
Method Article
Identifying Per- and Polyfluorinated Chemical Species with a Combined Targeted and Non-Targeted-Screening High-Resolution Mass Spectrometry Workflow
In This Article
Summary
Here, we present a protocol for the sequential targeted quantification and non-targeted analysis of fluorinated compounds in water by mass spectrometry. This methodology provides quantitative levels of known fluorochemical compounds and identifies unknown chemicals in related samples with semi-quantitative estimates of their abundance.
Abstract
Historical and emerging per- and polyfluoroalkyl substances (PFASs) have garnered significant interest from the public and government agencies from the local to federal levels. The continuing evolution of PFAS chemistries presents a challenge to the environmental monitoring, where ongoing development of targeted methods necessarily lags the discovery of new chemical compounds. There is a need, therefore, to have forward-looking methodologies that can detect emerging and unexpected compounds, monitor these species over time, and resolve details of their chemical structure to enable future work in human health. To this end, non-targeted analysis by high-resolution mass spectrometry offers a broad base detection approach that can be combined with almost any sample preparation scheme and provides significant capabilities for compound identification after detection. Herein, we describe a solid-phase extraction (SPE) based sample concentration method tuned for shorter chain and more hydrophilic PFAS chemistries, such as per fluorinated ether acids and sulfonates, and describe analysis of samples prepared in this fashion in both targeted and non-targeted modes. Targeted methods provide superior quantification when reference standards are available but are intrinsically limited to expected compounds when performing analysis. In contrast, a non-targeted approach can identify the presence of unexpected compounds and provide some information about their chemical structure. Information about chemical features can be used to correlate compounds across sample locations and track abundance and occurrence over time.
Introduction
The class of per- and polyfluoroalkyl substances (PFASs) are persistent organic pollutants with significant public health concern. The specific compounds perfluorooctanoic acid (PFOA) and perfluorooctanesulfonate (PFOS) have drinking water health advisory levels set by the EPA1,2 and their major US production ceased in the 2000s3,4. To gain a significant understanding for the properties of PFAS materials in the textile and consumer product manufacturing spheres, hundreds, if not thousands, of alternate PFAS chemistries have been developed to fill product niches, including replacements for the deprecated compounds5,6,7,8. There is an ongoing need to monitor the environmental levels of straight chain perfluorinated carboxylic acids and sulfonates such PFOS, PFOA, and their related homologous series, but emerging chemical compounds are not covered by established methods such as EPA Method 5379 and frequently lack analytical standards for traditional targeted analysis. The intention of this protocol is thus two-fold. It provides a pathway for the targeted LC-MS/MS analysis of fluorochemical species in water where analytical standards are available and details the seamless integration of a non-targeted, high-resolution mass spectrometry-based approach for data analysis that enables the detection of unknown or unexpected compounds in the same samples.
Solid-phase extraction (SPE) is an established technique for the sample cleanup and concentration with applications to many analytes and sample matrices10,11. For PFAS analysis, multiple solid retentive phases including non-polar, functionalized polar, and ion exchange columns have been used to varying extents for subclasses of fluorinated species in a wide variety of matrices9,12,13,14,15,16. Advances in SPE sample analysis using on-line setups greatly increase the throughput of the approach and improve the reproducibility of sample handling, but the fundamental process remains consistent17. Some efforts to remove the offline concentration of SPE using large volume injections have also been undertaken, but these require modifications to the chromatography that place them outside the realm of casual analysis18,19. Our sample analysis uses a polymeric weak anion exchange (WAX) retentive phase to thoroughly separate acidic PFAS materials from the traditional organic contaminants while achieving substantial sample concentration factors. This WAX phase is important to capture the short chain perfluorinated acids such as perfluorobutane sulfonate (PFBS) or perfluorinated ethers such as hexafluoropropylene oxide dimer acid (HFPO-DA) which are more polar than the longer chain legacy perfluorinated species20,21. As there has been a significant shift towards shorter fluorinated chains and ether inclusion in recent PFAS chemistry5, this phase selection enables more thorough recovery of novel compounds for MS analysis.
Targeted LC-MS/MS quantitation using authenticated standards and stable isotope labeled internal standards provides an unparalleled level of specificity and sensitivity for the quantitative analysis. While this approach is desirable in many situations, it is impractical for all-too-common situations in analysis. Targeted approaches work only for species that are expected in the sample, and for which methods have previously been established. For new and emerging compounds, this approach is incapable of even detecting species that may be of interest, regardless of their chemistry or concentration, and low-resolution mass spectrometers are nearly incapable of providing enough information to make unequivocal chemical assignments of unknown compounds. Consequently, the field of non-targeted analysis has arisen, leveraging the power of high-resolution modern mass spectrometers to analyze samples without a presupposed hypothesis and retroactively assign chemicals to detectable features in the sample. This approach has been used extensively in the fields of biology22,23,24 and environmental science25,26,27 on numerous classes of chemicals. Perfluorinated chemicals are particularly straightforward to identify in this method due to their unique mass spectral patterns, and hundreds of compounds have been described in just the past few years5,28.
The protocol discussed here is intended to align targeted LC-MS/MS PFAS quantitation with the need to identify and semi-quantitatively monitor emerging compounds of interest. The SPE phase selection and sample preparation techniques are intended to ensure capture of more hydrophilic emerging PFAS acids from water and may be less suited for longer chain polymeric species and non-ionic species. Further, the data generated by non-targeted analysis is dense and of high dimensionality, which necessitates the use of data analysis software. Such software packages are frequently vendor specific and require modification to operate between instrument platforms. Where possible, the analysis process has been described in a generic fashion and open source/freeware alternatives are referenced, but the efficiency and accuracy of any software approach must be assessed on an individual basis.
Protocol
1. Collection of water samples
- Preparation of PFAS Standard Stocks
- Prepare a PFAS standard mixture in methanol containing any targeted compounds of interest (e.g., PFOA, PFOS, HFPO-DA) at 1 ng/µL. This is the Native PFAS Mixture. Commercially prepared mixtures are also available (i.e., PFAC Mix A and Mix B).
- Prepare a standard mixture containing matched stable isotope labeled (SIL) PFAS compounds (e.g., 13C4-PFOA, 13C8-PFOS, 13C3-HFPO-DA) at 1 ng/µL. This is the IS PFAS Mixture. Commercially prepared mixtures are also available (i.e., MPAFC Mix A and Mix B).
NOTE: If an SIL version of the targeted PFAS is unavailable, a surrogate with similar structure and chain length can be used (e.g., 13C2-PFHxA for HFPO-DA)
- Preparation of Field Blank (FB), Spike Blank (SB) samples
- Fill two, clean high-density polypropylene (HDPE) or polypropylene (PP) bottles with 1,000 mL of laboratory deionized (DI) water, known to be PFAS free.
CAUTION: PFAS materials frequently have undefined toxicity and/or carcinogenicity. Care should be taken to avoid oral or skin exposure to standards or stock solutions. - Add a quantity of PFAS standard mixture to one of the bottles at a final concentration equivalent to the expected sample concentrations (e.g., 100 ng/L). This is the Spike Blank (SB).
- Add 5 mL of 35% nitric acid preservative to the Spike Blank.
- Carry both SB sample and the unspiked field blank to the sampling location as controls.
- Fill two, clean high-density polypropylene (HDPE) or polypropylene (PP) bottles with 1,000 mL of laboratory deionized (DI) water, known to be PFAS free.
- Field sampling
NOTE: Sample collector should wear nitrile gloves and sample from flowing systems where possible. Tap samples should be allowed to flow and equilibrate prior to sampling (2-3 min).- Collect 500-1,000 mL of water from the field location in a clean HDPE or PP bottle.
- Add 5 mL of 35% nitric acid preservative to sample bottles and field blank.
CAUTION: Nitric acid is corrosive and a strong oxidizer
2. Sample extraction
NOTE: PFAS are ubiquitous and persistent. Ensure that all solvents are of the highest grade and have been analyzed for low level PFAS contamination. Thoroughly rinse all laboratory equipment used for preparing standards before preparing blanks and samples.
- Sample pretreatment
- Pour each sample into a separate, pre-cleaned 1 L HDPE graduated cylinder and record the exact volume.
- Add 10 mL of methanol to the emptied sample bottle, cap it, and shake well to rinse adsorbed PFAS from the bottle interior.
- Return the measured water sample to the rinsed bottle with the methanolic rinse.
- Standard curve for quantitation
- Fill eight, 1 L HDPE/PP bottles with PFAS-free DI water.
- Select eight evenly spaced concentrations covering the desired quantitation range. For example: 10, 25, 50, 100, 250, 500, 750 and 1,000 ng/L for a range of 10-1,000 ng/L.
- Add a quantity of Native PFAS mix to each bottle to yield the final PFAS concentrations in 2.2.2 (e.g., 100 µL PFAS Mix A to 1L of DI water = 100 ng/L).
- Internal standard addition
NOTE: Addition of stable isotope labeled internal standard (IS) is necessary only if quantitative results are desired in addition to non-targeted analysis.- Add the IS PFAS mixture to each sample at a concentration approximating the midpoint of the calibration curve (e.g., 250 µL of IS PFAS mix = 250 ng/L)
- Filtration
- Filter samples through GF/A glass fiber filters (47 mm, 1.6 µm pore size) under gentle vacuum into a pre-cleaned 1 L HDPE vacuum flask.
- If particulate matter remains in the bottle, rinse with additional deionized water into the filter. Return the filtered water to the sample bottle or a new container for solid phase extraction.
- Solid phase extraction (SPE)
NOTE: Cartridge concentration described here uses a constant flow piston pump. Alternative methods of concentration using a vacuum manifold20 or using an on-line SPE-LC-MS17 setup are possible but not discussed.- Condition a weak anion exchange (WAX) cartridge with 25 mL of methanol.
- Condition the WAX cartridge with an additional 25 mL of deionized water.
- Position pump draw tubing in filtered sample bottles and label SPE cartridges with corresponding sample names.
- Pump 500 mL of sample water through the cartridge at a steady flow rate of 10 mL/min (500 mL total), discarding flow-through liquid to waste.
NOTE: Larger or smaller volumes can be concentrated depending on expected sample concentrations. - Remove the cartridge from piston pump for elution.
NOTE: If concentrating additional samples using the same pump, the piston pump should be flushed with 25 mL of methanol before installing the next cartridge for equilibration. - Transfer SPE cartridge to a vacuum manifold and equip with external glass reservoir.
- Flush SPE cartridge with 4 mL of 25 mM, pH 4.0 sodium acetate buffer under gentle vacuum. Discard flow through. Wash SPE cartridge with 4 mL of neutral methanol.
NOTE: Neutral wash fraction can be collected if specific nonionic polar analytes are expected. Otherwise, discard to waste - Place a 15 mL polypropylene centrifuge tube beneath each SPE cartridge to collect eluent. Elute sample with 4 mL of 0.1% ammonium hydroxide in methanol.
- Remove elution tube and reduce eluate volume to 500 - 1,000 µL by evaporation under dry nitrogen stream in a water bath at slightly elevated temperature (40 °C).
- Concentrated sample extracts can be stored prior to analysis at room temperature.
- Targeted LC-MS/MS quantitation
- Dilute 100 µL of sample extract with 300 µL of 2 mM ammonium acetate buffer into an HPLC sample vial.
- Calibrate and equilibrate an HPLC and MS systems according to manufacturer's instructions.
NOTE: Background PFAS are commonly detected due to the use of fluoropolymer components of most LC systems and in sample vial septa. Confirm that the detectable levels in blanks is negligible before use. Modification of the LC system to replace Teflon components is suggested where possible. The use of an analytical "hold-up" column adjacent to the LC mixing valve is also suggested29. - Prepare an analytical worklist consisting of the standard curve, samples, and an additional replicate of the standard curve to assess instrumental drift across the run. An example worklist is shown in Table 1.
- Analyze the samples using LC and MS methods established for the targeted compound(s) of interest. The example LC gradient is shown in Table 2 and MS method parameters are shown in Table 3 and Table 4. Further detailed discussion can be found in McCord et al.21.
- Generate a standard curve from the standard samples using the peak area ratio of the analyte to the internal standard versus the concentration of analyte. Generate a quadratic regression formula with 1/x weighting for concentration prediction9.
- Quantitate targeted analytes in each sample using the prepared standard curves and area ratio (standard area/IS area) for each measurement.
- If the concentration exceeds the calibration range, dilute the original sample with DI water spiked with the appropriate IS concentration and re-extract to bring the concentration into the appropriate range.
- Non-targeted LC-MS/MS data collection
- Dilute 100 µL of sample extract with 300 µL of 2 mM ammonium acetate buffer into an HPLC sample vial.
- Calibrate and equilibrate an HPLC and high-resolution MS according to manufacturer's instructions.
- Prepare an analytical worklist as in 2.6.2.
- Using the instrument software, collect LC-MS data in with a wide scan MS1 in data-dependent mode to collect MS/MS. Example LC gradient in Table 5. Further discussion of instrument settings can be found in Strynar et al.30 and Newton et al.31.
NOTE: For improved MS/MS quality data-dependent analysis can be carried out with a preferred ion list of a subset of features remaining after data processing in 2.8.1-2.8.8.
- Non-targeted data processing
NOTE: Data analysis can be performed with a wide range of software and these methods do not reflect the only, or best method for an arbitrary dataset. Where possible, steps provide a generic description that can be carried out in alternate software. Processing of the example data used in this manuscript was carried out using vendor specific software (Software 1 and Software 2) as detailed in Newton et al.31.- Perform molecular feature extraction of chemical features using one of several open source software packages32,33 or vendor software to identify monoisotopic masses, retention times, and integrated peak areas of chemical features.
- In Software 1, select Add/Remove Sample Files > Add Files and select the raw data from the non-targeted experiment, then hit OK.
- In Software 1 select Batch Recursive Feature Extraction > Open Method… to load a preestablished method, or manually edit software settings. Profinder settings for feature extraction are found in Table 6.
- In Software 1, after feature extraction, select File > Export as CSV…, File > Export as CEF…, or File > Export as PFA… for further processing. CEF files are assumed for the remainder of the description.
- In Software 2 (MPP) create a new experiment with Type Unidentified and Workflow type Data Import Wizard and click OK.
- In MPP Select Data Files and locate the exported Software 1 results (either CEF or PFA) to import; then click Next until Alignment Parameter options appear.
- In MPP, set the Compound Alignment values to 0.0 (alignment was already performed in the feature extraction of Software 1, step 2.8.1.2) then click Next through the steps until Finish is available.
- Filter identifications based on analytical reproducibility. Where multiple replicate samples are available, features should be present in >80% of individual replicates and have an analytical coefficient of variation (CV) of < 30%
- In MPP select Experimental Setup > Experiment Grouping and assign each raw file a group corresponding to its origin sample (i.e., replicates from the same source should be in the same group). Multiple groups can be created corresponding to nested variables (e.g., instrumental vs. technical replicates).
- In MPP select Experimental Setup > Create Interpretation then select the experiment parameter (i.e., group) and click Next until Finish is available. This will create a category that future filtering can operate on.
- In MPP select Quality Control > Filter by Frequency. Set Entity List to All Entities and the Interpretation to the sample Group(non-averaged) created in 2.8.2.2, then hit Next.
- For Input parameters, set entity retention at 80% of sampled in at least one condition then click Next until Finish is available. Name the list Frequency Filtered Features
- In MPP select Quality Control > Filter on Sample Variability. Set the Entity List to the Frequency Filtered Features from 2.8.2.4 and the interpretation to Group(non-averaged), then hit Next.
- Select the radio button for Raw Data and the Range of Interest to Coefficient of variation < 30%. Click Next > Finish and save the list as CV Filtered Features.
- Remove features where no samples have significantly higher (>3 fold) abundance than the Field Blank (FB) sample.
- In MPP select Analysis > Fold Change. Set Entity List to CV Filtered Features and the Interpretation to the sample Group then hit Next. Select the fold change option to All against single condition and select condition FB or whatever the group name for the blank processed sample was.
- On the following screen, set the Fold-Change cutoff to 3.0 and click through to the end of the prompts. Save the list as FC Filtered List.
- Perform binary comparisons of individual samples of interest against an appropriate background sample (e.g., upstream vs. downstream of a point source) to determine fold-changes for individual chemical features.
- In MPP select Analysis > Filter on Volcano Plot. Set the entity list to FC Filtered List and the Interpretation to Group.
- For the fold-change condition pair choose two samples for comparison (e.g., a paired upstream and downstream sample) and select test Mann-Whitney Unpaired.
- For preliminary analysis, do not select a value for multiple test correction on the following screen, click through to the result plot.
- On the results screen select a fold-change cutoff of 3.0 and a p-value cutoff to 0.1. Then Finish and export the list as Prelim Results.
- For each feature remaining after filtering, generate predicted chemical formula(s) from the exact mass and composite mass spectrum.
- In MPP, select Results Interpretation > IDBrowser Identification and the Prelim Results entity list.
- In the IDBrowser select Identify all compounds using molecular formula generator (MFG) as the identification method.
- In the Generate Formula options add F to the Elements column and set the Maximum to 50, then select Finish. Following formula generation select Save and Return to return to MPP.
- In MPP, right click the filtered and MFG matched Entity List and select Export List. Save the results.
- Examine the monoisotopic mass of species in the reduced significant chemical feature list for those containing mass defects indicative of fluorination; see Kind and Fiehn34.
- Note chemical series containing common polyfluorination motifs (CF2 (m/z 49.9968), CF2O (m/z 65.9917), CH2CF2O (m/z 80.0074), etc.) using a mass defect plot or software algorithm; see discussion section, Liu et al.17, Loos et al.35 , and Dimzon et al.36.
- Search predicted chemical formulas or neutral masses against the EPA Chemistry Dashboard database and/or other databases to return potential chemical structures.
- Open the EPA Comptox Chemicals Dashboard Batch Search tool (https://comptox.epa.gov/dashboard/dsstoxdb/batch_search) and paste the list of identifiers (either formulas or masses) into the identifier box, after selecting the identifier type (i.e., MS-ready Formula or Monoisotopic Mass).
- Select Download Chemical Data… and also select any physical/chemical/toxicology data desired for potential matches from the dropdown.
- Using chemical intuition and available reference data, remove unlikely matches from the potential chemical structure list for each formula based on feasibility due to chemical stability, physical properties such as ionizability or hydrophobicity, the presence of manufacturing chemicals from nearby sources, etc. In the absence of additional data, spectral feasibility can be ranked purely on the basis of literature prevalence; see McEachran et al.37.
- Confirm structures using available standards and/or targeted high-resolution MS/MS matching of fragments against spectra from databases, in silico theoretical spectra, or manual curation.
- Perform molecular feature extraction of chemical features using one of several open source software packages32,33 or vendor software to identify monoisotopic masses, retention times, and integrated peak areas of chemical features.
Results
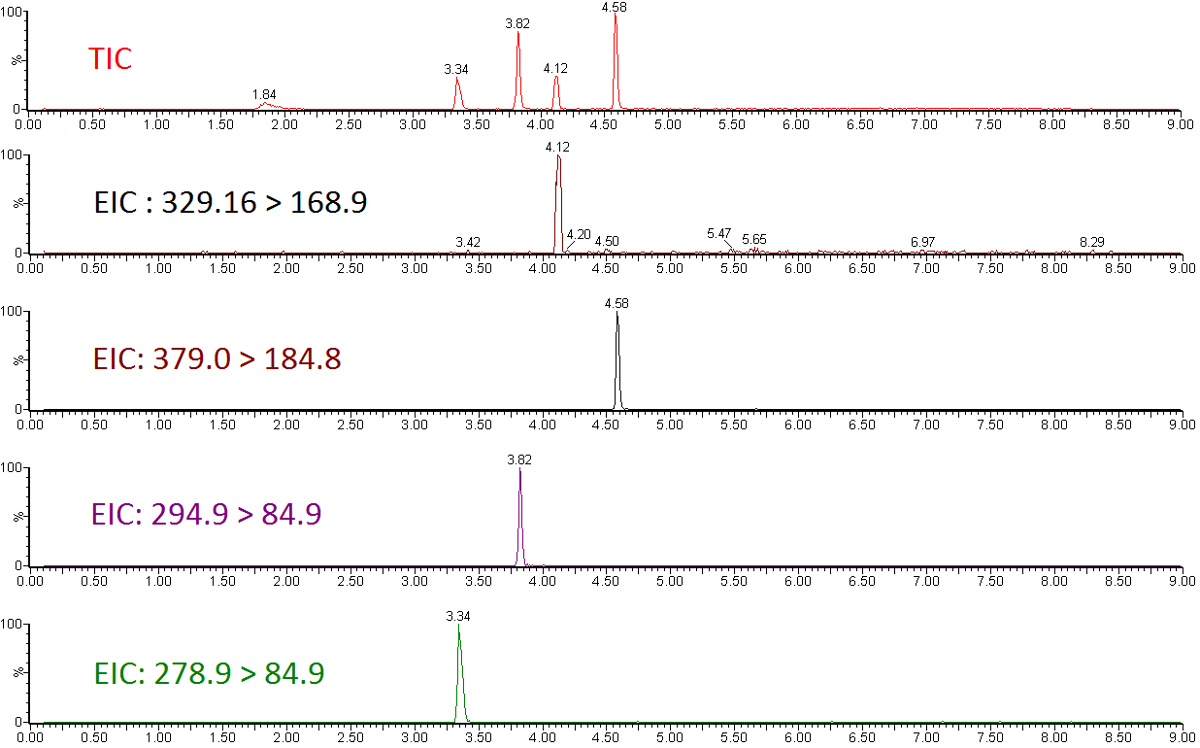
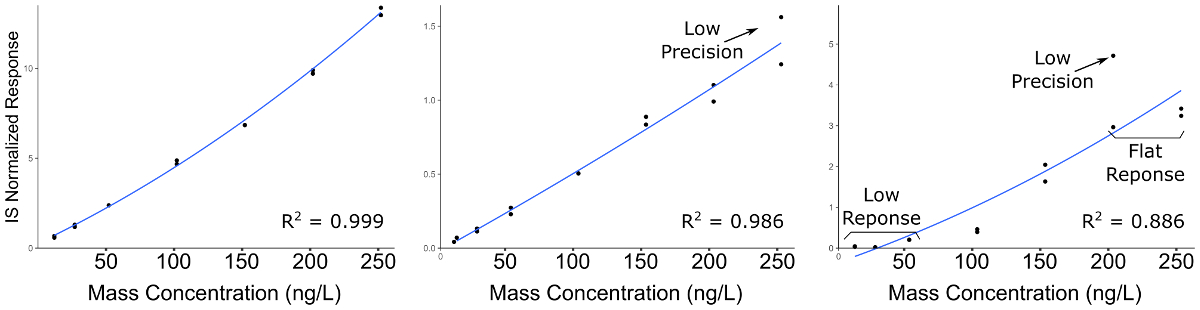
Quantitative LC-MS/MS results are in the form of ion-chromatograms for the total ion chromatogram (TIC) and the extracted ion chromatograms (EIC) of specific chemical transitions for measured chemicals (Figure 1). The integrated peak area of a chemical transition is related to the compound abundance and can be used to calculate the exact concentration using a calibration curve normalized to an internal standard (Figure 2). Low or flat response of individual analytes indicates that the calibration range is outside the linear range of the mass spectrometer, or that the instrument requires tuning/calibration. Poor precision of replicates indicates an issue with sample injection or inconsistent chromatography that requires modification of LC parameters.
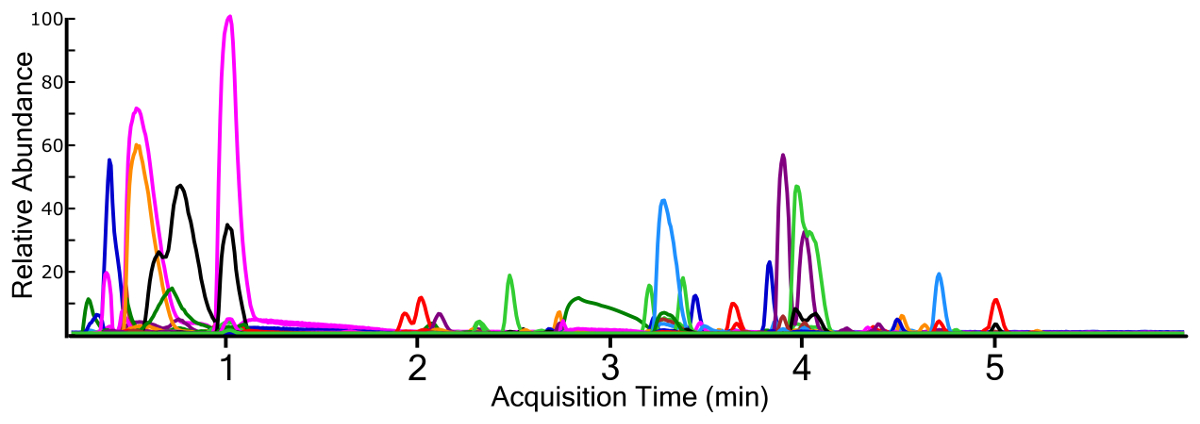
Non-targeted analysis using a full MS1 scan yields a TIC for samples (Figure 3), which allows for ad hoc generation of EICs for individual ions (Figure 4). Any given chromatographic time point contains signals for chemical species, and when using a high-resolution mass spectrometer, the isotopic fingerprint of the compound. Identifying compounds from the MS1 scan is performed programmatically by a peak-picking algorithm using one of several approaches38,39,40. Peak picking yields chemical features with a measured accurate mass and chromatographic retention time, as well as the mass spectrum of the ion and the chromatographic peak area. This information is typically stored in a digital database format for further processing and filtering, but the nested and interconnected nature of the data can be understood conceptually (Figure 5).
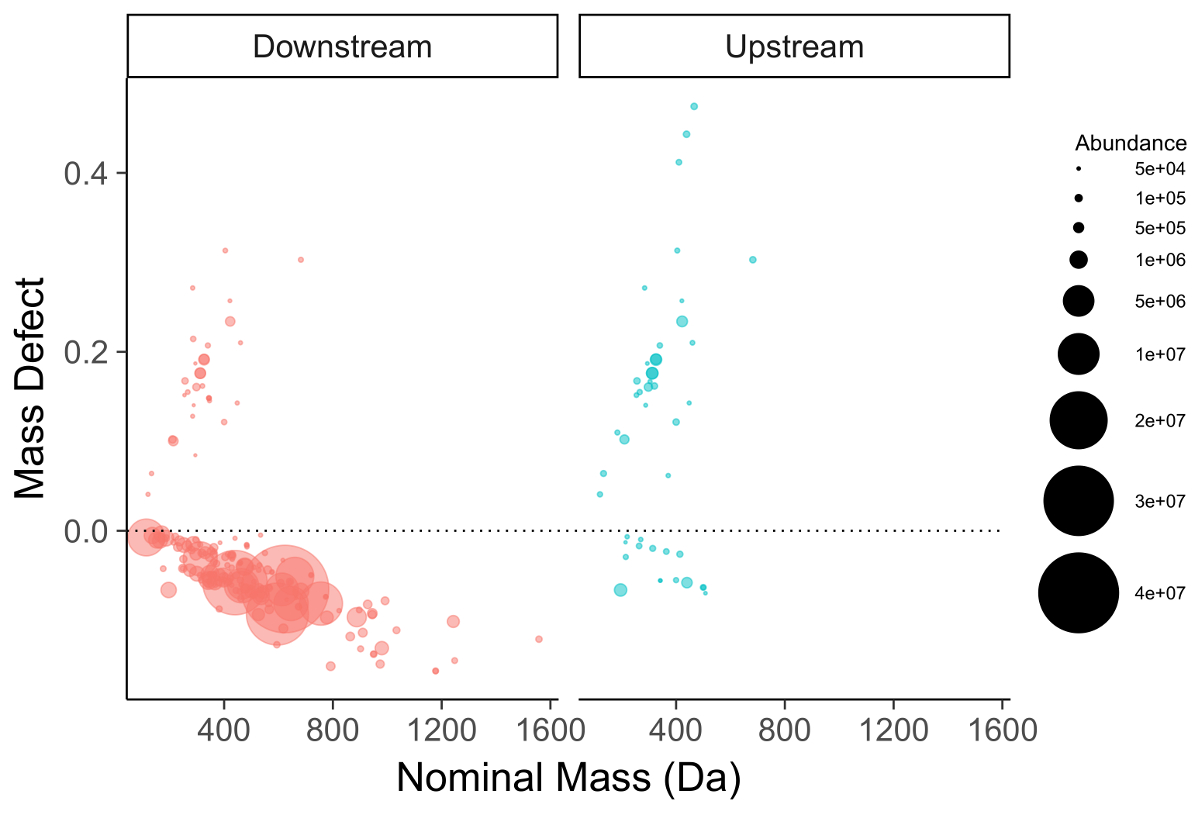
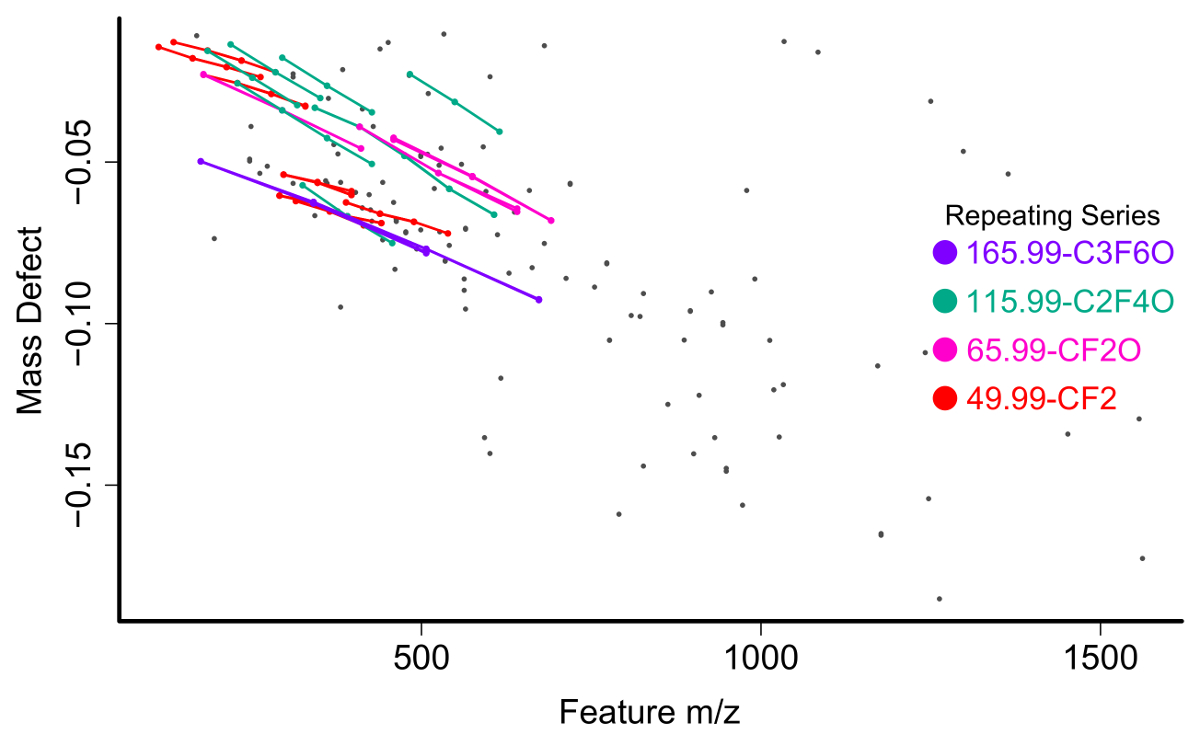
The feature list is filtered for compounds meeting one of several criteria to be selected for further investigation. The first and most straightforward is filtering by mass defect (the difference between the exact mass of a feature and its nominal mass). PFAS compounds have negative mass defects (Figure 6) due to their preponderance of fluorine atoms, and polyfluorinated compounds have positive, but substantially smaller mass defects than homologous organic materials31,34. A second method filtering step is to identify homologous series containing repeating units common to PFAS species, such as CF2 or CF2O. Identifying these can be done using Kendrick Mass defect plots17,36, or software packages such as R's nontarget package35 (Figure 7).
Following filtering, assignment of chemical identity on the shortlist of highly differentially observed and/or tentatively per/polyfluorinated species can begin. Accurate mass provides a relatively small list of potential chemical formulas for matching but is insufficient for identification without the addition of spectral matching to isotope pattern of the mass spectrum41. From high resolution MS1 data, one or more putative chemical formulas are matched against the isotopic fingerprint of the mass spectrum and scored (Figure 8). Formulas for matching can be generated ab initio using a defined pool of atoms or can be sourced from a combination of literature reported compounds and the contents of one or more databases. The US EPA Chemistry Dashboard (https://comptox.epa.gov/dashboard/) hosts a constantly updated list of PFAS compounds identified by the agency, as well as lists compiled by other organizations such as the NORMAN Network42.
Chemical formulas can be further confirmed, and some structural information can be garnered from MS/MS spectra (Figure 9). Candidate structures are available from large chemical databases such as the EPA chemistry dashboard, Pubchem, the CAS registry, etc. Predicted spectra can be generated or acquired using a variety of fragmentation programs and assigned,43 or MS/MS spectra can be interpreted manually.
An example data matrix is available in the Supplemental Information containing a whole feature matrix from ten samples (5 upstream, 5 downstream) collected upstream and downstream of a fluorochemical point source. Each row represents a chemical feature with associated retention time, neutral mass, mass spectrum, and raw abundance for each sample. (Supplemental Table, Sheet 1). Initial filtering (Supplemental Table, Sheet 2) for negative mass defect and statistical significance in an unpaired t-test between upstream and downstream reduces the number of "interesting" chemical features to ~120. Predicted chemical formulae were obtained from Agilent IDBrowser and searched against the EPA Comptox Chemicals Dashboard, which returned possible matches (Supplemental Table, Sheet 3). The "top-hit" for each chemical formula based on data sources37 was assigned (Supplemental Table, Sheet 4). Note that more than half of the remaining features do not have high quality matches. Identified features with no matches can be the result of in-source fragmentation/adduct formation, poor formula assignment, or the identification of PFASs not found in the source database. Interpretation of the raw spectra in order to validate assignments is beyond the scope of this manuscript but more information can be found in the works cited15,30,31,44,45.
| ID | Sample Name | Sample Type | Std Conc | Vial | LC Method | MS Method |
| 1 | DB_001 | Blank | 1:A,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 2 | DB_002 | Blank | 1:A,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 3 | DB_003 | Blank | 1:A,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 4 | DB_004 | Blank | 1:A,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 5 | DB_005 | Blank | 1:A,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 6 | FB | Blank | 1:A,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 7 | 10 std | Standard | 10 | 1:A,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 8 | 25 std | Standard | 25 | 1:A,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 9 | 50 std | Standard | 50 | 1:A,5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 10 | 100 std | Standard | 100 | 1:A,6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 11 | 250 std | Standard | 250 | 1:A,7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 12 | 500 std | Standard | 500 | 1:A,8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 13 | 750 std | Standard | 750 | 1:B,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 14 | 1000 std | Standard | 1000 | 1:B,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 15 | DB_006 | Blank | 1:B,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 16 | SB_DUP1 | Analyte | 1:B,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 17 | SB_DUP2 | Analyte | 1:B,5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 18 | SW Site 03 | Analyte | 1:B,6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 19 | SW Site 16 | Analyte | 1:B,7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 20 | SW Site 30 | Analyte | 1:B,8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 21 | DB_007 | Analyte | 1:C,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 22 | SW Site 19 | Analyte | 1:C,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 23 | SW Site 48 | Analyte | 1:C,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 24 | SW Site 49 | Analyte | 1:C,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 25 | SW Site 05 | Analyte | 1:C,5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 26 | SW Site 47 | Blank | 1:C,6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 27 | DB_008 | Analyte | 1:C,7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 28 | SW Site 19_DUP | Analyte | 1:C,8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 29 | SW Site 20 | Analyte | 1:D,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 30 | SW Site 21 | Analyte | 1:D,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 31 | SW Site 46 | Analyte | 1:D,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 32 | SW Site 47 | Analyte | 1:D,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 33 | DB_009 | Blank | 1:D,5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 28 | SW Site 32 | Analyte | 1:D,6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 29 | SW Site 50 | Analyte | 1:D,7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 30 | SW Site 25 | Analyte | 1:D,8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 31 | SW Site 21_DUP | Analyte | 1:E,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 32 | SW Site 52 | Analyte | 1:E,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 33 | DB_010 | Blank | 1:E,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 34 | FB | Blank | 1:A,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 35 | 10 std | Standard | 10 | 1:A,3 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 36 | 25 std | Standard | 25 | 1:A,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 37 | 50 std | Standard | 50 | 1:A,5 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 38 | 100 std | Standard | 100 | 1:A,6 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 39 | 250 std | Standard | 250 | 1:A,7 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 40 | 500 std | Standard | 500 | 1:A,8 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 41 | 750 std | Standard | 750 | 1:B,1 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 42 | 1000 std | Standard | 1000 | 1:B,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
| 43 | DB_011 | Blank | 1:B,2 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min | |
| 44 | DB_012 | Blank | 1:E,4 | PFAS grad 400uL/min - 9 min run | PFCMXA + HFPO-DA MS/MS - 9 min |
Table 1: Example worklist for Targeted Analysis and quantitation of PFAS using LC-MS/MS
| Time (min) 0 | % A (2.5mM Ammonium Acetate in 5% MeOH) 90 | % B (2.5mM Ammonium Acetate in 95% MeOH) 10 |
| 5 | 15 | 85 |
| 5.1 | 0 | 100 |
| 7 | 0 | 100 |
| 7.1 | 90 | 10 |
| 9 | 90 | 10 |
Table 2: Example gradient for LC separation in targeted analysis
| Capilary Voltage (kv) | 1.97 |
| Cone Voltage (V) | 15 |
| Extractor Voltage (V) | 3 |
| RF Lens (V) | 0.3 |
| Source Temp | 150 |
| Desolvation Temp | 40 |
| Desolvation Gas Flow (L/hr) | 300 |
| Cone Gas Flow (L/hr) | 2 |
Table 3: Ionization source parameters for targeted analysis
| Cmp | Precursor | Product | Dwell Time | Cone Voltage (V) | Collision Energy (eV) |
| PFBA | 212.80 | 168.75 | 0.01 | 15 | 10 |
| 13C4-PFBA IS | 216.80 | 171.75 | 0.01 | 15 | 10 |
| PFPeA | 262.85 | 218.75 | 0.01 | 15 | 9 |
| PFBS °1 | 298.70 | 79.90 | 0.01 | 40 | 30 |
| PFBS °2 | 298.70 | 98.80 | 0.01 | 40 | 28 |
| PFHxA °1 | 312.70 | 118.70 | 0.01 | 13 | 21 |
| PFHxA °2 | 312.70 | 268.70 | 0.01 | 13 | 10 |
| 13C2-PFHxA IS | 314.75 | 269.75 | 0.01 | 13 | 9 |
| HFPO-DA 1° | 329.16 | 168.90 | 0.01 | 10 | 12 |
| HFPO-DA 2° | 329.16 | 284.90 | 0.01 | 10 | 6 |
| HFPO-DA IS 1° | 332.16 | 168.90 | 0.01 | 10 | 12 |
| HFPO-DA IS 2° | 332.16 | 286.90 | 0.01 | 10 | 6 |
| PFHpA °1 | 362.65 | 168.65 | 0.01 | 14 | 17 |
| PFHpA °2 | 362.65 | 318.70 | 0.01 | 14 | 10 |
| PFHxS °1 | 398.65 | 79.90 | 0.01 | 50 | 38 |
| PFHxS °2 | 398.65 | 98.80 | 0.01 | 50 | 32 |
| 13C4-PFHxS IS | 402.65 | 83.90 | 0.01 | 50 | 38 |
| PFOA °1 | 412.60 | 168.70 | 0.01 | 15 | 18 |
| PFOA °2 | 412.60 | 368.65 | 0.01 | 15 | 11 |
| 13C4-PFOA IS | 416.75 | 371.70 | 0.01 | 15 | 11 |
| PFNA °1 | 462.60 | 218.75 | 0.01 | 15 | 17 |
| PFNA °2 | 462.60 | 418.60 | 0.01 | 15 | 11 |
| PFNA IS | 467.60 | 422.60 | 0.01 | 15 | 11 |
| PFOS °1 | 498.65 | 79.90 | 0.01 | 60 | 48 |
| PFOS °2 | 498.65 | 98.80 | 0.01 | 60 | 38 |
| 13C4-PFOS IS | 502.60 | 79.70 | 0.01 | 60 | 48 |
| PFDA °1 | 512.60 | 218.75 | 0.01 | 16 | 18 |
| PFDA °2 | 512.60 | 468.55 | 0.01 | 16 | 12 |
| 13C2 - PFDA IS | 514.60 | 469.55 | 0.01 | 16 | 12 |
Table 4: Example transition table and MS/MS parameters for the contents of PFAC-MXA, along with HFPO-DA
| Time (min) | % A (2.5mM Ammonium Acetate in 5% MeOH) | % B (2.5mM Ammonium Acetate in 95% MeOH) |
| 0 | 90 | 10 |
| 0.5 | 90 | 10 |
| 3 | 50 | 50 |
| 3.5 | 50 | 50 |
| 5.5 | 40 | 60 |
| 6 | 40 | 60 |
| 7 | 0 | 100 |
| 11 | 0 | 100 |
Table 5: Example gradient for LC separation in non-targeted analysis
| Profinder Parameter | Setting Value |
| Extraction Peak Height Filter | 800 counts |
| Permitted Ion(s) | -H/+H |
| Feature Extraction Isotope Model | Common organic molecules |
| Allowed Charge States | 2-Jan |
| Compound Ion Count Threshold | Two or more ions |
| Alignment RT Tolerance | 0.40min + 0.0% |
| Alignment Mass Tolerance | 20.00ppm + 2.0mDa |
| Post-Processing Absolute Height Filter | >= 10000 counts in one sample |
| Post-Processing MFE Score Filter | >= 75 in one sample |
| Peak Integration Algorithm | Agile 2 |
| Peak Integration Height Filter | >= 5000 counts |
| Find by Ion Absolute Height Filter | >= 7500 counts in one sample |
| Find by Ion Score Filter | >= 50.00 in one sample |
Table 6: Molecular feature extraction and alignment settings for Profinder software. All unlisted values retained their default settings for data processing.
| Ion Abundance Threshold | Feature Thresholds | Replicate Threshold (n =5) | Run Time | Features | Pass Replicate Threshold | Pass CV Threshold | Features to 90% of TIC |
| 1x S/N | 2000 | None | 8.15 | 987 | 505 | 421 | 91 |
| 2x S/N | 5000 | None | 5.02 | 707 | 357 | 313 | 93 |
| 3x S/N | 10000 | None | 2.3 | 308 | 249 | 230 | 93 |
| 1x S/N | 2000 | 100% | 3.3 | 603 | 339 | 297 | 92 |
| 2x S/N | 35000 | 100% | 1.58 | 310 | 248 | 229 | 93 |
| 3x S/N | 10000 | 100% | 1.45 | 202 | 190 | 182 | 92 |
Table 7: Comparison of sample processing time and chemical feature identifications for different feature extraction thresholds.

Figure 1: Total ion chromatogram and extracted ion chromatograms for a subset of perfluorinated ether standards. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative calibration curves for compounds demonstrating decreasing quality of analytical curve construction. Left-most panel indicates a high quality calibration; Middle panel indicates a compound with poor precision across preparation duplicates, particularly at the higher concentrations; Right Panel indicates a curve with poor precision and a low linear dynamic range, resulting in flat response at the high end of the calibration range, and no detectable signal at the lower end. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Overlaid total ion chromatograms (TIC) for surface water extracts collected upstream and downstream of a fluorochemical production site. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Extracted ion chromatograms (EIC) for all identified chemical features from a surface water sample containing multiple fluorochemical classes. Each chemical trace is a different color for differentiation. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Conceptual diagram of raw and predicted information for a chemical feature identified as hexafluoropropylene oxide dimer acid (HFPO-DA). Chemical features are compiled from software extraction of raw data from MS measurements and contain chromatographic (e.g., retention time (RT)) and mass spectrometry information. Predicted formula, structures, and chemical identities are generated from raw measurement data for each feature. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Mass defect plot for chemical features identified in a manufacturing outfall (red, left) and reference surface water (blue, right). Fluorinated compounds fall near and below the dashed zero line. Note the persistent PFOA/PFOS series in the background surface water sample (right). Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Mass vs mass defect plot for unidentified chemical features from a surface water sample with homologous series identified and labeled by the nontarget R package. Please click here to view a larger version of this figure.
{kind=link}

Figure 8: Mass spectrum of an unknown chemical features with predicted isotopic intensities of three possible chemical formula with the same monoisotopic mass. Please click here to view a larger version of this figure.
{kind=link}

Figure 9: Fragmentation spectrum of a perfluorinated ether compound with annotated fragment peaks. Please click here to view a larger version of this figure.
{kind=link}

Figure 10: Graphical representation of filtering thresholds. From left to right, ion abundance threshold for chemical feature mass spectra, feature abundance threshold for extracted chromatographic features, and replicate threshold for feature detection frequency in a triplicate injection experiment. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Sample Handling and Preparation
The inclusion of reference/spike standards are of paramount importance to any targeted analysis, as they provide a backstop for checking analytical validity. Lack of QC samples prevents any assessment of the accuracy of the results; the ubiquitous nature of fluorochemicals means that chance contamination of field samples, processing materials, or LC-MS system is not uncommon and must be accounted for. Further, it allows for the validation of the protocol regardless of variation in the day-to-day sample processing, as many of the steps can be highly variable, particularly the SPE and sample concentration steps. The extraction of both legacy and novel perfluorinated chemicals can be heavily influenced by the choice of stationary phase for concentration, and components of the source samples, such as pH and salinity46. The influence of sample conditions should be considered if particular classes of pefluorinated chemicals are of interest. Alternative sample preparation schemes for water extracts can be used if the laboratory setup is available and the downstream data analysis remains similar.
Targeted Data Analysis
For compounds with available standards and matched, stable isotope labeled internal standards, the primary concerns for data analysis are instrumental and determination of method detection limits and suitable reporting ranges can be determined on a laboratory-by-laboratory basis using standard approaches, such as signal-to-noise ratio from low-level standard spikes47. In the absence of matched internal standards errors from mismatched matrix effects can occur, and accurate back-prediction of spiked samples can be used to estimate the accuracy of the measurements. When lacking standards to prepare a curve, a quantitative estimate of an unknown can be made by treating it identically to a closely matched standard compound, but errors in the estimate are on the order of 10+ fold with limited ability to quantify the uncertainty, see McCord, Newton, and Strynar21. In these cases, trend data can still be collected, but concentration estimates are inherently unreliable.
Non-targeted Data Analysis
Peak picking settings have a substantial impact on the number of chemical features identified, but the quality of feature selection is also heavily impacted. The decisions of interest in peak picking are 1) intensity of individual masses to be included in spectra, the ion abundance threshold 2) the intensity of extracted chromatogram peaks to be considered features, the feature abundance threshold 3) feature detection frequency, the replicate threshold, and 4) analytical variation, the CV threshold (Figure 10).
Setting unrealistically low thresholds for peak picking results in an exponential increase in sample time to resolve additional features of increasingly low abundance (Table 7). The ion-abundance threshold filters mass spectral features where enough of the individual isotope abundances do not pass the threshold. This ideally selects only for features with quality MS spectra, ensuring they are real chemical features rather than instrumental noise, and allowing for formula prediction in downstream processing. An appropriate threshold is based on instrumental noise, ideally at least 3x the noise threshold for MS1 scans. Feature abundance threshold filters chemical features based on the intensity or area of the chromatographic feature extracted. This step enables rejection of low abundance peaks, which are typically of poor chromatographic quality, have high variances, or are the result of other poor software extraction. An appropriate threshold must be determined per experiment, and per matrix based on an acceptable level of poor feature generation (e.g., features below the threshold exhibit unacceptably poor chromatography). Further analytical QC can be used to reject features at the chromatographic level based on inconsistent identification in analytical and/or preparatory replicates (replicate threshold) or based on poor reproducibility across replicates (CV threshold). Appropriate levels depend on the quality of the peak integration software used and the chemical entities under investigation. For water soluble perfluorinated compounds and lightly optimized integration protocols, features should be identified in 80+% of analytical replicates and CVs are expected to fall below 30%, as detailed in the methods section.
The peaks detected from non-targeted analysis do not yield quantitative estimates of the concentrations of the materials detected. Further, the identity of true unknowns can be difficult to confirm because novel compounds are absent from publicly available databases. Novel structural determination requires extensive analysis with multiple methods and requires expertise in both mass spectrometry and chemistry. However, normalizing the peak areas of chemical features can provide semi-quantitative estimates of concentrations of unknowns from known species21. If consistent sampling and preparation steps are employed, time trend information for individual species can be generated to monitor the persistence of a chemical into the future as the response for an individual species should be consistent barring large variations in the matrix21.
The primary benefit of this method is the extensibility of the sample treatment to allow both targeted and nontargeted analysis. While targeted analysis provides equivalent or superior quantitative information, it greatly lacks breadth of analysis desired when dealing with new and emerging materials, as well as their relationship to matrix materials. Applying a targeted methodology, or even a suspect screening method based only on known materials and limited databases is completely blind to previously unobserved species, even if they may have significant health effects. As software improves and databases become more robust, the accuracy of unknown identification will continue to increase, with a concomitant decrease in the time investment and level of expertise necessary to analyze the multidimensional data generated by this approach. Nevertheless, data generated presently is of significant future value because data banking allows for post-hoc analysis with newly developed software and enables comparison across time even if the identity of a detected compound is currently unknown.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The U.S. Environmental Protection Agency, through its Office of Research and Development, funded and managed the research described here. This document has been reviewed by the U.S. Environmental Protection Agency, Office of Research and Development, and approved for publication. The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. This research was supported in part by an appointment to the Postdoctoral Research Program at the National Exposure Research Laboratory administered by the Oak Ridge Institute for Science and Education through Interagency Agreement DW89992431601 between the U.S. Department of Energy and the U.S. Environmental Protection Agency.
Materials
| Name | Company | Catalog Number | Comments |
| Acqity ultra-high performance liquid chromatography system | Waters Corporation | Modified with PFCs analysis kit (176001744); equivalent UPLC system is acceptible if PFAS background is checked and confirmed to be low | |
| Ammonium acetate | Fluka | 17836 | Mass spectrometry grade >99% pure |
| Ammonium Hydroxide | Sigma-Aldrich | 338818 | |
| Balance | Mettler | AB204S | |
| BEH C18 reverse phase UPLC column, 2.1×50 mm, 1.7 μm | Waters Corporation | 186002350 | |
| Dual piston syringe pump | Waters Corporation | SPC10-C | |
| Glacial Acetic Acid | Sigma-Aldrich | ARK2183 | |
| Glass Microfiber Filters | Whatman | 1820-070 | |
| High density polyethelye sample bottle | Nalgene | 2189-0032 | |
| High Resolution Mass Spectrometer | Various | Mass Spectrometer should be capable of providing accurate mass to <10ppm and collecting MS/MS data. Agilent 6530 qTOF and Thermo Fisher Orbitrap Fusion were used in this work | |
| Methanol | Sigma-Aldrich | ||
| Nitric Acid (35% w/w) | Thermo Fisher Scientific | SVCN-5-1 | Can be prepared in house using concentrated nitric acid and reagent water |
| Polypropylene Buchner funnel | ACE Glass | 12557-09 | |
| Polypropylene cenitrfuge tube and cap | BD Falcon | 352096 | |
| Polypropylene Vacuum Flask (1 L) | Nalgene | DS4101-1000 | |
| Quattro Premier XE triple quadrupole mass spectrometer | Waters Corporation | Equivalent triple-quadrupole or better system can be used instead, should provide high sensitivity and stability for targeted analysis | |
| Reagent Water | Any source determined to be PFAS free | ||
| Sodium Acetate | Sigma-Aldrich | W302406 | |
| TurboVap nitrogen evaporator | Caliper Life Sciences | 103198 | Equivalent systems or rotary vacuum evaporator may be used instead |
| Weak anion exchange SPE cartridge (Oasis WAX Plus) | Waters Corporation | 186003519 | |
| Standard Solutions | |||
| 2,3,3,3-Tetrafluoro-2-(1,1,2,2,3,3,3-heptafluoropropoxy)propanoic acid (HFPO-DA) | Wellington | HFPO-DA | |
| Additional targeted compound standards of interest | to be determined based on preliminary analysis and standard availability | ||
| Mass labeled HFPO-DA | Wellington | M2HFPO-DA | |
| Native PFCA/PFAS Mixture (2 ug/mL) | Wellington | PFAC-MXA | or PFAC-MXB; or individually prepared mixture containing compounds of interest |
| Stable Isotope Labeled PFCA/PFAS Mixture (2 ug/mL) | Wellington | MPFAC-MXA | or MPFAC-MXB; or individually prepared mixture containing compounds of interest as appropriate for Native PFASs |
| Software | |||
| Mass Profiler Professional | Agilent | Or open source software packages | |
| Profinder | Agilent | Or open source software packages |
References
- . Provisional Health Advisories for Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS). United States Environmental Protection Agency. , (2009).
- . Lifetime Health Advisories and Health Effects Support Documents for Perfluorooctanoic Acid and Perfluorooctane Sulfonate. United States Environmental Protection Agency. , 33250-33251 (2016).
- . Fact Sheet: 2010/2015 PFOA Stewardship Program Available from: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-20102015-pfoa-stewardship-program (2006)
- EPA and 3M Announce phase out of PFOS. Environmental Protection Agency Available from: https://yosemite.epa.gov/opa/admpress.nsf/0/33aa946e6cb11f35852568e1005246b4 (2000)
- Wang, Z., Cousins, I. T., Scheringer, M., Hungerbühler, K. Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors. Environment International. 60, 242 (2013).
- Scheringer, M., et al. Helsingør Statement on poly- and perfluorinated alkyl substances (PFASs). Chemosphere. 114, 337-339 (2014).
- Wang, Z., DeWitt, J. C., Higgins, C. P., Cousins, I. T. A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs). Environmental Science & Technology. 51 (5), 2508-2518 (2017).
- Xiao, F., Golovko, S. A., Golovko, M. Y. Identification of novel non-ionic, cationic, zwitterionic, and anionic polyfluoroalkyl substances using UPLC-TOF-MSE high-resolution parent ion search. Analytica Chimica Acta. 988, 41-49 (2017).
- Shoemaker, J., Grimmett, P., Boutin, B. Method 537. Determination of selected perfluorinated alkyl acids in drinking water by solid phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS). US Environmental Protection Agency. , (2009).
- Poole, C. F., Gunatilleka, A. D., Sethuraman, R. Contributions of theory to method development in solid-phase extraction. Journal of Chromatography A. 885 (1), 17-39 (2000).
- Ahrens, L. Polyfluoroalkyl compounds in the aquatic environment: a review of their occurrence and fate. Journal of Environmental Monitoring. 13 (1), 20-31 (2011).
- Higgins, C. P., Field, J. A., Criddle, C. S., Luthy, R. G. Quantitative Determination of Perfluorochemicals in Sediments and Domestic Sludge. Environmental Science & Technology. 39 (11), 3946-3956 (2005).
- Szostek, B., Prickett, K. B., Buck, R. C. Determination of fluorotelomer alcohols by liquid chromatography/tandem mass spectrometry in water. Rapid Communications in Mass Spectrometry. 20 (19), 2837-2844 (2006).
- Alzaga, R., Bayona, J. M. Determination of perfluorocarboxylic acids in aqueous matrices by ion-pair solid-phase microextraction-in-port derivatization-gas chromatography-negative ion chemical ionization mass spectrometry. Journal of Chromatography A. 1042 (1-2), 155-162 (2004).
- Schaider, L. A., et al. Fluorinated Compounds in U.S. Fast Food Packaging. Environmental Science & Technology Letters. 4 (3), 105-111 (2017).
- Lindstrom, A. B., Strynar, M. J., Libelo, E. L. Polyfluorinated Compounds: Past, Present, and Future. Environmental Science & Technology. 45 (19), 7954 (2011).
- Liu, Y., Pereira, A. D. S., Martin, J. W. Discovery of C5-C17 Poly-and Perfluoroalkyl Substances in Water by In-Line SPE-HPLC-Orbitrap with In-Source Fragmentation Flagging. Analytical Chemistry. 87 (8), 4260 (2015).
- Backe, W. J., Day, T. C., Field, J. A. Zwitterionic, Cationic, and Anionic Fluorinated Chemicals in Aqueous Film Forming Foam Formulations and Groundwater from U.S. Military Bases by Nonaqueous Large-Volume Injection HPLC-MS/MS. Environmental Science & Technology. 47 (10), 5226-5234 (2013).
- Mazzoni, M., Rusconi, M., Valsecchi, S., Martins, C. P. B., Polesello, S. An on-line solid phase extraction-liquid chromatography-tandem mass spectrometry method for the determination of perfluoroalkyl acids in drinking and surface waters. Journal of Analytical Methods in Chemistry. 2015, 942016 (2015).
- Li, F., et al. Method development for analysis of short- and long-chain perfluorinated acids in solid matrices. International Journal of Environmental Analytical Chemistry. 91 (12), 1117-1134 (2011).
- McCord, J., Newton, S., Strynar, M. Validation of quantitative measurements and semi-quantitative estimates of emerging perfluoroethercarboxylic acids (PFECAs) and hexfluoroprolyene oxide acids (HFPOAs). J Chromatoqr A. , (2018).
- Wang, Y., Liu, S., Hu, Y., Li, P., Wan, J. -. B. Current state of the art of mass spectrometry-based metabolomics studies - a review focusing on wide coverage, high throughput and easy identification. RSC Advances. 5 (96), 78728-78737 (2015).
- Cajka, T., Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Analytical Chemistry. 88 (1), 524-545 (2016).
- Mann, M., Kelleher, N. L. Precision proteomics: The case for high resolution and high mass accuracy. Proceedings of the National Academy of Sciences of the United States of America. 105 (47), 18132-18138 (2008).
- Sobus, J. R., et al. Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. Journal of Exposure Science & Environmental Epidemiology. , (2017).
- Bletsou, A. A., Jeon, J., Hollender, J., Archontaki, E., Thomaidis, N. S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trends in Analytical Chemistry. 66, 32-44 (2015).
- Viant, M. R., Sommer, U. Mass spectrometry based environmental metabolomics: a primer and review. Metabolomics. 9 (1), 144-158 (2013).
- Xiao, F. Emerging poly- and perfluoroalkyl substances in the aquatic environment: A review of current literature. Water Research. 124, 482-495 (2017).
- Nakayama, S. F., Strynar, M. J., Reiner, J. L., Delinsky, A. D., Lindstrom, A. B. Determination of perfluorinated compounds in the Upper Mississippi River Basin. Environmental Science & Technology. 44 (11), 4103 (2010).
- Strynar, M., et al. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environmental Science & Technology. 49 (19), 11622 (2015).
- Newton, S., et al. Novel Polyfluorinated Compounds Identified Using High Resolution Mass Spectrometry Downstream of Manufacturing Facilities near Decatur, Alabama. Environmental Science & Technology. 51 (3), 1544-1552 (2017).
- Forsberg, E. M., et al. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nature Protocols. 13, 633 (2018).
- Sturm, M., et al. OpenMS - An open-source software framework for mass spectrometry. BMC Bioinformatics. 9 (1), 163 (2008).
- Kind, T., Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 8, 105-105 (2007).
- Loos, M., Singer, H. Nontargeted homologue series extraction from hyphenated high resolution mass spectrometry data. Journal of Cheminformatics. 9, 12 (2017).
- Dimzon, I. K., et al. High Resolution Mass Spectrometry of Polyfluorinated Polyether-Based Formulation. Journal of the American Society for Mass Spectrometry. 27, 309 (2016).
- McEachran, A. D., Sobus, J. R., Williams, A. J. Identifying known unknowns using the US EPA's CompTox Chemistry Dashboard. Analytical and Bioanalytical Chemistry. 409 (7), 1729-1735 (2017).
- French, W. R., et al. Wavelet-Based Peak Detection and a New Charge Inference Procedure for MS/MS Implemented in ProteoWizard's msConvert. Journal of Proteome Research. 14 (2), 1299-1307 (2015).
- Tautenhahn, R., Böttcher, C., Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 9, 504 (2008).
- Rafiei, A., Sleno, L. Comparison of peak-picking workflows for untargeted liquid chromatography/high-resolution mass spectrometry metabolomics data analysis. Rapid Communications in Mass Spectrometry. 29 (1), 119-127 (2015).
- Kind, T., Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics. 7, 234-234 (2006).
- Brack, W., Dulio, V., Slobodnik, J. The NORMAN Network and its activities on emerging environmental substances with a focus on effect-directed analysis of complex environmental contamination. Environmental Sciences Europe. 24 (1), 29 (2012).
- Blaženović, I., et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: database boosting is needed to achieve 93% accuracy. Journal of Cheminformatics. 9, 32 (2017).
- Rager, J. E., et al. Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environment International. 88, 269-280 (2016).
- Munoz, G., et al. Environmental Occurrence of Perfluoroalkyl Acids and Novel Fluorotelomer Surfactants in the Freshwater Fish Catostomus commersonii and Sediments Following Firefighting Foam Deployment at the Lac-Mégantic Railway Accident. Environmental Science & Technology. 51 (3), 1231-1240 (2017).
- Brumovský, M., Bečanová, J., Karásková, P., Nizzetto, L. Retention performance of three widely used SPE sorbents for the extraction of perfluoroalkyl substances from seawater. Chemosphere. 193, 259-269 (2018).
- Definition, Definition and Procedure for the Determination of the Method Detection Limit (Revision 2). Environmental Protection Agency. , (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved