
Method Article
Perfil proteómico de EPS-Urine a través de la Digestión FASP y análisis independiente de datos
En este artículo
Resumen
Aquí, presentamos un protocolo de digestión en filtro optimizado con información detallada sobre lo siguiente: digestión de proteínas, purificación de péptidos y análisis de adquisición independiente de datos. Esta estrategia se aplica al análisis de muestras de secreciones prostáticas-orina expresadas y permite una alta cobertura de proteoma y un perfil sin etiquetas de bajo valor del proteoma urinario.
Resumen
El protocolo de muestra asistido por filtros (FASP) es ampliamente utilizado para la preparación de muestras de proteómica porque permite concentrar muestras diluidas y es compatible con una amplia variedad de detergentes. Los flujos de trabajo proteómicos ascendentes, como FASP, dependen cada vez más de los métodos LC-MS/MS realizados en modo de análisis independiente de datos (DIA), un método de escaneo que permite una cobertura profunda del proteoma y una baja incidencia de valores que faltan.
En este informe, proporcionaremos los detalles de un flujo de trabajo que combina un protocolo FASP, un paso de purificación de doble punta de escenario y LC-MS/MS en modo DIA para la cartografía de proteoma urinario. Como muestra modelo, analizamos las secreciones prostáticas expresadas (EPS) -orina, una muestra recogida después de un examen rectal digital (DRE), que es de interés en los estudios de descubrimiento de biomarcadores de cáncer de próstata.
Introducción
La evolución constante de las tecnologías proteómicas promete tener un impacto considerable en ayudar al diagnóstico de la enfermedad y la predicción de la respuesta al tratamiento proporcionando mapas de alta resolución de los factores clave de efectores moleculares presentes en una amplia variedad de muestras como tejidos y biofluidos. Desde el punto de vista analítico, la orina ofrece varias ventajas como la facilidad de recolección y la estabilidad importante del proteoma con respecto a otros biofluidos1. El análisis proteómico de la orina es de especial interés en estudios de descubrimiento de biomarcadores sobre cánceres urológicos, ya que permite el muestreo no invasivo en proximidad a los tejidos de interés2. En particular, una muestra que parece ser prometedora para estudiar patologías relacionadas con la próstata es la EPS-orina3,4 (es decir, una muestra de orina recogida después de un examen rectal digital (DRE)). Esta última operación antes de la recolección de muestras enriquece la orina con proteínas específicas de la próstata. La EPS-orina es un buen candidato para investigar trastornos relacionados con la glándula prostática5, incluido el cáncer de próstata (PCa), ya que a través de DRE, las proteínas secretadas por el tumor se pueden verter en la muestra de orina, aumentando la posibilidad de detectar proteínas específicas del tejido canceroso.
Un papel crucial en permitir la detección y cuantificación de biomarcadores potenciales de proteínas es desempeñado por espectrometría de masas (EM). En las últimas dos décadas, los protocolos basados en LA para el análisis proteómico han permitido detectar un número cada vez mayor de proteínas en una sola ejecución de LC-MS/MS gracias a mejoras continuas en la instrumentación de EM y en el software de análisis de datos6.
La preparación de muestras proteómicas basada en MS generalmente implica digestión enzimática de la mezcla proteica, que se puede lograr a través de una amplia variedad de protocolos tales como: digestión en solución, MStern blotting7,captura de suspensión (trampa de S)8,preparación de muestras mejorada en fase sólida (SP3)9,digestión en etapa10 y preparación de muestras asistida por filtro (FASP)11. Todos los protocolos se pueden utilizar para la proteómica urinaria, aunque los resultados pueden variar con respecto al número de proteínas y péptidos identificados y en términos de reproducibilidad12.
En este trabajo, nuestra atención se centró en el análisis de eps-orina por el protocolo FASP. El protocolo FASP fue diseñado originalmente para analizar proteínas extraídas de tejidos y cultivos celulares, pero su uso se amplió al análisis de otros tipos de muestras, como la orina13. Con respecto a la digestión directa en solución FASP es un enfoque proteómico más flexible14,ya que al lograr la eliminación efectiva de detergentes y otros contaminantes como sales de la mezcla proteica antes de la digestión enzimática15,permite la elección de condiciones óptimas de solubilización de proteínas. Además, una característica adicional de FASP es que proporciona un medio para la concentración de muestras. Esto es de particular interés para el análisis proteómico urinario, ya que permite comenzar a partir de volúmenes de muestra relativamente grandes (cientos de microlitros). A la luz del potencial del protocolo FASP, varios estudios han centrado la atención en la automatización del flujo de trabajo, con el objetivo de reducir la variabilidad experimental y procesar un número elevado de muestras en paralelo16.
En nuestro flujo de trabajo, FASP es seguido por la adquisición de LC-MS/MS en el análisis independiente de datos (DIA), que proporciona una alta cobertura de proteoma, una buena precisión cuantitativa y una baja incidencia de valores que faltan. El enfoque DIA es un método confidencial donde se seleccionan todos los iones para eventos MS/MS, opuestos a lo que sucede en el análisis dependiente de datos (DDA) donde solo se fragmentan los iones con la intensidad más alta. El espectrómetro de masas, que funciona en modo DIA, realiza ciclos de escaneo con diferentes anchos de aislamiento que cubren toda la gama de precursores m/z. Este enfoque permite detectar reproduciblemente un alto número de péptidos por unidad de tiempo, proporcionando una instantánea proteómica de la muestra17. Además, los datos generados por DIA tienen otra característica interesante: la posibilidad de un análisis posteriori 18. Los datos DIA son más complejos que los obtenidos por DDA, ya que los espectros de EM/EM en DIA resultan del co-aislamiento de varios iones precursores dentro de cada ventana m/z 19. La desenredada de los espectros compuestos de EM/EM en señales de péptidos distintas y específicas se logra utilizando dos elementos fundamentales: una biblioteca espectral y un software dedicado para el análisis de datos. La biblioteca espectral es generada por un experimento dependiente de los datos, que generalmente implica fraccionamiento de péptidos para maximizar la cobertura de proteoma, que proporciona una lista de miles de iones precursores determinados experimentalmente y espectros de MS/EM de péptidos detectables en la muestra en consideración. El software de análisis de datos, en su lugar, utiliza la información contenida en la biblioteca espectral para interpretar los datos DIA mediante la generación de cromatogramas de iones extraídos específicos que permiten la detección y cuantificación de péptidos. Si bien el análisis de datos DIA sin bibliotecas es ahora factible, DIA basada en bibliotecas todavía proporciona mejores resultados en términos de cobertura de proteoma20.
El protocolo de preparación de muestras aquí descrito (Figura 1) consiste en los siguientes pasos: un paso de centrifugación (para eliminar los desechos celulares), digestión FASP, purificación de la punta de escenario21,cuantificación de proteínas y análisis DIA. Este protocolo ha sido diseñado para el análisis de EPS-orina en el contexto del descubrimiento del biomarcador de cáncer de próstata, pero se puede aplicar al análisis proteómico de cualquier muestra de orina.
Protocolo
El estudio fue aprobado por el Comité Ético Institucional de la Universidad Magna Graecia de Catanzaro, RP 41/2018. Se obtuvo el consentimiento informado por escrito de todos los pacientes inscritos en el estudio.
1. Preparación de muestras
- Centrífuga muestras de EPS-orina dentro de 2 h de recolección a 2.100 x g durante 10 minutos a temperatura ambiente (RT). Guarde el sobrenadante a -80 °C hasta su uso.
2. Descongelación de muestras
- Transfiera la muestra de -80 °C a -20 °C el día anterior a la digestión.
- Antes del procesamiento de muestras, transfiera la muestra durante 15-20 minutos a 4 °C y luego a RT.
3. Preparación de reactivos para fasp
- El mismo día, prepare las siguientes soluciones: búfer de urea, iodoacetamida de 50 mM (IAA) en búfer de urea, bicarbonato de trietillammonio de 50 mM y ditiotiotioteitol de 50 mM (TDT).
- Preparar el buffer de urea (urea 8 M, 100 mM Tris-HCl pH 8): para 10 ml, pesar 4.800 mg de urea y disolverlo en la cantidad adecuada de agua HPLC después de añadir 1 ml de 1 M Tris pH 8. Se necesita un volumen de 800 μL de urea para cada muestra.
- Preparar 500 mM dithiothreitol (TDT): disolver 38,5 mg de TDT en 500 μL de agua HPLC. Para cada muestra, se necesitan 66,7 μL de TDT.
- Preparar 50 mM IAA en Urea Buffer: disolver 9,25 mg de AIEA en 1 ml de búfer de urea. Para cada muestra se necesitan 50 μL de AIEA.
- Preparar bicarbonato de trietillammonio de 50 mM (TEAB). Añadir 150 μL de 1 M TEAB a 2,85 ml de agua HPLC. Para cada muestra se necesitan 460 μL de TEAB.
- Preparar una solución de trippsina (100 ng/μL) en agua HPLC. Esta solución se puede almacenar a -80 °C y descongelarse inmediatamente antes de su uso. Para cada muestra, se necesitan 2 μL (200 ng).
4. Digestión de proteínas por FASP
- Diluir 500 μL de cada muestra de EPS-orina con 66,7 μL de sulfato de dodecilo sódico (SDS) del 10% (w/v), 66,7 μL de TDT de 500 mM y 33,3 μL de 1 M Tris pH 8 para solubilizar proteínas y reducir los enlaces de disulfuro. Incubar 10 minutos a 95 °C con suave agitación.
- Añadir 300 μL de muestra de EPS-orina diluida en la unidad de filtro (10 kDa) y centrífuga a 14.000 x g durante 20 minutos.
- Añadir 200 μL de búfer de urea al filtro y centrífuga a 14.000 x g durante 15 minutos. Repita este paso y, a continuación, descarte el flujo.
- Añadir 50 μL de solución y centrífuga del AIEA a 6.000 x g durante 25 minutos.
- Añadir 200 μL de búfer de urea y centrífuga a 14.000 x g durante 20 minutos. Repita este paso y, a continuación, descarte el flujo.
- Añadir 200 μL de 50 mM trillalammonium bicarbonato buffer (TEAB) y centrífuga a 14.000 x g durante 20 minutos. Repita este paso.
- Transfiera la unidad de filtro a un nuevo tubo de recogida.
- Añadir 60 μL de 50 mM de tampón TEAB y 200 ng de trippsina e incubar la muestra en un termo-mezclador a 37 °C durante la noche.
NOTA: Para evitar la evaporación de la muestra durante la incubación nocturna, envuelva cada unidad de filtro con una capa de papel de aluminio y luego una capa de parafilm. - Después de la incubación de trypsina, añadir 140 μL de agua y centrifugar el vial a 14.000 x g durante 25 minutos para recoger el volumen de digestión (180-200 μL).
5. Preparación de reactivos para la purificación SCX
- Preparar 1 ml de lavado 1 (0,5% ácido fórmico (FA) y 20% acetonítrilo (ACN)) de la siguiente manera: añadir 50 μL de 10% FA y 200 μL de 100% ACN a 750 μL de agua HLPC. Para cada muestra, se necesitan 100 μL de lavado 1.
- Preparar 1,5 ml de lavado 2 (0,5% FA y 80% de ACN) de la siguiente manera: añadir 75 μL del 10 % FA y 1,2 ml de 100% ACN a 225 μL de agua HPLC. Para cada muestra se necesitan 190 μL de lavado 2.
- Preparar 100 μL de solución de eluting (acetato de amonio de 500 mM (AA) y 20% de ACN): añadir 25 μL de 2 M AA y 20 μL de 100% ACN a 55 μL de agua HPLC.
- Prepare las sugerencias de etapas de la siguiente manera.
- Empaquete una punta de pipeta de 200 μL con una capa de resina SCX con una aguja de jeringa de extremo contundente (calibre 16). Inserte la punta de escenario en una tapa de microcentrífuga que haya sido perforada previamente para acomodar la microcolumn.
- Monte la tapa en una microcentrífuga de 2 ml de la que se extrajo la tapa original. El dispositivo centrífugo micro-SPE que acaba de montarse debe caber en una centrífuga de sobremesa. Dado que las tapas perforadas son tediosas para prepararse, se pueden utilizar varias veces.
6. Purificación SCX
- Diluir 30 μL de cada muestra a 120 μL usando lavado 2.
- Condicionar la punta de escenario añadiendo 50 μL de lavado 1 encima de la resina de extracción y centrífuga a 400 x g durante 2 minutos. Dado que la resistencia al flujo depende de la fuerza con la que la resina de extracción se embaló en la punta de la pipeta, es posible que sea necesario ajustar la velocidad de la centrífuga para obtener un caudal de 20-30 μL/min. Trate de no dejar la punta seca durante la carga y el lavado de la muestra.
- Equilibrar la punta de escenario con 50 μL de lavado 2 y centrífuga a 400 x g durante 2 minutos.
- Cargue la muestra diluida (120 μL) utilizando una velocidad de giro más baja.
- Lave la punta de escenario con 50 μL de lavado 2 y centrífuga a 400 x g durante 2 minutos. Repita con 50 μL de lavado 1.
NOTA: Después de este lavado la resina debe estar completamente seca. - Mezcla de péptido eluto con 7 μL de solución de eluato (acetato de amonio de 500 mM (AA) y 20% de ACN) utilizando lentamente una velocidad de giro más baja.
- Añadir 27 μL de 0.1% FA para diluir AA y obtener una muestra para la inyección preliminar de LC-MS/MS. La dilución final a 34 μL dará lugar a una relación volumétrica de 1:1 entre la orina y la solución de péptido (1 μL de digestión purificada corresponderá a 1 μL de muestra de orina original).
7. Cuantificación de proteínas por norma externa mediante análisis DDA (Figura 2)
- Determine la cantidad de proteínas en las muestras inyectando 2 μL del resumen de péptidos resultante en LC-MS/MS e interpolando el área total resultante del péptido con una curva de calibración construida de la siguiente manera:
- Preparar cinco soluciones de resumen HeLa diferentes (1 ng/μL, 2,5 ng/μL, 7,5 ng/μL, 25 ng/μL, 75 ng/μL) utilizando un caldo de resumen HeLa (100 μg/μL).
- Inyecte cada solución de Hela en duplicado (2 μL).
- Establezca el método LC-MS/MS.
- Inyecte 2 μL de las cinco mezclas de péptidos HeLa y separe péptidos a través de un gradiente lineal de 75 minutos a un caudal de 230 nL/minuto en una columna i.d de 15 cm y 75 μm llena de partículas de sílice C18 de 3 μm. Utilice un degradado binario constituido por la fase móvil A (0,1% FA, 2% ACN) y la fase móvil B (0,1% FA y 80% de ACN).
- Realice la elución de gradiente aumentando el contenido de fase B móvil del 6% al 42% en 60 minutos y del 42% al 100% en 8 minutos adicionales. Mantenga durante 5 minutos 100% B y luego vuelva al 0% B en dos minutos.
- Opere en modo DDA utilizando un método top-12 y establezca el rango de escaneo completo de EM en 350-1800 m/z,resolución 70.000, objetivo ACG 1e6 y tiempo máximo de inyección de 50 ms. Ajuste la ventana de masa para el aislamiento de iones precursores en 1,6 m/z,resolución 35.000, objetivo ACG 1e5, tiempo máximo de inyección 120 ms, fragmentación de HCD a partir de 25 NCE (energía de colisión normalizada) y exclusión dinámica 15 segundos.
- Analice los archivos sin procesar utilizando Sequest como motor de búsqueda y la secuencia de proteínas COMPLETA HUMAN Uniprot como base de datos. En los experimentos presentados aquí, la base de datos se descargó en marzo de 2016 y contenía 42.013 secuencias.
- Utilice los siguientes ajustes: Tolerancia MS 15 ppm, TOLERANCIA MS/MS 0.02 Da, trypsin como enzima estableciendo dos sitios de escote perdidos. Establecer la carbamidometilación de lisina, serina, trionina, tirosina (+57.021) y la oxidación de las metioninas como modificaciones dinámicas y la carbamidometilación de cisteínas (+57.021) como modificaciones estáticas. Utilice el corte de la tasa de descubrimiento falso (FDR) para la identificación de péptidos y proteínas a 0,01 y filtrar por percolador.
- Calcule el área pico para todas las señales de péptido MS1. Calcule el área total de los péptidos.
- Inyecte 2 μL de mezclas de péptidos obtenidas de muestras de EPS-orina utilizando el mismo método LC-MS/MS utilizado para las muestras de HeLa y analice el archivo sin procesar utilizando los mismos parámetros utilizados para el procesamiento de datos HeLa.
- Calcule la intensidad total de las señales de péptido resumiendo los valores de área de todos los péptidos identificados e interpolando la curva estándar externa. Esto proporcionará una estimación aceptable de la cantidad de péptidos presentes en el resumen de proteínas EPS-orina.
8. Preparación de reactivos para el protocolo C18 StageTip
- Preparar 500 μL de la solución A (0,1% ácido trifluoroacético (TFA), 50% ACN): añadir 250 μL de 100% ACN y 10 μL de 5% de TFA a 240 μL de agua HPLC.
- Preparar 2 ml de solución B (0,1 % TFA): añadir 40 μL de 5% de TFA a 1960 μL de agua HPLC.
- Preparar 100 μL de solución de eluting (0,1% FA, 50% ACN): añadir 1 μL del 10% FA y 50 μL del 100% ACN a 49 μL de agua HPLC.
- Prepare las sugerencias de etapas como se describió anteriormente. En este caso, se utilizan discos de extracción C18.
9. Protocolo de la punta de escenario SCX/C18
NOTA: Purifique los resúmenes eps-orina mediante el protocolo C18 StageTip para eliminar las sales.
- A partir de 2 μg de péptidos (cuantificados por la inyección preliminar). Diluir la solución de digestión 4 veces en lavado 2 SCX y proceder como se describió anteriormente (en la sección "Purificación SCX"). Elute la mezcla de péptidos con 7 μL de solución de eluato (acetato de amonio de 500 mM (AA) y 20% de ACN) utilizando lentamente una velocidad de giro más baja (caudal de 5 μL/min).
- Acidificar el eluato SCX con 150 μL del 0,1% del ácido trifluoroacético (TFA) para lograr un pH inferior a 3 y una concentración de disolvente orgánico por debajo del 5%.
- Condiciona la punta de escenario C18 con 50 μL de solución A y centrífuga a 400 x g durante 2 minutos. Equilibrio C18 StageTip con 50 μL de solución B y centrífuga a 400 x g durante 2 minutos.
- Cargue la muestra en la punta del escenario lentamente utilizando una velocidad de giro más baja.
- Lave la punta de escenario C18 con 50 μL de solución B y centrífuga.
- Elicuente lentamente la mezcla de péptidos con 10 μL de solución de elución utilizando una velocidad de giro más baja.
- Seque el eluato (10 μL) a 30 °C en una centrífuga de vacío durante 3 minutos. Añadir 47 μL de 0.1% FA. La concentración esperada de péptidos será de 40 ng/ μL. Analizar por LC-MS/MS en modo DIA.
10. Análisis DIA (Figura 3)
- separación de nanoLC: Separe la mezcla de péptidos utilizando un gradiente lineal de 140 minutos a un caudal de 230 nL/min en una columna de identificación de 15 cm y 75 μm llena de partículas de sílice C18 de 3 μm.
- Realice la elución de gradiente a un caudal de 230 nL/minuto y aumente el contenido de fase B móvil del 3% al 25% en 90 minutos, luego del 25% al 40% en 30 minutos y finalmente del 40% al 100% en 8 minutos.
- Después de 10 minutos al 100% B, equilibra la columna con la fase móvil A durante 15 min.
- Parámetros espectrométricos de masa: realizar análisis DIA utilizando un método que emplea el siguiente ciclo de escaneo: (i) un evento ms de análisis completo a una resolución de 17.500 (objetivo 1e6 de AGC, tiempo máximo de inyección 50 ms) seguido de (ii) 20 ventanas a 20 m/z de ancho de aislamiento, a partir de m/z 350, (ii) 5 ventanas a 50 m/z de ancho de aislamiento y (iv) 1 ventana a 200 m/z de ancho de aislamiento, terminando el rango de escaneo de DIA a 1200 m/z. Realice exploraciones DIA a la resolución 35.000 (objetivo 5e5 de AGC, tiempo máximo de inyección 120 ms y 25 NCE).
11. Fraccionamiento de fase C18 invertido de alto pH para la generación de bibliotecas
NOTA: Pool EPS-urine muestras representativas en una cantidad superior a 10 μg con el fin de construir una biblioteca dependiente de datos para el análisis DIA mediante el siguiente procedimiento:
- Acidifique la mezcla de péptidos (11 μg) en un 0,2% de TFA y cargue la solución resultante en una punta de escenario C18 repleta de dos capas de discos de extracción para permitir una mayor capacidad. Estimamos que la capacidad de carga de un solo micro disco (a partir de una aguja de jeringa de calibre 16) estaba en el rango de 5-10 μg.
- Condición y equilibrio de la fase estacionaria como ya se ha descrito para la purificación C18.
- Llevar a cabo el fraccionamiento de péptidos por elución escalonada (n=10) desde la fase alta de pH invertido C18 utilizando soluciones de elución que contengan el 0,2% del hidróxido de amonio, TEAB de 10 mM y concentraciones crecientes de V/v de ACN (4%, 8%, 12%, 16%, 20%, 24%, 28%, 32%, 40%, 80%).
- Analice las 10 fracciones utilizando los mismos parámetros cromatográficos del método DIA. Utilice el espectrómetro de masas en modo DDA utilizando los ajustes adoptados anteriormente para el análisis preliminar (consulte "Cuantificación de proteínas por norma externa mediante el análisis DDA").
12. Análisis de datos
- Genere la biblioteca espectral importando los resultados de identificación de proteínas obtenidos de los experimentos DDA LC-MS/MS de fraccionamiento C18 de fase alta invertida en un software dedicado al análisis DIA.
- Establezca el número mínimo y máximo de iones de fragmento utilizados para la identificación y cuantificación en 3 y 6, respectivamente. Filtre los resultados obtenidos por un valor Q de 0,01.
- Importe los archivos sin procesar DIA y asocie a cada archivo sin procesar el mismo archivo FASTA utilizado para crear la biblioteca espectral.
- Establezca el número mínimo y máximo de péptidos únicos utilizados para la cuantificación de proteínas en 1 y 10, respectivamente.
- Calcule la intensidad de cada proteína sumando el área pico de iones de fragmento.
- Generar una matriz con la intensidad de las proteínas cuantificadas en cada muestra.
Resultados
Este protocolo para el análisis proteómico urinario incluye los siguientes pasos: digestión FASP, estimación de la cantidad de proteínas a través de calibración estándar externa, purificación de doble punta de escenario (SCX y C18)y análisis LC-MS/MS en modo DIA.
Después de la digestión de proteínas, se realizan inyecciones preliminares después de la purificación stagetip SCX de los péptidos resultantes. Los archivos raw LC-MS/MS se procesan para obtener el número de péptidos identificados, el número de proteínas identificadas y el área de péptidos detectados. El área total obtenida resumiendo todos los péptidos identificados se utiliza para estimar el contenido de proteínas a través de la interpolación a un estándar externo: un digesto de proteínas HeLa inyectado a cinco cantidades diferentes (2, 5, 15, 50, 150 ng, respectivamente). La cantidad de proteínas para las seis muestras analizadas en este estudio varió de una muestra a una muestra, mostrando un valor medio de 78 ng/μL.
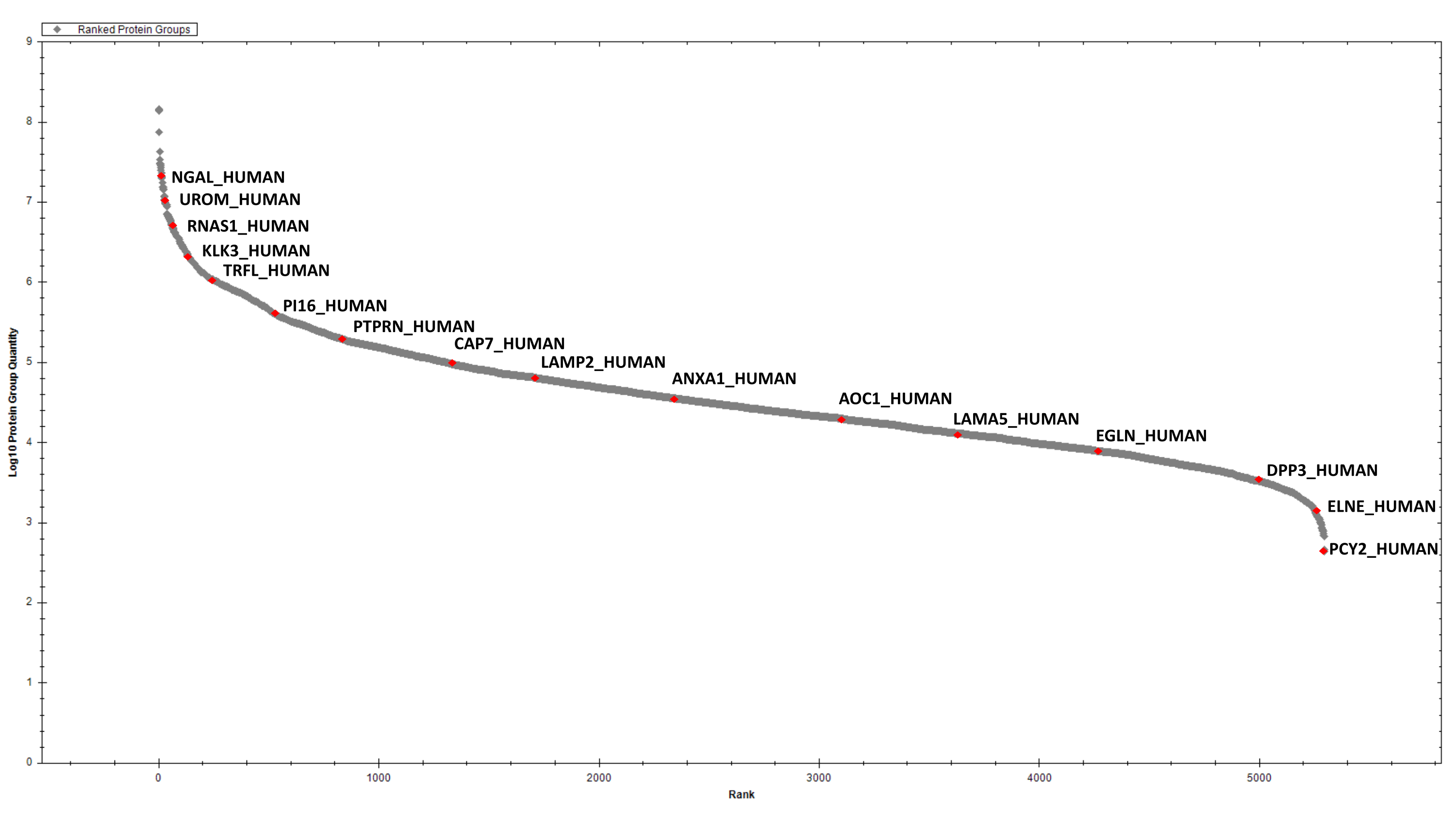
Después de la estimación de proteínas, 2 μg de proteínas digeridas de cada muestra se purifican mediante SCX secuencial y C18 StageTip antes del análisis DIA. La biblioteca espectral para buscar datos DIA se crea después del fraccionamiento en fase invertida de alto pH y el análisis DDA LC-MS/MS de una muestra representativa (por ejemplo, un grupo de muestras). Utilizando los parámetros descritos anteriormente, hemos identificado y cuantificado, acumulativamente, 2387 grupos proteicos en las seis muestras de EPS-orina que se están considerando(Tabla suplementaria 1 y Figura 4).
Con el fin de evaluar desde un punto de vista cualitativo la relevancia de la lista de proteínas identificadas y cuantificadas, la matriz obtenida se comparó con una lista de 624 proteínas previamente identificadas en la EPS22directa, una muestra recogida en un procedimiento más invasivo, que ha demostrado ser una fuente de interesantes biomarcadores candidatos al cáncer de próstata. En total, 508 de las 624 proteínas fueron detectadas con éxito por nuestro protocolo FASP/DIA en EPS-urine.

Figura 1: Flujo de trabajo proteómico. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Representación de nuestro diseño experimental para cuantificación de proteínas basado en estándar externo (HeLa digest). Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Pasos clave del análisis DIA: i) elaboración de la biblioteca espectral a través de fraccionamientos en fase invertida de alto pH y análisis DDA, (ii) análisis de muestras utilizando el enfoque DIA, (iii) análisis de datos por Spectronaut. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Gráfica clasificada para las proteínas EPS-urinarias detectadas. Algunos éxitos seleccionados están etiquetados. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Cuadro complementario 1: Tabla representativa de proteínas cuantificadas en seis muestras de EPS-orina en Spectronaut. Haga clic aquí para descargar esta tabla.
Discusión
En este trabajo, se presenta una estrategia para analizar muestras de EPS-orina. El protocolo FASP es una opción ideal para la proteómica urinaria, ya que permite la concentración de muestras antes de la digestión enzimática. De hecho, utilizando este flujo de trabajo, varios cientos de microlitros de orina se pueden cargar en un solo filtro y procesarse. Además, la digestión en filtro ofrece una relativa libertad en la elección de condiciones de desnaturalización. En nuestro trabajo, la desnaturalización de proteínas se logra diluyendo muestras de orina en un tampón que contiene Tris, SDS y TDT (concentración final: Tris de 50 mM, 1% SDS, 50 mM TDT). Las proteínas se desnaturalizan eficientemente inmediatamente después de descongelar la muestra, con el fin de evitar la degradación no deseada por proteasas activas. El protocolo FASP original se ha mejorado aumentando el volumen de lavados de 100 μL a 200 μL. De esta manera, se logra una mejor eliminación de residuos, especialmente detergentes del filtro.
Después de la digestión enzimática, la estimación de proteínas por norma externa utilizando un método rápido LC-MS/MS, dependiente de los datos se realiza antes de los últimos pasos del protocolo, que comprenden la purificación de doble punta de escenario y el análisis LC-MS/MS en modo DIA, en el mismo material inicial para todas las muestras (2 μg).
La versatilidad de FASP se asocia con el enfoque DIA, un método sensible que proporciona un bajo número de valores que faltan17. En nuestro trabajo, se identificaron y cuantificaron 2387 proteínas, lo que permitió extraer un perfil proteómico detallado de eps-orina. La identificación y cuantificación de 2387 proteínas fue posible a través de la generación de una rica biblioteca espectral, obtenida a través de un fraccionamiento invertido de péptidos de alto pH, y por nuestro método DIA, dirigido a una amplia gama de precursores m/z. Este flujo de trabajo identificó más del 80% de las proteínas encontradas anteriormente en el análisis directo-EPS, lo que demuestra que una fracción considerable del proteoma EPS-urinario se deriva de secreciones prostáticas expresadas, por lo tanto es una rica fuente de proteínas específicas del tejido prostático26.
En conclusión, nuestro diseño experimental combina la versatilidad de FASP con la sensibilidad de DIA con el fin de obtener un rico mapa del proteoma urinario. Esta estrategia se recomienda para analizar muestras de EPS-orina, pero su uso se puede extender a la proteómica urinaria en general, o incluso a otros tipos de muestras.
Divulgaciones
Todos los autores no declaran conflicto de intereses.
Agradecimientos
Este trabajo fue apoyado por MIUR (Ministero Università Ricerca, PRIN 2017 to MG) y por POR Calabria FESR 2014-2020, acción 1.2.2, "INNOPROST".
Materiales
| Name | Company | Catalog Number | Comments |
| 1 M Tris HCL pH 8.0 | Lonza | 51238 | |
| 2-iodoacetamide for synthesis | Merck | 8047440100 | |
| Acetonitrile for HPLC LC-MS grade | VWR | 83640.290 | |
| Ammonium acetate | Fluka | 9690 | |
| ammonium hydroxide solution | Sigma | 30501 | |
| ammonium hydroxide volumetric standard, 5N solution in water | Merck | 318612-500ML | |
| Dithiothreitol | Amresco | 0281-25G | |
| Empore Cation 47mm Extraction Disks | Microcolumn | 2251 | |
| Empore Disk C18 | Varian | 12145004 | |
| Formic acid optima | Fisher Scientific | A117-50 | |
| Hela Protein Digest Standard | Fisher Scientific | 88329 | |
| Microcon-10kDa Centrifugal Filter Unit with Ultracel-10 membrane | MERCK | MRCPRT010 | |
| sodium dodecyl sulfate solution | Merck | 71736-500ML | |
| Thiethylammonium bicarbonate buffer | Merck | T7408-100ML | |
| trifluoroacetic acid | Riedel-de Haën | 34957 | |
| Trypsin from porcine pancreas | Merck | T6567-1MG | |
| Urea | Merck | U6504-500G | |
| Water HPLC gradient grade | Fisher Scientific | W/0106/17 | |
| Proteome Discoverer 1,4 | Thermo Fisher Scientific | ||
| Spectronaut 14.0 | Biognosys |
Referencias
- Hüttenhain, R., Soste, M., Selevsek, N., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 10 (2013).
- Roobol, M. J., Carlsson, S. V. Risk stratification in prostate cancer screening. Nature Reviews Urology. 10 (1), 38-48 (2013).
- Principe, S., et al. Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. Journal of Proteome Research. 11 (4), 2386-2396 (2012).
- Kim, Y., et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nature Communications. 7, 1-10 (2016).
- Drake, R. R., et al. Clinical collection and protein properties of expressed prostatic secretions as a source for biomarkers of prostatic disease. Journal of Proteomics. 72 (6), 907-917 (2009).
- Macklin, A., Khan, S., Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clinical Proteomics. 17 (1), 1-25 (2020).
- Berger, S. T., et al. MStern blotting-high throughput polyvinylidene fluoride (PVDF) membrane-based proteomic sample preparation for 96-well plates. Molecular and Cellular Proteomics. 14 (10), 2814-2823 (2015).
- Zougman, A., Selby, P. J., Banks, R. E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 14 (9), 1006 (2014).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10 (10), 757 (2014).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11 (3), 319-324 (2014).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6 (5), 359-362 (2009).
- Ding, H., et al. Urine Proteomics: Evaluation of Different Sample Preparation Workflows for Quantitative, Reproducible, and Improved Depth of Analysis. Journal of Proteome Research. 19 (4), 1857-1862 (2020).
- Zhao, M., et al. A comprehensive analysis and annotation of human normal urinary proteome. Scientific Reports. 7 (1), 1-13 (2017).
- Wiśniewski, J. R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Analytical Chemistry. 88 (10), 5438-5443 (2016).
- Wiśniewski, J. R., Zielinska, D. F., Mann, M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Analytical Biochemistry. 410 (2), 307-309 (2011).
- Yu, Y., et al. Urine sample preparation in 96-well filter plates for quantitative clinical proteomics. Analytical Chemistry. 86 (11), (2014).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Molecular and Cellular Proteomics. 11 (6), 1-17 (2012).
- Liu, Y., Hüttenhain, R., Collins, B., Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Review of Molecular Diagnostics. 13 (8), 811-825 (2013).
- Gallien, S., Duriez, E., Demeure, K., Domon, B. Selectivity of LC-MS/MS analysis: Implication for proteomics experiments. Journal of Proteomics. 81, 148-158 (2013).
- Muntel, J., et al. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. Journal of Proteome Research. 14 (11), 4752-4762 (2015).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Kim, Y., et al. Identification of Differentially Expressed Proteins in Direct Expressed Prostatic Secretions of Men with Organ-confined Versus Extracapsular Prostate Cancer. Molecular & Cellular Proteomics. 11 (12), 1870-1884 (2012).
- Decramer, S., et al. Urine in clinical proteomics. Molecular and Cellular Proteomics. 7 (10), 1850-1862 (2008).
- Thongboonkerd, V. Practical points in urinary proteomics. Journal of Proteome Research. 6 (10), 3881-3890 (2007).
- Konvalinka, A., Scholey, J. W., Diamandis, E. P. Searching for new biomarkers of renal diseases through proteomics. Clinical Chemistry. 58 (2), 353-365 (2012).
- Drake, R. R., et al. In-Depth Proteomic Analyses of Direct Expressed Prostatic Secretions. Journal of Proteome Research. 9 (5), 2109-2116 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados