors for 15 ml tubes | |||
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer C | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 µMacs columns. |
| Ultra-fine scale | Mettler Toledo | ML204T | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
| DynaMag-2 Magnet-1 each | Life Technologies | 12321D | |
| RNaseZap | Sigma | R2020 | Optional |
| TruSeq stranded total RNA library prep kit | Illumina | RS-122-2201 | Or equivalent. For T cells we used 400 ng 4sU and Total RNA with 11 cycles for PCR amplification. rRNA depletion is included in this kit |
| Nanodrop | Thermo Scientific | use a Nanodrop or equivalent instrument to measure RNA concentration | |
| Ribosome Profiling | |||
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPYSC12116 (Yeast) | |
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPHMR12126 (Mammalian) | |
| Illustra MicroSpin S-400 HR Columns | GE Healthcare | 27-5140-01 | |
| RNA Clean & Concentrator-25 kit | Zymo Research | R1017 | |
| RNA Clean & Concentrator-5 kit | Zymo Research | R1015 | |
| Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) | Illumina | MRZG126 or MRZG12324 | |
| (High Sensitivity DNA Kit) | Agilent Technologies | 5067-4626 | Already needed for 4sU-seq |
| All other consumables and equipment are listed in the User guide | !!! | Carefully read the user guide and order required consumables in advance (consider a long delivery time for some consumables e.g. gels) | |
| ChIP | |||
| 10 mM Tris-HCl (pH 8.0) | gereral lab supplier | ||
| 100 bp Plus Marker | Thermo Fisher | SM0323 | |
| 16 % Formaldehyde | Thermo Fisher | 28908 | Add to a final concentration of 1 % |
| 70% EtOH | gereral lab supplier | Always prepare fresh | |
| Agarose | gereral lab supplier | ||
| Agencourt RNAClean XP beads | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| ChIP library preparation kit | KapaBiosystems | KK8504 | Or use the kit of your choice |
| DNA low bind microcentrifuge tubes | Eppendorf | Z666548-250EA | or equivalent |
| Dynabeads Protein G | Invitrogen | 10004D | Use these superparamagnetic beads coupled to protein G in step 4.3.1.; Bring to RT before use |
| Glycine | gereral lab supplier | Prepare a 2M stock solution | |
| Glycogen | Roche | 10-901-393-001 | |
| MinElute PCR Purification Kit | Qiagen | 28004 | Use this kit (or equivalent) to purify chromatin in step 4.2.4. |
| Phosphatase Inhibitor (PhosStop) | Roche | 4906837001 | Add freshly to the buffer and keep on ice |
| Power SYBRgreen Master mix | Thermo Fisher | 4367659 | |
| Protease Inhibitor (cOmplete, EDTA-free) | Roche | 11873580001 | Add freshly to the buffer and keep on ice |
| Proteinase K | Invitrogen | 25530049 | |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32851 | Use this kit for quantifying DNA quantity in step 4.6.4. on a Qubit Fluorometer |
| Rnase, DNase free | Roche | 11-119915001 | |
| Salmon sperm (sonicated to around 100bp) | Sigma | D1626 | |
| TE pH 8.0 | gereral lab supplier | ||
| Antibodies (ChIP grade if possible) | |||
| anti-RNA Pol II [8WG16] | abcam | ab817 | |
| anti-Histon H3K36me3 | abcam | ab9050 | |
| or antibody of interest | |||
| Buffers | |||
| Binding/Blocking buffer | Store at RT | PBS with 0.5 % BSA and 0.5 % Tween 20 | |
| Cell-Lysis buffer | Store at RT | 5 mM Pipes [pH 8.0], 85 mM KCl, and 0.5 % NP40 | |
| ChIP IP buffer | Store at RT | 0.01 % SDS; 1. 1% Triton X-100;1.2 mM EDTA; 16.7 mM Tris-HCl, pH 8.1; 16.7 mM NaCl | |
| Elution buffer | Store at RT up to 6 months | 10 mM Tris-HCl (pH 8.0), 5 mM EDTA (pH 8.0), 300 mM NaCl and 0.5 % SDS | |
| Nuclei-Lysis buffer | Store at RT | 50 mM Tris [pH 8.0], 10 mM EDTA, and 1 % SDS | |
| Wash buffer I | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20mM Tris-HCL pH 8.1; 150 mM NaCl | |
| Wash buffer II | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20 mM Tris-HCL pH 8.1; 500 mM NaCl | |
| Wash buffer III | Store at RT | 0.25 M LiCl; 1% NP-40; 1 mM EDTA; 10 mM Tris-HCl, pH 8.1 | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | use this instrument for electrophoretical analysis |
| Nanodrop | Thermo Scientific | ||
| Bioruptor TBX microtubes 1.5 ml | Diagenode | C30010010 | |
| or tubes special for your sonication device | |||
| Bioruptor sonication device or sonication device of your choice | Sonication of T cells with Bioruptor: 20 - 25 cycles (30 s on, 30 s off at high in two 1.5 ml bioruptor microtubes with 500 µl each tube) | ||
| Magnetic stand for tubes | |||
| Thermomixer | |||
| Agarose gel electrophoresis | |||
| Qubit Fluorometer | Thermo Scientific | Use this Fluorometer for quantifying low amounts of RNA/DNA |
Referências
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 5 (7), 621-628 (2008).
- Chavez, S., Garcia-Martinez, J., Delgado-Ramos, L., Perez-Ortin, J. E. The importance of controlling mRNA turnover during cell proliferation. Curr Genet. 62 (4), 701-710 (2016).
- Rutkowski, A. J., et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun. 6, 7126 (2015).
- Ozsolak, F., Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 12 (2), 87-98 (2011).
- Carninci, P., et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 38 (6), 626-635 (2006).
- Churchman, L. S., Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 469 (7330), 368-373 (2011).
- Core, L. J., Waterfall, J. J., Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 322 (5909), 1845-1848 (2008).
- Hah, N., et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 145 (4), 622-634 (2011).
- Dolken, L., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14 (9), 1959-1972 (2008).
- Paulsen, M. T., et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67 (1), 45-54 (2014).
- Davari, K., et al. Rapid Genome-wide Recruitment of RNA Polymerase II Drives Transcription, Splicing, and Translation Events during T Cell Responses. Cell Rep. 19 (3), 643-654 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 22 (10), 2031-2042 (2012).
- Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 10 (10), 669-680 (2009).
- Blecher-Gonen, R., Barnett-Itzhaki, Z., Jaitin, D., Amann-Zalcenstein, D., Lara-Astiaso, D., Amit, I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc. 8 (3), 539-554 (2013).
- Schwanhausser, B., et al. Global quantification of mammalian gene expression control. Nature. 473 (7347), 337-342 (2011).
- Larsson, O., Tian, B., Sonenberg, N. Toward a genome-wide landscape of translational control. Cold Spring Harb Perspect Biol. 5 (1), a012302 (2013).
- Brar, G. A., Weissman, J. S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 16 (11), 651-664 (2015).
- Hafner, M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Radle, B., Rutkowski, A. J., Ruzsics, Z., Friedel, C. C., Koszinowski, U. H., Dolken, L. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. (78), (2013).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10-12 (2011).
- Bonfert, T., Kirner, E., Csaba, G., Zimmer, R., Friedel, C. C. ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics. 16, 122 (2015).
- Li, H., Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26 (5), 589-595 (2010).
- Liao, Y., Smyth, G. K., Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30 (7), 923-930 (2014).
- Zhang, Y., et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9 (9), R137 (2008).
- Guo, Y., Mahony, S., Gifford, D. K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 8 (8), e1002638 (2012).
- Team, R. D. C. . R: A Language and Environment for Statistical Computing. , (2016).
- Huber, W., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nature Methods. 12 (2), 115-121 (2015).
- Eisenberg, E., Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29 (10), 569-574 (2013).
- Friedel, C. C., Dolken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol Biosyst. 5 (11), 1271-1278 (2009).
- Burger, K., et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 10 (10), 1623-1630 (2013).
- Duffy, E. E., Rutenberg-Schoenberg, M., Stark, C. D., Kitchen, R. R., Gerstein, M. B., Simon, M. D. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 59 (5), 858-866 (2015).
- Parekh, S., Ziegenhain, C., Vieth, B., Enard, W., Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 6, 25533 (2016).
- Baudrimont, A., et al. Multiplexed gene control reveals rapid mRNA turnover. Sci Adv. 3 (7), e1700006 (2017).
- Rabani, M., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 29 (5), 436-442 (2011).
- Schlackow, M., Nojima, T., Gomes, T., Dhir, A., Carmo-Fonseca, M., Proudfoot, N. J. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol Cell. 65 (1), 25-38 (2017).
- Mukherjee, N., Calviello, L., Hirsekorn, A., de Pretis, S., Pelizzola, M., Ohler, U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. 24 (1), 86-96 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados