
Method Article
Identificar por-polifluorado química especies y con un flujo de trabajo combinado dirigida y no dirigida-detección de alta resolución espectrometría de masas
En este artículo
Resumen
Aquí, presentamos un protocolo para la cuantificación específica secuencial y análisis no dirigidos de compuestos fluorados en agua por espectrometría de masas. Esta metodología proporciona niveles cuantitativos de fluoroquímicos conocidos compuestos e identifica sustancias químicas desconocidas en muestras relacionadas con cálculos semicuantitativo de su abundancia.
Resumen
Históricos y emergentes por- y sustancias perfluorados (PFAS) han cosechado gran interés de las agencias públicas y de gobierno desde lo local a nivel federal. La continua evolución de la química de caídas presenta un desafío para el monitoreo ambiental, donde el desarrollo de métodos específicos queda necesariamente el descubrimiento de nuevos compuestos químicos. Por lo tanto, es necesario, que las metodologías que pueden detectar compuestos emergentes e inesperados, monitorear estas especies con el tiempo y resolver detalles de su estructura química para permitir futuras de trabajo en la salud humana. Con este fin, no se desea tratar análisis por espectrometría de masas de alta resolución ofrece un enfoque de detección de base amplia que puede combinarse con casi cualquier esquema de preparación de la muestra y proporciona capacidades significativas para la identificación de compuestos después de la detección. Adjunto, describimos un método de concentración muestra extracción en fase sólida (SPE) basado diseñado para la cadena más corta y más químicos hidrofílicos de caídas, como por los ácidos éter fluorado y sulfonatos y describir el análisis de muestras preparadas de esta manera en modos dirigidos y no dirigidos. Métodos específicos proporcionan cuantificación superior cuando los estándares de referencia están disponibles pero son intrínsecamente limitados a compuestos esperados al realizar el análisis. En cambio, un enfoque dirigido no puede identificar la presencia de compuestos inesperados y proporcionar alguna información sobre su estructura química. Información sobre características químicas puede utilizarse para correlacionar compuestos a través de ubicaciones de muestra y seguimiento de abundancia y ocurrencia en el tiempo.
Introducción
La clase de por- y sustancias perfluorados (PFAS) son contaminantes orgánicos persistentes con problema de salud pública significativo. El ácido de compuestos específicos perfluorooctanoico (PFOA) y perfluorooctanesulfonate (PFOS) tienen niveles Asesor de salud de agua potable por la EPA1,2 y su mayor producción en Estados Unidos cesó en la década de 20003,4 . Para tener una comprensión significativa de las propiedades de las propuestas de materiales en el producto textil y fabricación de esferas, cientos, si no miles, de la química de propuestas alternativa se han desarrollado para llenar nichos de productos, incluyendo reemplazos para los compuestos obsoleto5,6,7,8. Hay un curso necesita monitorizar los niveles ambientales de ácidos carboxílicos de cadena recta perfluorados y sulfonatos tal PFOS, PFOA y sus homólogos series relacionadas, pero compuestos químicos emergentes no están cubiertos por métodos establecidos como EPA Método 5379 y con frecuencia falta estándares analíticos para el análisis objetivo tradicional. La intención de este protocolo es pues doble. Proporciona una vía para el análisis de LC-MS/MS selectivo de especies de fluoroquímicos en agua donde los estándares analíticos están disponibles y detalles de la integración de un enfoque no dirigido, de alta resolución basados en espectrometría de masa, para análisis de datos permite la detección de compuestos desconocidos o inesperados en las mismas muestras.
Extracción en fase sólida (SPE) es una técnica establecida para la limpieza de la muestra y la concentración con las aplicaciones para muchos analitos y muestra matrices10,11. Para el análisis de caídas, varias fases sólidas de retentivas como no polar, funcionalizados polar y columnas de intercambio iónico se han utilizado en diferentes grados para las subclases de las especies fluoradas en una amplia variedad de matrices9,12, 13,14,15,16. Avances en análisis de muestras SPE mediante configuraciones en línea mucho aumentan el rendimiento del enfoque y mejoran la reproducibilidad de la manipulación de la muestra, pero el proceso fundamental permanece constante17. También se han realizado algunos esfuerzos para eliminar la concentración offline ECE usando inyecciones de gran volumen, pero estos requieren modificaciones a la cromatografía que los ponen fuera del ámbito de análisis casual18,19 . Nuestro análisis de la muestra utiliza una fase retentiva poliméricos anión débil intercambio (cera) bien separar materiales ácidos de caídas de los tradicionales contaminantes orgánicos logrando factores de concentración muestra substancial. Esta fase de la cera es importante captar los ácidos de cadena corta perfluorados como sulfonato de perfluorobutane (PFBS) o éteres de polieter tales como ácido de dímero de oxido de hexafluoropropileno (HFPO-DA) que son más polares que los perfluorados legado de cadena más especies20,21. Como ha habido un cambio significativo hacia la inclusión de éter y cadenas más cortas de fluorados en recientes caídas química5, esta selección de fase permite la recuperación más completa de compuestos novedosos para el análisis de MS.
Cuantificación específica de LC-MS/MS usando había autenticado estándares y etiquetado como estándares internos de isótopos estables proporciona un incomparable nivel de especificidad y sensibilidad para el análisis cuantitativo. Aunque este método es deseable en muchas situaciones, no es práctico para situaciones muy común en el análisis. Enfoques específicos trabajan solamente para las especies que se espera que en la muestra, y para que los métodos se han establecido previamente. Para los compuestos nuevos y emergentes, este enfoque no es capaz de detectar incluso especies que pueden ser de interés, independientemente de su química o concentración, y espectrómetros de masas de baja resolución son casi incapaces de proporcionar suficiente información para hacer tareas de química inequívocas de compuestos desconocidos. En consecuencia, ha surgido el campo de análisis orientada a no, aprovechar el potencial de los espectrómetros de masas moderna alta resolución para analizar muestras sin una hipótesis presupuesta y retroactivo asignar productos químicos características detectables en la muestra. Este enfoque se ha utilizado extensivamente en los campos de la biología22,23,24 y ciencias ambientales25,26,27 en numerosas clases de productos químicos. Productos químicos perfluorados son particularmente fácil de identificar en este método debido a sus patrones de masa espectrales únicas y cientos de compuestos se han descrito en apenas los últimos pocos años5,28.
El protocolo aquí pretende alinear cuantificación de LC-MS/MS propuestas específica con la necesidad de identificar y controlar semi-cuantitativamente compuestos emergentes de interés. La fase SPE selección y técnicas de preparación de la muestra se pretenden asegurar la captura de más hidrofílicos ácidos emergentes de caídas de agua y puede ser menos adecuado para más especies poliméricas de cadena y no iónicos. Además, los datos generados por el análisis objetivo no están densos y de alta dimensionalidad, que hace necesario el uso de software de análisis de datos. Tales paquetes de software son con frecuencia proveedor específico y requieren modificación para operar entre las plataformas de instrumento. Donde sea posible, el proceso de análisis se ha descrito en forma genérica y alternativas de fuente abierta/freeware se hace referencia, pero la eficiencia y la exactitud de cualquier enfoque de software deben ser evaluados de forma individual.
Protocolo
1. colección de muestras de agua
- Preparación de propuestas de acciones estándar
- Preparar una mezcla estándar de PFAS en metanol que contiene compuestos específicos de interés (por ejemplo, PFOA, PFOS, HFPO-DA) 1 ng/μl. Esto es la mezcla de propuestas indígenas. También existen mezclas preparadas comercialmente (es decir, experimentada mezcla A y B de la mezcla).
- Preparar una mezcla estándar que contiene isótopos estables emparejado marcado compuestos (SIL) de caídas (p. ej., 13C4- PFOA, 13C8- PFOS, 13C3- HFPO-DA) en 1 ng/μl. Se trata de la mezcla es de caídas. También existen mezclas preparadas comercialmente (es decir, mezcla de MPAFC A y B de la mezcla).
Nota: Si está disponible una versión SIL de las propuestas específicas, un sustituto con similar estructura y cadena de longitud puede utilizarse (por ejemplo, 13C2- PFHxA para HFPO-DA)
- Preparación de campo en blanco (FB), muestras de punto en blanco (SB)
- Llenar dos, polipropileno limpio de alta densidad (HDPE) o polipropileno (PP) frascos con 1.000 mL de laboratorio desionizada (DI), conocido por ser libre de caídas.
PRECAUCIÓN: Los materiales caídas con frecuencia han undefined toxicidad y carcinogenicidad. Debe tenerse cuidado para evitar la exposición oral o de la piel a normas o soluciones. - Añadir una cantidad de mezcla estándar caídas a una de las botellas en una concentración final equivalente a las concentraciones esperadas de la muestra (por ejemplo, 100 ng/L). Este es el punto en blanco (SB).
- Añadir 5 mL de ácido nítrico al 35% conservante a la espiga en blanco.
- Llevar muestra de SB y el campo antes en blanco a la ubicación del muestreo como controles.
- Llenar dos, polipropileno limpio de alta densidad (HDPE) o polipropileno (PP) frascos con 1.000 mL de laboratorio desionizada (DI), conocido por ser libre de caídas.
- Muestreo de campo
Nota: Colector de muestra debe usar guantes de nitrilo y muestra de sistemas de flujos siempre que sea posible. Toma de muestras se debe fluir y alcancen antes de toma de muestras (2-3 min).- Recoger 500-1.000 mL de agua de la ubicación del campo en una botella limpia de HDPE o PP.
- Añadir 5 mL de ácido nítrico al 35% conservante a botellas de la muestra y el campo en blanco.
PRECAUCIÓN: el ácido nítrico es corrosivo y un oxidante fuerte
2. extracción de muestra de
Nota: Caídas son ubicuo y persistente. Asegúrese de que todos los disolventes son de la más alta calidad y han sido analizados bajo nivel contaminación de caídas. Enjuague a fondo todos los equipos de laboratorio utilizados para la preparación de las normas antes de preparar muestras y espacios en blanco.
- Pretratamiento de la muestra
- Vierta cada muestra en un cilindro de polietileno de alta densidad graduada 1 L separado, previamente limpio y registro el volumen exacto.
- Añadir 10 mL de metanol al frasco vacio muestra, tápalo y agitar bien para aclarar propuestas fijado por adsorción desde el interior de la botella.
- Devolver la muestra de agua medido a la botella de lavado con el enjuague metanólico.
- Curva estándar para cuantificación
- Llenar ocho, 1 botellas HDPE/PP L con agua desionizada libre de caídas.
- Seleccione ocho concentraciones separadas uniformemente cubriendo el rango de cuantificación deseada. Por ejemplo: 10, 25, 50, 100, 250, 500, 750 y 1.000 ng/L para una gama de 10-1.000 ng/L.
- Añadir una cantidad de mezcla nativa caídas a cada botella para obtener las concentraciones finales de PFAS en 2.2.2 (por ejemplo, 100 μl caídas mezcla A 1L de agua desionizada = 100 ng/L).
- Adición de estándar interno
Nota: Además del isótopo estable que etiqueta estándar interno (IS) es necesario sólo si se desean que los resultados cuantitativos además de análisis orientada a no.- Agregue la mezcla de propuestas es a cada muestra a una concentración aproximadamente la mitad de la curva de calibración (por ejemplo, 250 μl de mezcla de caídas es = 250 ng/L)
- Filtración
- Filtrar las muestras a través de filtros de fibra de vidrio GF/A (47 mm, 1,6 μm tamaño de poro) bajo vacío suave en un matraz de vacío previamente limpiado 1 L polietileno de alta densidad.
- Si partículas permanece en la botella, enjuague con agua desionizado en el filtro. Vertiendo el agua filtrado en la botella de muestra o un nuevo contenedor para extracción en fase sólida.
- Extracción en fase sólida (SPE)
Nota: Concentración de cartucho descrito aquí utiliza una bomba de pistón de flujo constante. Métodos alternativos de concentración usando un colector de vacío20 o usando una configuración on-line de17 SPE-LC-MS son posibles pero no discutido.- Condición de un cartucho de intercambio (cera) de aniones débilmente con 25 mL de metanol.
- Estado del cartucho de cera con un adicional 25 mL de agua desionizada.
- Posición bomba drenaje tubo en botellas de la muestra filtrada y cartuchos SPE con nombres correspondientes de la muestra de la etiqueta.
- 500 mL de muestra de agua a través del cartucho con un caudal constante de 10 mL/min (total 500 mL), desechando a través de flujo de líquido a la bomba.
Nota: Volúmenes más grandes o más pequeñas pueden ser concentrados dependiendo de las concentraciones de la muestra prevista. - Extraiga el cartucho de la bomba de pistón para la elución.
Nota: Si la concentración de muestras adicionales utilizando la misma bomba, la bomba de pistón debe aclararse con 25 mL de metanol antes de instalar el cartucho siguiente para conseguir el equilibrio. - Transferencia de cartucho SPE a un colector de vacío y equipar con depósito externo de cristal.
- Limpie el cartucho SPE con 4 mL de 25 mM, tampón de acetato de sodio 4.0 pH bajo vacío suave. Deseche el flujo a través. Cartucho de SPE lavar con 4 mL de metanol neutro.
Nota: Neutro Lave fracción puede ser recopilada si se espera que los analitos polares no iónicos específicos. De lo contrario, descarte de desechos - Coloque un tubo de centrífuga de polipropileno de 15 mL por debajo de cada cartucho SPE a recoger el eluyente. Eluir la muestra con 4 mL de hidróxido de amonio 0.1% en metanol.
- Retire el tubo de elución y reducir el volumen de eluido a 500-1.000 μl por evaporación bajo corriente de nitrógeno seco en un baño de agua a temperatura ligeramente elevada (40 ° C).
- Concentrado muestra extractos pueden almacenar antes del análisis a temperatura ambiente.
- Objetivo cuantificación LC-MS/MS
- Diluir 100 μl de muestra se extrae con 300 μL de tampón de acetato de amonio de 2 mM en un frasco de muestra HPLC.
- Calibrar y equilibrar un sistema HPLC y MS según instrucciones del fabricante.
Nota: Comúnmente se detectan antecedentes caídas debido al uso de componentes de fluoropolímero de la mayoría de sistemas de LC y en la septa del vial de muestra. Confirme los niveles detectables en los espacios en blanco insignificantes antes de su uso. Modificación del sistema LC para reemplazar componentes de Teflon se sugiere siempre que sea posible. El uso de una columna analítica de "hold-up" junto a la válvula mezcladora de LC es también sugerido29. - Preparar un worklist analítica de la curva estándar, las muestras y una repetición adicional de la curva estándar para evaluar la deriva instrumental a través de la gestión. Una lista de tareas de ejemplo se muestra en la tabla 1.
- Analizar las muestras mediante métodos LC y MS para establecidos específico de interés. El gradiente de LC de ejemplo se muestra en la tabla 2 y MS parámetros del método se muestran en la tabla 3 y tabla 4. Más detallada puede encontrarse en McCord et al.21.
- Generar una curva estándar de las muestras estándar utilizando la proporción del área pico del analito a la norma interna frente a la concentración del analito. Generar una fórmula de regresión cuadrática con 1 / x ponderación para concentración predicción9.
- Cuantificar los analitos específicos en cada muestra utilizando las curvas estándar preparadas y proporción del área (área estándar es área) para cada medición.
- Si la concentración supera el intervalo de calibración, diluir la muestra original con agua DI añadió la concentración es adecuada y volver a extraer para la concentración en el rango adecuado.
- Recopilación de datos de LC-MS/MS no dirigidos
- Diluir 100 μl de muestra se extrae con 300 μL de tampón de acetato de amonio de 2 mM en un frasco de muestra HPLC.
- Calibrar y equilibre un HPLC y MS alta resolución según las instrucciones del fabricante.
- Preparar una lista de tareas analítica como en 2.6.2.
- Usando el software del instrumento, recoger datos de LC-MS con una amplia exploración MS1 en modo dependiente de datos para recoger gradiente MS/MS. ejemplo LC en la tabla 5. Discusión adicional de opciones puede encontrarse en Strynar et al.30 y31de Newton et al.
Nota: Para mejor MS/MS análisis de datos dependiente de calidad pueden realizarse con una lista de ion preferido de un subconjunto de características restante después de procesar los datos en 2.8.1-2.8.8.
- Tratamiento de datos orientada a no
Nota: Se pueden realizar análisis de datos con una amplia gama de software y estos métodos no reflejan el método único o mejor para un conjunto de datos arbitrario. Siempre que sea posible, los pasos proporcionan una descripción genérica que se puede hacer en software alternativo. Procesamiento de los datos del ejemplo utilizados en este manuscrito se llevó a cabo utilizando el software específico del vendedor (Software 1 y 2 de Software) como se indica en Newton et al.31.- Realizar extracción molecular característica de características químicas usando uno de varios software paquetes32,33 o proveedor de software de código abierto para identificar masas monoisotopic, tiempos de retención y áreas de pico integrado de química Funciones.
- En 1 de Software, seleccione Agregar o quitar archivos de muestra > Agregar archivos y seleccione los datos en bruto en el experimento no dirigido, entonces golpear OK.
- En Software 1 Seleccione Batch recursivo característica extracción > método abierto... un método preestablecido, o manualmente editar la configuración de software. Profinder ajustes para la extracción de la característica se encuentran en la tabla 6.
- En el Software 1, después de la extracción de la característica, seleccione archivo > Exportar como CSV..., archivo > Exportar como CEF..., o archivo > Exportar como PFA... para su posterior procesamiento. Archivos CEF se asumen para el resto de la descripción.
- En Software 2 (MPP) crear un nuevo experimento con tipo no identificado y el tipo de flujo de trabajo Asistente de importación de datos y haga clic en Aceptar.
- En MPP Seleccione los archivos de datos y localizar los resultados exportados del Software 1 (CEF o PFA) para importar; luego haga clic en siguiente hasta que aparezcan opciones de Parámetros de alineación .
- En MPP, establezca los valores de alineación compuesto a 0.0 (alineación ya fue realizada en la extracción de la característica de Software 1, paso 2.8.1.2) luego haga clic en siguiente a través de los pasos hasta que termine .
- Filtro de identificación basados en análisis reproducibilidad. Donde hay múltiples muestras replicadas, las características deben estar presentes en > 80% del individuo replica y tiene un coeficiente de análisis de variación (CV) de < 30%
- En Seleccione MPP disposición Experimental > experimento agrupación y asignar a cada archivo raw un grupo correspondiente a su muestra de origen (es decir, deben ser repeticiones de la misma fuente en el mismo grupo). Pueden crear varios grupos correspondientes a variables anidadas (p. ej., instrumental vs repeticiones técnicas).
- En Seleccione MPP disposición Experimental > interpretación crear Seleccione el parámetro de experimento (es decir, de grupo) y haga clic en siguiente hasta que termine . Que se creará una categoría futuras filtrado puede operar en.
- En Seleccione MPP Control de calidad > filtro de frecuencia. Establecer entidades en Todas las entidades y la interpretación a la muestra Group(non-averaged) había creado en 2.8.2.2, luego del golpe siguiente.
- Para los parámetros de entrada, sistema de retención de la entidad en el 80% de la muestra en al menos una condición y haga clic en siguiente hasta que termine . Nombre de la lista de características de filtrado de frecuencia
- En Seleccione MPP Control de calidad > filtro sobre la muestra variabilidad. Encuentra la lista de personas a las funciones de filtrado de frecuencia de 2.8.2.4 y la interpretación a Group(non-averaged), luego del golpe siguiente.
- Seleccione el botón de radio para los Datos crudos y el rango de interés de coeficiente de variación < 30%. Haga clic en siguiente > acabado y guardar la lista como características de filtrado de CV.
- Quitar características donde no muestras tienen significativamente más alta (> 3 veces) la abundancia de la muestra en blanco (campo de FB).
- En Seleccione MPP análisis > cambio doble. Lista entidad a las Características de filtrado de CV y la interpretación de la muestra grupo luego del golpe a siguiente. Seleccione la opción de cambiar de doblez y contra única condición condición seleccionar FB o lo que era el nombre del grupo de la muestra procesada en blanco.
- En la pantalla siguiente, establece el corte doblez-cambio en 3.0 y haga clic al final de las instrucciones. Guarde la lista como Lista de filtrado de FC.
- Realizar comparaciones binarias de muestras individuales de interés contra una muestra de fondo apropiado (por ejemplo, aguas arriba y aguas abajo de un punto de origen) para determinar cambios de doblez de determinadas características químicas.
- En Seleccione MPP análisis > filtro en volcán parcela. Establecer la lista de personas en Lista de filtrado de FC y la interpretación al grupo.
- Para la condición de doble cambio par elegir dos muestras de comparación (por ejemplo, una sincronizado aguas arriba y aguas abajo muestra) y selecciona prueba Mann-Whitney no apareados.
- Para el análisis preliminar, no seleccione un valor para la corrección de pruebas múltiples en la pantalla siguiente, haga clic en a la parcela de resultado.
- En la pantalla de resultados Seleccione un atajo de doble cambio de 3.0 y un corte del valor de p en 0,1. Acabado y exportar la lista de Resultados Prelim.
- Para cada característica restante después de filtrar, generar representación química predicha de la masa exacta y espectro de masa compuesto.
- En el MPP, seleccione interpretación de resultados > IDBrowser identificación y la lista de personas Resultados Prelim .
- En el IDBrowser seleccionar identificar todos los compuestos con fórmula molecular generador (MFG) como método de identificación.
- En la fórmula de generar opciones Añadir F a la columna de elementos y definir el máximo a 50, a continuación, seleccione terminar. Tras generación fórmula seleccione Guardar y volver a regresar al MPP.
- En MPP, haga clic derecho en el filtrado y MFG emparejado de entidades y seleccione Exportar lista. Guardar los resultados.
- Examinar la masa monoisotopic de especies en la lista reducida característica química importante para las personas con defectos de masa de fluoración; ver tipo y Fiehn34.
- Nota la serie de química que contienen motivos de polyfluorination comunes (CF2 (m/z 49.9968), CF2O (m/z 65.9917) CH2CF2O (m/z 80.0074), etc.) mediante un algoritmo de software o parcela de defecto de masa; ver sección discusión, Liu et al.17, Loos et al35 y Dimzon et al36.
- Buscar fórmulas químicas previstas o neutro masas contra la base de datos de EPA química Dashboard u otras bases de datos para devolver estructuras químicas posibles.
- Abra la herramienta de búsqueda de lotes de EPA Comptox productos químicos Dashboard (https://comptox.epa.gov/dashboard/dsstoxdb/batch_search) y pegar la lista de identificadores (fórmulas o masas) en el cuadro identificador, después de seleccionar el tipo de identificador (es decir, MS-listo Fórmula o masa de Monoisotopic).
- Seleccione Descargar datos químicos... y también seleccionar los datos de física/química/toxicología para posibles partidos de la lista desplegable.
- Usando la intuición química y datos de referencia disponibles, retire poco probable acerca de los partidos de la potencial lista de estructura química de cada formulación a base de viabilidad debido a la estabilidad química, características físicas tales como ionizability o hidrofobicidad, la presencia de la fabricación de productos químicos de fuentes cercanas, etcetera. En ausencia de datos adicionales, viabilidad espectral puede ser alineado puramente sobre la base de la prevalencia de la literatura; Ver McEachran et al37.
- Confirmar estructuras utilizando estándares disponibles o MS/MS de alta resolución dirigida de fragmentos contra espectros de bases de datos, en silico los espectros teóricos o curación manual.
- Realizar extracción molecular característica de características químicas usando uno de varios software paquetes32,33 o proveedor de software de código abierto para identificar masas monoisotopic, tiempos de retención y áreas de pico integrado de química Funciones.
Resultados
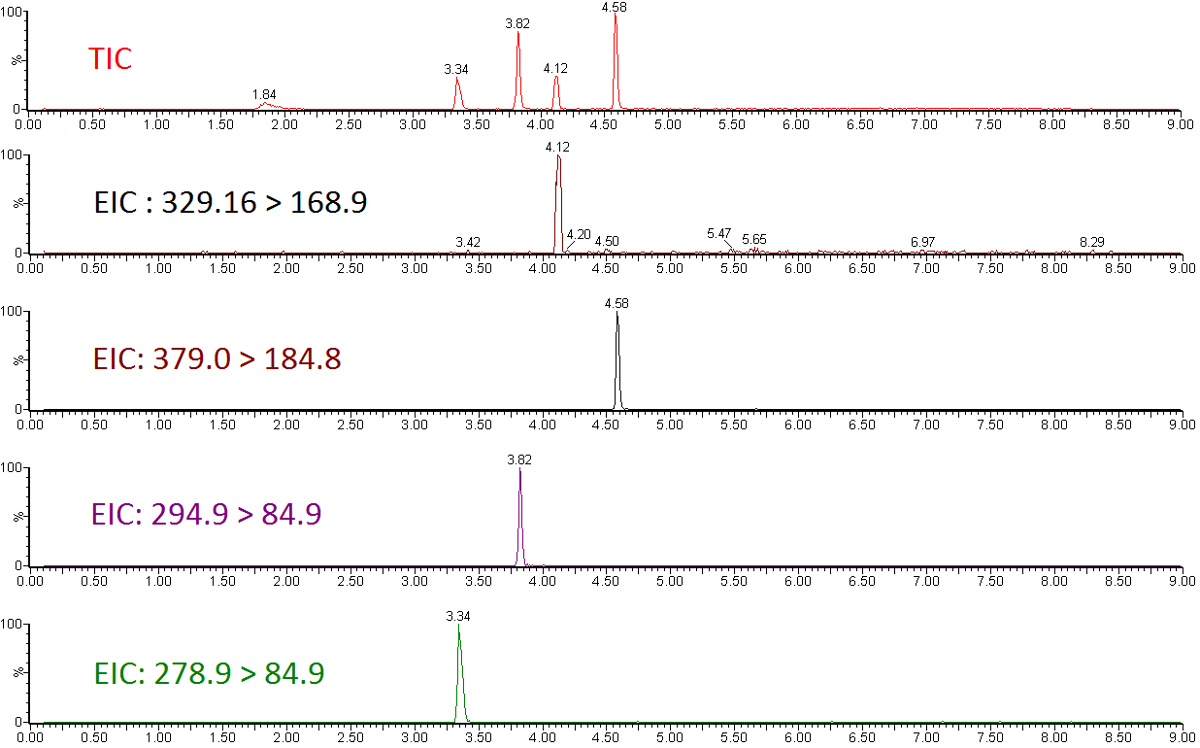
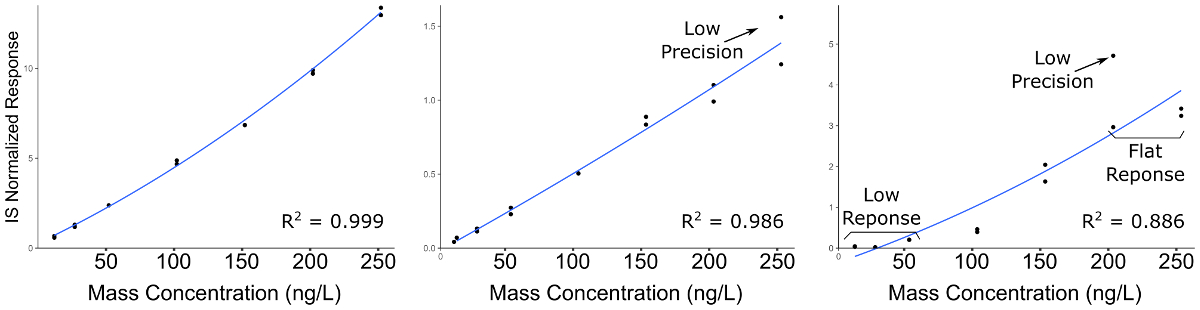
Resultados cuantitativos de LC-MS/MS se encuentran en forma de ion-cromatogramas para el cromatograma de iones totales (TIC) y los cromatogramas de iones extraídos (EIC) de transiciones específicas química productos químicos medidos (figura 1). El área de pico integrado de una transición química se relaciona con la abundancia compuesta y puede ser utilizado para calcular la concentración exacta utilizando una curva de calibración normalizada a un estándar interno (figura 2). Respuesta baja o plana de analitos individuales indica que el intervalo de calibración está fuera del rango lineal del espectrómetro de masas, o que el instrumento requiere de ajuste y calibración. Mala precisión de repeticiones indica un problema con la inyección de la muestra o cromatografía inconsistente que requiere modificación de los parámetros de la LC.
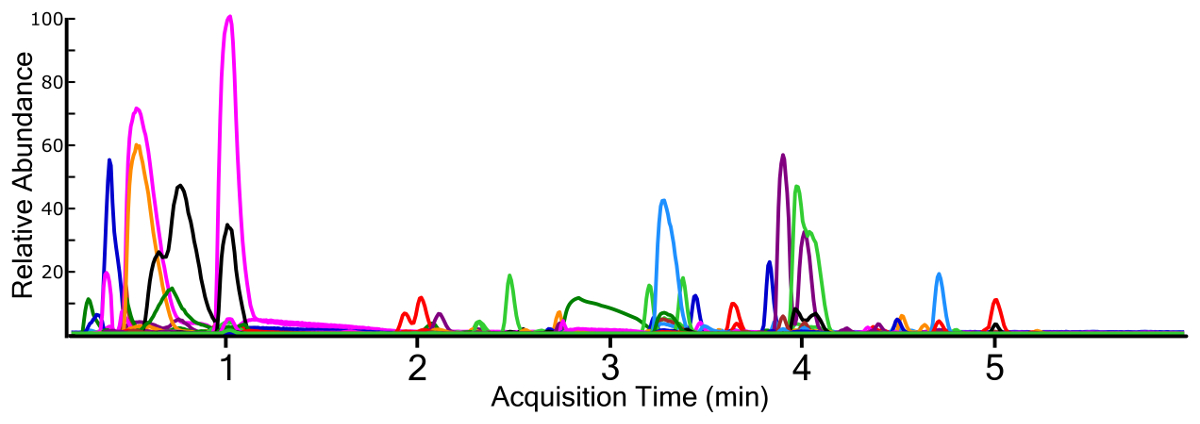
Análisis no-dirigido usando un análisis completo de MS1 produce un TIC para muestras (figura 3), que permite la generación ad hoc de EIC para los iones individuales (figura 4). Cualquier punto del tiempo cromatográfico dado contiene señales de especies químicas y cuando se utiliza un espectrómetro de masas alta resolución, la huella isotópica del compuesto. Identificación de compuestos a partir de la exploración de MS1 se realiza mediante programación de un algoritmo de peak-picking utilizando uno de varios enfoques38,39,40. Picking de picos rinde características químicas con una masa exacta medida y tiempo de retención cromatográficos, así como el espectro de masas de los iones y el área del pico cromatográfico. Esta información se almacena normalmente en un formato de base de datos digital para posterior procesamiento y filtrado, pero la naturaleza anidada e interconectada de los datos puede entenderse conceptualmente (figura 5).
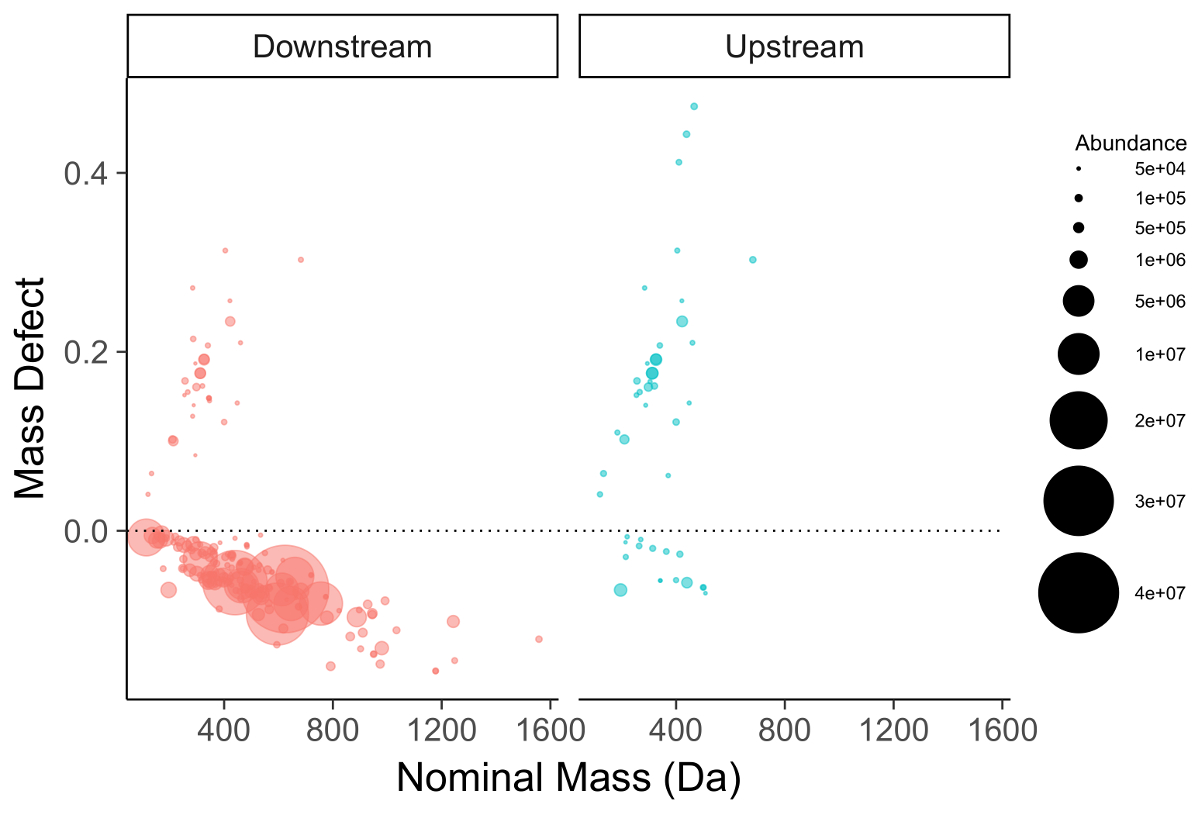
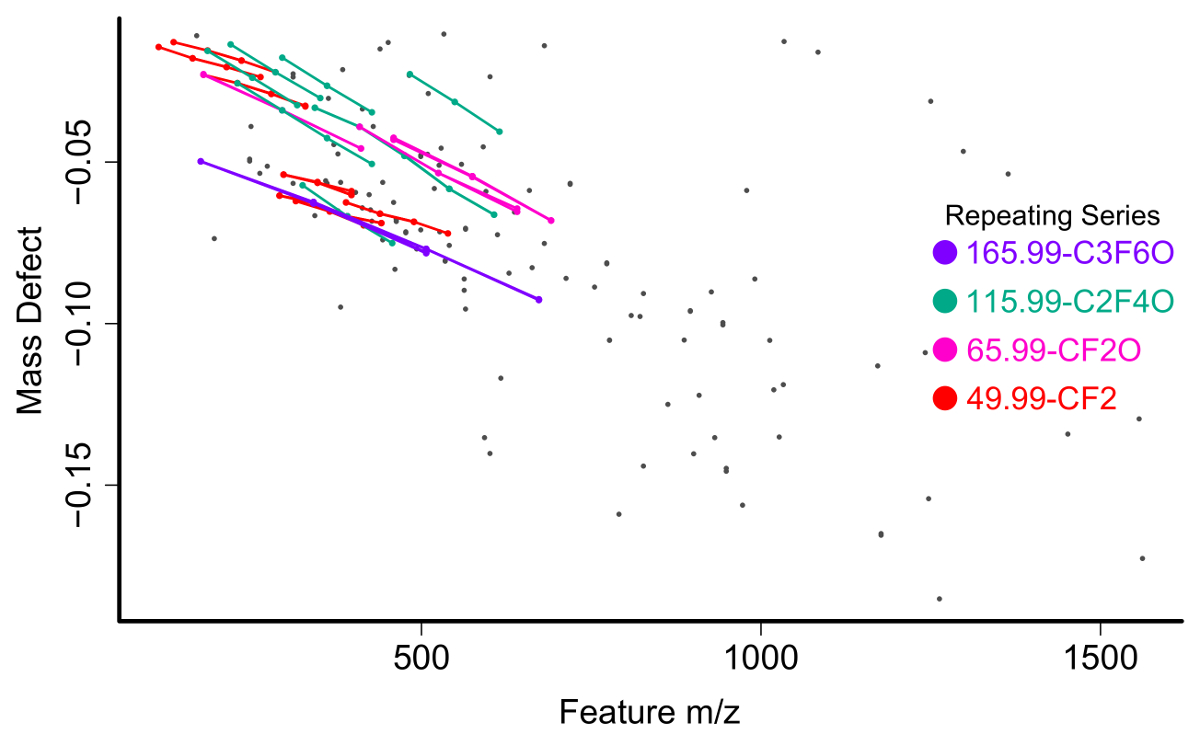
Se filtra la lista de funciones para la reunión de varios criterios para ser seleccionado para la posterior investigación de compuestos. La primera y más sencilla es filtrado por defecto de masa (la diferencia entre la masa exacta de una función y su masa nominal). PFAS compuestos tienen defectos de masa negativo (figura 6) debido a su preponderancia de átomos de flúor y compuestos polifluorado defectos de masa positivas, pero substancialmente más pequeños que materiales orgánicos homólogo31,34 . Un segundo método de filtrado paso es identificar serie homóloga que contiene unidades que se repiten comunes a especies de caídas, como CF2 o CF2O. identificación de éstos se puede hacer con masa de Kendrick defecto parcelas17,36, o paquetes de software como R nontarget paquete35 (figura 7).
Después de filtrado, asignación de identidad química de la lista de observados altamente diferenciado o tentativamente por / polifluorado especies pueden comenzar. Masa exacta proporciona una lista relativamente pequeña de fórmulas químicas posibles para emparejar pero es insuficiente para la identificación sin la adición de coincidencia espectral al patrón de isótopos del espectro de masas41. De alta resolución MS1 datos, uno o más supuestos fórmulas químicas se compara con la huella isotópica del espectro de masas y anotó (figura 8). Fórmulas para emparejar puede ser generadas usando ab initio un grupo definido de átomos o puede ser origen de una combinación de literatura informó compuestos y el contenido de una o más bases de datos. Los anfitriones nos EPA química Dashboard (https://comptox.epa.gov/dashboard/) una lista constantemente actualizada de PFAS compuestos identificados por la agencia, así como las listas compilada por otras organizaciones como la de NORMAN Network42.
Fórmulas químicas se pueden confirmar aún más, y cierta información estructural se puede garnered de espectros MS/MS (figura 9). Estructuras del candidato son de química bases de datos grandes tales como el tablero de instrumentos de química EPA, Pubchem, el registro de CAS, etcetera. Espectros previstos pueden ser generados o adquirieron usando una variedad de programas de fragmentación y asignado,43 o espectros MS/MS pueden interpretarse manualmente.
Una matriz de datos de ejemplo está disponible en la información complementaria que contiene una matriz de toda característica de diez muestras (5 arriba, 5 abajo) recopilada aguas arriba y aguas abajo de una fuente puntual de fluoroquímicos. Cada fila representa una característica química con tiempo de retención asociada, masa neutra, espectro de masa y abundancia de materia prima para cada muestra. (Tabla suplementaria, hoja 1). Inicial de filtrado (Tabla suplementaria, hoja 2) para el defecto de masa negativa y significación estadística en un t-test no pareado entre aguas arriba y aguas abajo reducen el número de características "interesantes" química ~ 120. Fórmulas químicas previstas fueron obtenidos de Agilent IDBrowser y búsquedas contra el EPA Comptox productos químicos tablero de mandos, que volvió posible coincide con (Tabla suplementaria, hoja 3). El "top-hit" para cada fórmula química basada en fuentes de datos37 fue asignado (Tabla suplementaria, hoja 4). Tenga en cuenta que más de la mitad de las restantes características no tienen partidos de alta calidad. Características identificadas con ninguna coincidencia pueden ser el resultado de la formación en fuente fragmentación/aducción, pobre asignación fórmula, o la identificación de PFAS no se encuentra en la base de datos de origen. Interpretación de los espectros crudos con el fin de validar las tareas está fuera del alcance de este manuscrito, pero más información se puede encontrar en las obras citadas15,30,31,44, 45.
| ID | Nombre de la muestra | Tipo de muestra | STD Conc | Frasco | Método de LC | Método de MS |
| 1 | DB_001 | En blanco | 1:, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 2 | DB_002 | En blanco | 1:, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 3 | DB_003 | En blanco | 1:, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 4 | DB_004 | En blanco | 1:, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 5 | DB_005 | En blanco | 1:, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 6 | FB | En blanco | 1:, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 7 | 10 enfermedades de transmisión sexual | Estándar | 10 | 1:, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 8 | 25 enfermedades de transmisión sexual | Estándar | 25 | 1:, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 9 | 50 std | Estándar | 50 | 1:, 5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 10 | 100 std | Estándar | 100 | 1:, 6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 11 | 250 std | Estándar | 250 | 1:, 7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 12 | 500 std | Estándar | 500 | 1:, 8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 13 | 750 std | Estándar | 750 | 1:B, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 14 | 1000 std | Estándar | 1000 | 1:B, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 15 | DB_006 | En blanco | 1:B, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 16 | SB_DUP1 | Analito | 1:B, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 17 | SB_DUP2 | Analito | 1:B, 5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 18 | Sitio SW 03 | Analito | 1:B, 6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 19 | Sitio SW 16 | Analito | 1:B, 7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 20 | Sitio SW 30 | Analito | 1:B, 8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 21 | DB_007 | Analito | 1: C, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 22 | Sitio SW 19 | Analito | 1, 2: C | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 23 | Sitio SW 48 | Analito | 1: C, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 24 | SW sitio 49 | Analito | 1: C, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 25 | Sitio SW 05 | Analito | 1, 5: C | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 26 | Sitio SW 47 | En blanco | 1: C, 6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 27 | DB_008 | Analito | 1: C, 7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 28 | 19_DUP sitio de SW | Analito | 1: C, 8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 29 | Sitio SW 20 | Analito | 1, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 30 | Sitio SW 21 | Analito | 1, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 31 | SW sitio 46 | Analito | 1, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 32 | Sitio SW 47 | Analito | 1, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 33 | DB_009 | En blanco | 1, 5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 28 | Sitio SW 32 | Analito | 1, 6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 29 | Sitio SW 50 | Analito | 1, 7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 30 | Sitio SW 25 | Analito | 1, 8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 31 | 21_DUP sitio de SW | Analito | 1:E, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 32 | Sitio SW 52 | Analito | 1:E, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 33 | DB_010 | En blanco | 1:E, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 34 | FB | En blanco | 1:, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 35 | 10 enfermedades de transmisión sexual | Estándar | 10 | 1:, 3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 36 | 25 enfermedades de transmisión sexual | Estándar | 25 | 1:, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 37 | 50 std | Estándar | 50 | 1:, 5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 38 | 100 std | Estándar | 100 | 1:, 6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 39 | 250 std | Estándar | 250 | 1:, 7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 40 | 500 std | Estándar | 500 | 1:, 8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 41 | 750 std | Estándar | 750 | 1:B, 1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 42 | 1000 std | Estándar | 1000 | 1:B, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 43 | DB_011 | En blanco | 1:B, 2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 44 | DB_012 | En blanco | 1:E, 4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
Tabla 1: Ejemplo worklist para el análisis objetivo y cuantificación de las propuestas utilizando LC-MS/MS de
| hora (min) 0 | % A (acetato de amonio 2,5 mM en MeOH 5%) 90 | % B (acetato de amonio 2,5 mM en el 95% MeOH) 10 |
| 5 | 15 | 85 |
| 5.1 | 0 | 100 |
| 7 | 0 | 100 |
| 7.1 | 90 | 10 |
| 9 | 90 | 10 |
Tabla 2: Gradiente de ejemplo para la separación de la LC en análisis específicos
| Capilary voltaje (kv) | 1.97 |
| Cono de voltaje (V) | 15 |
| Extractor de voltaje (V) | 3 |
| Lente de RF (V) | 0.3 |
| Temp de la fuente | 150 |
| Desolvation Temp | 40 |
| Desolvation Gas caudal (L/hr) | 300 |
| Flujo de Gas del cono (L/hr) | 2 |
Tabla 3: Parámetros de fuente de ionización para el análisis objetivo
| CMP | Precursor | Producto | Tiempo de permanencia | Cono de voltaje (V) | Energía de la colisión (eV) |
| PFBA | 212.80 | 168.75 | 0.01 | 15 | 10 |
| C 13 4-PFBA ES | 216.80 | 171.75 | 0.01 | 15 | 10 |
| PFPeA | 262.85 | 218.75 | 0.01 | 15 | 9 |
| PFBS ° 1 | 298.70 | 79.90 | 0.01 | 40 | 30 |
| PFBS ° 2 | 298.70 | 98.80 | 0.01 | 40 | 28 |
| PFHxA ° 1 | 312.70 | 118.70 | 0.01 | 13 | 21 |
| PFHxA ° 2 | 312.70 | 268.70 | 0.01 | 13 | 10 |
| 13C 2-PFHxA es | 314.75 | 269.75 | 0.01 | 13 | 9 |
| HFPO-DA 1° | 329.16 | 168.90 | 0.01 | 10 | 12 |
| HFPO-DA 2° | 329.16 | 284.90 | 0.01 | 10 | 6 |
| HFPO-DA ES DE 1° | 332.16 | 168.90 | 0.01 | 10 | 12 |
| HFPO-DA ES 2° | 332.16 | 286.90 | 0.01 | 10 | 6 |
| PFHpA ° 1 | 362.65 | 168.65 | 0.01 | 14 | 17 |
| PFHpA ° 2 | 362.65 | 318.70 | 0.01 | 14 | 10 |
| PFHxS ° 1 | 398.65 | 79.90 | 0.01 | 50 | 38 |
| PFHxS ° 2 | 398.65 | 98.80 | 0.01 | 50 | 32 |
| C 13 4-PFHxS es | 402.65 | 83.90 | 0.01 | 50 | 38 |
| PFOA ° 1 | 412.60 | 168.70 | 0.01 | 15 | 18 |
| PFOA ° 2 | 412.60 | 368.65 | 0.01 | 15 | 11 |
| C 13 4-PFOA ES | 416.75 | 371.70 | 0.01 | 15 | 11 |
| PFNA ° 1 | 462.60 | 218.75 | 0.01 | 15 | 17 |
| PFNA ° 2 | 462.60 | 418.60 | 0.01 | 15 | 11 |
| PFNA ES | 467.60 | 422.60 | 0.01 | 15 | 11 |
| PFOS ° 1 | 498.65 | 79.90 | 0.01 | 60 | 48 |
| PFOS ° 2 | 498.65 | 98.80 | 0.01 | 60 | 38 |
| C 13 4-PFOS ES | 502.60 | 79.70 | 0.01 | 60 | 48 |
| PFDA ° 1 | 512.60 | 218.75 | 0.01 | 16 | 18 |
| PFDA ° 2 | 512.60 | 468.55 | 0.01 | 16 | 12 |
| 13 2 - PFDA ES | 514.60 | 469.55 | 0.01 | 16 | 12 |
Tabla 4: Tabla de transición del ejemplo y parámetros de MS/MS para el contenido de experimentada-MXA, junto con HFPO-DA
| hora (min) | % A (acetato de amonio 2,5 mM en MeOH 5%) | % B (acetato de amonio 2,5 mM en el 95% MeOH) |
| 0 | 90 | 10 |
| 0.5 | 90 | 10 |
| 3 | 50 | 50 |
| 3.5 | 50 | 50 |
| 5.5 | 40 | 60 |
| 6 | 40 | 60 |
| 7 | 0 | 100 |
| 11 | 0 | 100 |
Tabla 5: Gradiente de ejemplo para la separación de la LC en análisis no dirigidos
| Parámetro de profinder | Valor de ajuste |
| Extracción máxima altura filtro | 800 cuentas |
| Ion(s) permitido | -H / H |
| Modelo de isótopo de extracción característica | Moléculas orgánicas comunes |
| Estados de carga permitida | 2 - Jan |
| Compuesto iónico cuenta umbral | Dos o más iones |
| Tolerancia de alineación RT | 0,40 min + 0.0% |
| Tolerancia total alineación | 20,00 ppm + 2.0mDa |
| Post-Processing de altura absoluta de filtro | > = 10000 puntos en una muestra |
| Procesamiento posterior filtro de puntuación de MFE | > = 75 en una muestra |
| Algoritmo de integración de pico | 2 ágil |
| Máxima integración altura filtro | > = 5000 puntos |
| Buscar por filtro de altura absoluta de Ion | > = 7500 puntos en una muestra |
| Buscar por filtro de puntuación de Ion | > = 50.00 en una muestra |
Tabla 6: Característica Molecular extracción alineación configuración y software Profinder. Todos los valores conservan su configuración predeterminada para el procesamiento de datos.
| Umbral de la abundancia de iones | Umbrales de la característica | Umbral de replicar (n = 5) | Tiempo de ejecución | Funciones | Pasar el umbral de replicar | Pasar el umbral de CV | Características hasta el 90% de las TIC |
| 1 x S/N | 2000 | Ninguno | 8.15 | 987 | 505 | 421 | 91 |
| 2 x S/N | 5000 | Ninguno | 5.02 | 707 | 357 | 313 | 93 |
| 3 x S/N | 10000 | Ninguno | 2.3 | 308 | 249 | 230 | 93 |
| 1 x S/N | 2000 | 100% | 3.3 | 603 | 339 | 297 | 92 |
| 2 x S/N | 35000 | 100% | 1,58 | 310 | 248 | 229 | 93 |
| 3 x S/N | 10000 | 100% | 1.45 | 202 | 190 | 182 | 92 |
Tabla 7: Comparación del tiempo de procesamiento de la muestra e identificaciones de característica química para umbrales de extracción de características diferentes.

Figura 1 : Total cromatograma de iones y cromatogramas de iones extraídos de un subconjunto de estándares de éter perfluorado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2 : Curvas de calibración representante para compuestos demostrando calidad decreciente de la curva analítica construcción. Panel de la izquierda indica una calibración de alta calidad; El panel central indica un compuesto con mala precisión en duplicados de preparación, particularmente en concentraciones más altas; Panel derecho indica una curva con mala precisión y un bajo rango dinámico lineal, dando por resultado una respuesta plana en el extremo superior de la gama de calibración y no hay señal detectable en el extremo inferior. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 : Overlaid cromatogramas de iones totales (TIC) para aguas superficiales extractos recogidos aguas arriba y aguas abajo de un sitio de producción de fluoroquímicos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4 : Cromatogramas de iones extraídos (EIC) para todos identificaron características química de una muestra de agua superficial que contiene múltiples clases de fluoroquímicos. Cada rastro químico es un color diferente para la diferenciación de. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5 : Diagrama conceptual de la información cruda y prevista para una característica química identificada como ácido de dímero de oxido de hexafluoropropileno (HFPO-DA). Características químicas se compilan de extracción del software de datos de MS medidas y contienen cromatográficos (p. ej., tiempo de retención (RT)) e información de espectrometría de masas. Fórmula prevista, estructuras e identidades químicas se generan a partir de mediciones de materia prima para cada función. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6 : Parcela de defecto de masa de elementos químicos identificados en un emisario de fabricación (rojo, izquierda) y agua de la superficie de referencia (azul, derecha). Compuestos fluorados caen cerca y por debajo de la punteada línea cero. Tenga en cuenta la persistente serie PFOS/PFOA en la muestra de agua de la superficie de fondo (derecha). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7 : Parcela de defecto de masa masa vs no identificados características química de una muestra de agua superficial con serie homóloga identificado y etiquetado por el nontarget paquete R. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8 : Espectro de masa de un desconocido características químicas con intensidades isotópicas previstas de tres fórmula química posible con el mismo monoisotopic Massachusetts Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 9 : Espectro de fragmentación de un éter perfluorado compuesto con anotado fragmento picos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 10 : Representación gráfica de los umbrales de filtrado. De izquierda a derecha, umbral de abundancia de ion para espectros de masas característica química, cuentan con umbral de abundancia para las características cromatográficas extraídos y replicar el umbral para la frecuencia de detección de función en un experimento de inyección por triplicado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Preparación y manejo de muestra
La inclusión de normas de referencia/punto son de vital importancia para cualquier análisis objetivo, ya que proporcionan un respaldo para la comprobación de validez analítica. Falta de muestras QC impide cualquier evaluación de la exactitud de los resultados; la naturaleza ubicua de fluoroquímicos significa que posibilidad contaminación de muestras de campo, materiales de procesamiento, sistema LC-MS no es infrecuente y debe explicarse. Además, permite la validación del protocolo independientemente de la variación en la muestra día a día de procesamiento, como muchos de los pasos pueden ser muy variables, particularmente el SPE y muestra pasos de concentración. La extracción de ambos productos químicos perfluorados obsoleta y novedoso puede ser influenciada por la elección de la fase estacionaria para la concentración y componentes de las muestras de la fuente, tales como pH y salinidad46. La influencia de las condiciones de la muestra se debe considerar si son clases particulares de pefluorinated químicos de interés. Esquemas de preparación de muestra alternativas para los extractos de agua pueden utilizarse si la configuración de laboratorio está disponible y el análisis de datos downstream sigue siendo similar.
Análisis de datos específicos
Compuestos con estándares disponibles y emparejado, estable isótopo marcado estándares internos, las principales preocupaciones para el análisis instrumental y determinación de límites de detección del método y rangos de información adecuados se pueden determinar en un base de laboratorio por laboratorio mediante métodos estándar, tales como relación señal a ruido de bajo nivel estándar picos de47. En ausencia de estándares internos coincidentes errores de efectos de matriz pueden ocurrir, y back-predicción precisa de las muestras inoculadas se puede utilizar para estimar la exactitud de las mediciones. Cuando carece de normas para realizar una curva, una estimación cuantitativa de un desconocido puede ser hecha por el tratamiento idénticamente a un estándar estrechamente emparejado compuesta, pero son errores en la estimación del orden de 10 veces con una capacidad limitada para cuantificar la incertidumbre, ver McCord, Newton y Strynar21. En estos casos, aún se pueden recoger datos de tendencia, pero las estimaciones de concentración son inherentemente poco confiables.
Análisis de datos no orientados
Configuración de selección de pico tiene un impacto sustancial en el número de elementos químicos identificados, pero la calidad de la selección es también fuertemente afectada. Las decisiones de interés en cosecha pico son 1) intensidad de masas individuales para ser incluidos en los espectros, el umbral de la abundancia de iones 2) la intensidad del cromatógrama extraído picos a tener en cuenta las características, la característica abundancia umbral 3) detección frecuencia, el umbral de replicar y 4) variación analítica, el umbral de la CV (figura 10).
Configuración de umbrales irrealmente bajos resultados de recolección de pico en un incremento exponencial en el tiempo de la muestra para resolver características adicionales de la abundancia cada vez más bajo (Tabla 7). Los filtros de umbral de la abundancia de iones de masa espectrales características donde lo suficiente de la abundancia de cada isótopo no pasa el umbral. Idealmente Esto selecciona sólo para características con los espectros de MS calidad, asegurando que son características real químicos en lugar de ruido instrumental y permitiendo para la predicción de la fórmula en el proceso aguas abajo. Un umbral adecuado se basa en ruido instrumental, idealmente por lo menos 3 x el umbral de ruido de MS1 explora. Umbral de abundancia característica filtra características químicas basados en la intensidad o el área de la función cromatográfica extraída. Este paso permite el rechazo de los picos de baja abundancia, que suelen ser de mala calidad cromatográfica, tienen varianzas altas o son el resultado de otra extracción pobre software. Un umbral adecuado debe determinarse por experimento y partiendo de un nivel aceptable de generación de función deficiente (p. ej., características abajo la cromatografía inaceptablemente pobre de exposición umbral) de la matriz. Más control de calidad analítico puede ser utilizada para rechazar características nivel cromatográfico basado en identificación incompatible en repeticiones analíticas y preparatorias (umbral de replicación) o basada en la pobre reproducibilidad de repeticiones (umbral de CV). Los niveles apropiados dependen de la calidad de los programas de integración de pico utilizados y de las entidades químicas bajo investigación. Compuestos perfluorados de soluble en agua y ligeramente optimizado integración protocolos, deben identificarse características en 80% de analítica replica y CVs deben caer por debajo del 30%, como se detalla en la sección de métodos.
Los picos detectados análisis orientada a no no den estimaciones cuantitativas de las concentraciones de los materiales detectados. Además, la identidad de verdaderos desconocidos puede ser difícil de confirmar porque faltan nuevos compuestos de bases de datos públicamente disponibles. Nueva determinación estructural requiere un análisis extenso con varios métodos y requiere experiencia en espectrometría de masas y de la química. Sin embargo, la normalización de las áreas pico de características químicas puede proporcionar estimaciones semicuantitativa de las concentraciones de incógnitas de las especies conocidas21. Si se emplean el muestreo constante y los pasos de preparación, información de tendencia del tiempo para las especies individuales puede generarse para controlar la persistencia de una sustancia química en el futuro como la respuesta de una especie individual debe ser coherente salvo grandes variaciones en la matriz21.
La principal ventaja de este método es la extensibilidad de la tratamiento de muestra para permitir análisis específicos y descartando. Específicas análisis proporciona información cuantitativa equivalente o superior, grandemente carece de amplitud de análisis deseado cuando se trata con materiales nuevos y emergentes, así como su relación con materiales de matriz. Aplicación de una metodología específica, o incluso un sospechoso método de detección basado sólo en materiales conocidos y bases de datos limitadas es totalmente ciego a especies no observadas previamente, aunque pueden tener efectos significativos para la salud. Mejora de software y bases de datos se convierten en más robustos, la exactitud de identificación desconocida seguirá aumentando, con una disminución concomitante en la inversión de tiempo y nivel de conocimientos necesarios para analizar los datos multidimensionales generados por este enfoque. Sin embargo, los datos generados en la actualidad son de significativo valor futuro porque datos bancarios permite análisis post-hoc con software recién desarrollado y permite la comparación a través del tiempo aunque se desconoce la identidad de un compuesto detectado.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
La Agencia de protección ambiental de Estados Unidos, a través de su oficina de investigación y desarrollo, había financiado y gestionado la investigación descrita aquí. Este documento ha sido revisado por la Agencia de protección ambiental, oficina de investigación y desarrollo y aprobado para su publicación. Las opiniones expresadas en este artículo son las de los autores y no representan necesariamente las opiniones o políticas de la Agencia de protección ambiental de Estados Unidos. Esta investigación fue apoyada en parte por una cita para el programa de Investigación Postdoctoral en el laboratorio de investigación exposición nacional administrado por el Instituto Oak Ridge para la ciencia y la educación a través de DW89992431601 de acuerdo interinstitucional entre el Departamento de energía de Estados Unidos y la Agencia de protección ambiental de Estados Unidos.
Materiales
| Name | Company | Catalog Number | Comments |
| Acqity ultra-high performance liquid chromatography system | Waters Corporation | Modified with PFCs analysis kit (176001744); equivalent UPLC system is acceptible if PFAS background is checked and confirmed to be low | |
| Ammonium acetate | Fluka | 17836 | Mass spectrometry grade >99% pure |
| Ammonium Hydroxide | Sigma-Aldrich | 338818 | |
| Balance | Mettler | AB204S | |
| BEH C18 reverse phase UPLC column, 2.1×50 mm, 1.7 μm | Waters Corporation | 186002350 | |
| Dual piston syringe pump | Waters Corporation | SPC10-C | |
| Glacial Acetic Acid | Sigma-Aldrich | ARK2183 | |
| Glass Microfiber Filters | Whatman | 1820-070 | |
| High density polyethelye sample bottle | Nalgene | 2189-0032 | |
| High Resolution Mass Spectrometer | Various | Mass Spectrometer should be capable of providing accurate mass to <10ppm and collecting MS/MS data. Agilent 6530 qTOF and Thermo Fisher Orbitrap Fusion were used in this work | |
| Methanol | Sigma-Aldrich | ||
| Nitric Acid (35% w/w) | Thermo Fisher Scientific | SVCN-5-1 | Can be prepared in house using concentrated nitric acid and reagent water |
| Polypropylene Buchner funnel | ACE Glass | 12557-09 | |
| Polypropylene cenitrfuge tube and cap | BD Falcon | 352096 | |
| Polypropylene Vacuum Flask (1 L) | Nalgene | DS4101-1000 | |
| Quattro Premier XE triple quadrupole mass spectrometer | Waters Corporation | Equivalent triple-quadrupole or better system can be used instead, should provide high sensitivity and stability for targeted analysis | |
| Reagent Water | Any source determined to be PFAS free | ||
| Sodium Acetate | Sigma-Aldrich | W302406 | |
| TurboVap nitrogen evaporator | Caliper Life Sciences | 103198 | Equivalent systems or rotary vacuum evaporator may be used instead |
| Weak anion exchange SPE cartridge (Oasis WAX Plus) | Waters Corporation | 186003519 | |
| Standard Solutions | |||
| 2,3,3,3-Tetrafluoro-2-(1,1,2,2,3,3,3-heptafluoropropoxy)propanoic acid (HFPO-DA) | Wellington | HFPO-DA | |
| Additional targeted compound standards of interest | to be determined based on preliminary analysis and standard availability | ||
| Mass labeled HFPO-DA | Wellington | M2HFPO-DA | |
| Native PFCA/PFAS Mixture (2 ug/mL) | Wellington | PFAC-MXA | or PFAC-MXB; or individually prepared mixture containing compounds of interest |
| Stable Isotope Labeled PFCA/PFAS Mixture (2 ug/mL) | Wellington | MPFAC-MXA | or MPFAC-MXB; or individually prepared mixture containing compounds of interest as appropriate for Native PFASs |
| Software | |||
| Mass Profiler Professional | Agilent | Or open source software packages | |
| Profinder | Agilent | Or open source software packages |
Referencias
- . Provisional Health Advisories for Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS). United States Environmental Protection Agency. , (2009).
- . Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluorooctane Sulfonate. United States Environmental Protection Agency. , 33250-33251 (2016).
- . Fact Sheet: 2010/2015 PFOA Stewardship Program Available from: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-20102015-pfoa-stewardship-program (2006)
- EPA and 3M Announce phase out of PFOS. Environmental Protection Agency Available from: https://yosemite.epa.gov/opa/admpress.nsf/0/33aa946e6cb11f35852568e1005246b4 (2000)
- Wang, Z., Cousins, I. T., Scheringer, M., Hungerbühler, K. Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors. Environment International. 60, 242 (2013).
- Scheringer, M., et al. Helsingør Statement on poly- and perfluorinated alkyl substances (PFASs). Chemosphere. 114, 337-339 (2014).
- Wang, Z., DeWitt, J. C., Higgins, C. P., Cousins, I. T. A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs). Environmental Science & Technology. 51 (5), 2508-2518 (2017).
- Xiao, F., Golovko, S. A., Golovko, M. Y. Identification of novel non-ionic, cationic, zwitterionic, and anionic polyfluoroalkyl substances using UPLC-TOF-MSE high-resolution parent ion search. Analytica Chimica Acta. 988, 41-49 (2017).
- Shoemaker, J., Grimmett, P., Boutin, B. Method 537. Determination of selected perfluorinated alkyl acids in drinking water by solid phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS). US Environmental Protection Agency. , (2009).
- Poole, C. F., Gunatilleka, A. D., Sethuraman, R. Contributions of theory to method development in solid-phase extraction. Journal of Chromatography A. 885 (1), 17-39 (2000).
- Ahrens, L. Polyfluoroalkyl compounds in the aquatic environment: a review of their occurrence and fate. Journal of Environmental Monitoring. 13 (1), 20-31 (2011).
- Higgins, C. P., Field, J. A., Criddle, C. S., Luthy, R. G. Quantitative Determination of Perfluorochemicals in Sediments and Domestic Sludge. Environmental Science & Technology. 39 (11), 3946-3956 (2005).
- Szostek, B., Prickett, K. B., Buck, R. C. Determination of fluorotelomer alcohols by liquid chromatography/tandem mass spectrometry in water. Rapid Communications in Mass Spectrometry. 20 (19), 2837-2844 (2006).
- Alzaga, R., Bayona, J. M. Determination of perfluorocarboxylic acids in aqueous matrices by ion-pair solid-phase microextraction-in-port derivatization-gas chromatography-negative ion chemical ionization mass spectrometry. Journal of Chromatography A. 1042 (1-2), 155-162 (2004).
- Schaider, L. A., et al. Fluorinated Compounds in U.S. Fast Food Packaging. Environmental Science & Technology Letters. 4 (3), 105-111 (2017).
- Lindstrom, A. B., Strynar, M. J., Libelo, E. L. Polyfluorinated Compounds: Past, Present, and Future. Environmental Science & Technology. 45 (19), 7954 (2011).
- Liu, Y., Pereira, A. D. S., Martin, J. W. Discovery of C5-C17 Poly-and Perfluoroalkyl Substances in Water by In-Line SPE-HPLC-Orbitrap with In-Source Fragmentation Flagging. Analytical Chemistry. 87 (8), 4260 (2015).
- Backe, W. J., Day, T. C., Field, J. A. Zwitterionic, Cationic, and Anionic Fluorinated Chemicals in Aqueous Film Forming Foam Formulations and Groundwater from U.S. Military Bases by Nonaqueous Large-Volume Injection HPLC-MS/MS. Environmental Science & Technology. 47 (10), 5226-5234 (2013).
- Mazzoni, M., Rusconi, M., Valsecchi, S., Martins, C. P. B., Polesello, S. An on-line solid phase extraction-liquid chromatography-tandem mass spectrometry method for the determination of perfluoroalkyl acids in drinking and surface waters. Journal of Analytical Methods in Chemistry. 2015, 942016 (2015).
- Li, F., et al. Method development for analysis of short- and long-chain perfluorinated acids in solid matrices. International Journal of Environmental Analytical Chemistry. 91 (12), 1117-1134 (2011).
- McCord, J., Newton, S., Strynar, M. Validation of quantitative measurements and semi-quantitative estimates of emerging perfluoroethercarboxylic acids (PFECAs) and hexfluoroprolyene oxide acids (HFPOAs). J Chromatoqr A. , (2018).
- Wang, Y., Liu, S., Hu, Y., Li, P., Wan, J. -. B. Current state of the art of mass spectrometry-based metabolomics studies - a review focusing on wide coverage, high throughput and easy identification. RSC Advances. 5 (96), 78728-78737 (2015).
- Cajka, T., Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Analytical Chemistry. 88 (1), 524-545 (2016).
- Mann, M., Kelleher, N. L. Precision proteomics: The case for high resolution and high mass accuracy. Proceedings of the National Academy of Sciences of the United States of America. 105 (47), 18132-18138 (2008).
- Sobus, J. R., et al. Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. Journal of Exposure Science & Environmental Epidemiology. , (2017).
- Bletsou, A. A., Jeon, J., Hollender, J., Archontaki, E., Thomaidis, N. S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trends in Analytical Chemistry. 66, 32-44 (2015).
- Viant, M. R., Sommer, U. Mass spectrometry based environmental metabolomics: a primer and review. Metabolomics. 9 (1), 144-158 (2013).
- Xiao, F. Emerging poly- and perfluoroalkyl substances in the aquatic environment: A review of current literature. Water Research. 124, 482-495 (2017).
- Nakayama, S. F., Strynar, M. J., Reiner, J. L., Delinsky, A. D., Lindstrom, A. B. Determination of perfluorinated compounds in the Upper Mississippi River Basin. Environmental Science & Technology. 44 (11), 4103 (2010).
- Strynar, M., et al. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environmental Science & Technology. 49 (19), 11622 (2015).
- Newton, S., et al. Novel Polyfluorinated Compounds Identified Using High Resolution Mass Spectrometry Downstream of Manufacturing Facilities near Decatur, Alabama. Environmental Science & Technology. 51 (3), 1544-1552 (2017).
- Forsberg, E. M., et al. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nature Protocols. 13, 633 (2018).
- Sturm, M., et al. OpenMS - An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Kind, T., Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 8, 105-105 (2007).
- Loos, M., Singer, H. Nontargeted homologue series extraction from hyphenated high resolution mass spectrometry data. Journal of Cheminformatics. 9, 12 (2017).
- Dimzon, I. K., et al. High Resolution Mass Spectrometry of Polyfluorinated Polyether-Based Formulation. Journal of the American Society for Mass Spectrometry. 27, 309 (2016).
- McEachran, A. D., Sobus, J. R., Williams, A. J. Identifying known unknowns using the US EPA's CompTox Chemistry Dashboard. Analytical and Bioanalytical Chemistry. 409 (7), 1729-1735 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard's msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Tautenhahn, R., Böttcher, C., Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 9, 504 (2008).
- Rafiei, A., Sleno, L. Comparison of peak-picking workflows for untargeted liquid chromatography/high-resolution mass spectrometry metabolomics data analysis. Rapid Communications in Mass Spectrometry. 29 (1), 119-127 (2015).
- Kind, T., Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics. 7, 234-234 (2006).
- Brack, W., Dulio, V., Slobodnik, J. The NORMAN Network and its activities on emerging environmental substances with a focus on effect-directed analysis of complex environmental contamination. Environmental Sciences Europe. 24 (1), 29 (2012).
- Blaženović, I., et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: database boosting is needed to achieve 93% accuracy. Journal of Cheminformatics. 9, 32 (2017).
- Rager, J. E., et al. Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environment International. 88, 269-280 (2016).
- Munoz, G., et al. Environmental Occurrence of Perfluoroalkyl Acids and Novel Fluorotelomer Surfactants in the Freshwater Fish Catostomus commersonii and Sediments Following Firefighting Foam Deployment at the Lac-Mégantic Railway Accident. Environmental Science & Technology. 51 (3), 1231-1240 (2017).
- Brumovský, M., Bečanová, J., Karásková, P., Nizzetto, L. Retention performance of three widely used SPE sorbents for the extraction of perfluoroalkyl substances from seawater. Chemosphere. 193, 259-269 (2018).
- Definition, Definition and Procedure for the Determination of the Method Detection Limit (Revision 2). Environmental Protection Agency. , (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados