
Method Article
Proteomic Profile of EPS-Urine through FASP Digestion and Data-Independent Analysis
In This Article
Summary
Here, we present an optimized on-filter digestion protocol with detailed information about the following: protein digestion, peptide purification and data independent acquisition analysis. This strategy is applied to the analysis of expressed prostatic secretions-urine samples and allows high proteome coverage and low missing value label-free profiling of the urinary proteome.
Abstract
Filter-aided sample protocol (FASP) is widely used for proteomics sample preparation because it allows to concentrate diluted samples and it is compatible with a wide variety of detergents. Bottom-up proteomics workflows like FASP increasingly rely on LC-MS/MS methods performed in data-independent analysis (DIA) mode, a scanning method that allows deep proteome coverage and low incidence of missing values.
In this report, we will provide the details of a workflow that combines a FASP protocol, a double StageTip purification step and LC-MS/MS in DIA mode for urinary proteome mapping. As a model sample, we analyzed expressed prostatic secretions (EPS)-urine, a sample collected after a digital rectal exam (DRE), which is of interest in prostate cancer biomarker discovery studies.
Introduction
The constant evolution of proteomic technologies promises to have a considerable impact in helping disease diagnosis and prediction of response to treatment by providing high resolution maps of key molecular effectors present in a wide variety of samples such as tissues and biofluids. From an analytical point of view, urine offers several advantages such as ease of collection and major stability of the proteome with respect to others biofluids1. Proteomic analysis of urine is of special interest in biomarker discovery studies on urological cancers, since it allows noninvasive sampling in proximity to the tissues of interest2. In particular, a sample that seems to be promising for studying prostate-related pathologies is the EPS-urine3,4 (i.e., a urine sample collected after a digital rectal exam (DRE)). This latter operation prior to sample collection enriches urine with prostate specific proteins. EPS-urine is a good candidate to investigate disorders related to the prostate gland5 including prostate cancer (PCa), since through DRE, proteins secreted by the tumor can be poured into the urine sample, increasing the chance of detecting cancer tissue-specific proteins.
A crucial role in allowing detection and quantification of potential protein biomarkers is played by mass spectrometry (MS). Over the last two decades, MS-based protocols for proteomic analysis have allowed an ever-increasing number of proteins to be detected in a single LC-MS/MS run thanks to continuous improvements in MS instrumentation and in data analysis software6.
MS-based proteomic sample preparation generally involves enzymatic digestion of the protein mixture, which can be achieved via a wide variety of protocols such as: in-solution digestion, MStern blotting7, suspension trapping (S-trap)8, solid-phase-enhanced sample preparation (SP3)9, in-StageTip digestion10 and filter aided sample preparation (FASP)11. All protocols can be used for urinary proteomics, even though results may vary with respect to the number of identified proteins and peptides and in terms of reproducibility12.
In this work, our attention was focused on the analysis of EPS-urine by the FASP protocol. The FASP protocol was originally designed to analyze proteins extracted from tissues and cell cultures, but its use was then expanded to the analysis of other sample types, such as urine13. With respect to straightforward in-solution digestion FASP is a more flexible proteomic approach14, since by achieving effective removal of detergents and other contaminants such as salts from the protein mixture before enzymatic digestion15, it allows the choice of optimal protein solubilization conditions. Moreover, an additional characteristic of FASP is that it provides a means for sample concentration. This is of particular interest for urinary proteomic analysis, because it allows to start from relatively large sample volumes (hundreds of microliters). In the light of the potential of the FASP protocol, several studies have focused the attention on workflow automation, with the aim of reducing experimental variability and processing an elevated number of samples in parallel16.
In our workflow, FASP is followed by LC-MS/MS acquisition in data-independent analysis (DIA), which provides high proteome coverage, good quantitative precision and low incidence of missing values. The DIA approach is a sensitive method where all ions are selected for MS/MS events, opposite to what happens in data-dependent analysis (DDA) where only ions with the highest intensity are fragmented. The mass spectrometer, operating in DIA mode, performs scan cycles with different isolation width covering the whole m/z precursor range. This approach allows to reproducibly detect a high number of peptides per unit time, providing a proteomics snapshot of the sample17. Moreover, data generated by DIA have another interesting characteristic: the possibility of a posteriori analysis18. DIA data are more complex than those obtained by DDA, because MS/MS spectra in DIA result from the co-isolation of several precursor ions within each m/z window19. Disentangling the composite MS/MS spectra into distinct and specific peptide signals is achieved by using two fundamental elements: a spectral library and a dedicated software for data analysis. The spectral library is generated by a data-dependent experiment, usually involving peptide fractionation to maximize proteome coverage, which provides a list of thousands of experimentally determined precursor ions and MS/MS spectra of peptides detectable in the sample under consideration. The data analysis software, instead, uses the information contained in the spectral library to interpret the DIA data by generating specific extracted ion chromatograms which allow peptide detection and quantification. While library-free DIA data analysis is now feasible, library-based DIA still provides better results in terms of proteome coverage20.
The sample preparation protocol here described (Figure 1) consists of the following steps: a centrifugation step (to remove cell debris), FASP digestion, StageTip purification21, quantification of proteins and DIA analysis. This protocol has been designed for the analysis of EPS-urine in the context of prostate cancer biomarker discovery, but it can be applied to proteomic analysis of any urine sample.
Protocol
The study was approved by the Institutional Ethical Committee of the Magna Graecia University of Catanzaro, RP 41/2018. Written informed consent was obtained from all patients enrolled in the study.
1. Sample preparation
- Centrifuge EPS-urine samples within 2 h of collection at 2,100 x g for 10 minutes at room temperature (RT). Store the supernatant at -80 °C until use.
2. Sample thawing
- Transfer the sample from -80 °C to -20 °C the day before digestion.
- Prior to sample processing, transfer the sample for 15-20 minutes at 4 °C and then at RT.
3. Reagent preparation for FASP
- On the same day, prepare the following solutions: urea buffer, 50 mM iodoacetamide (IAA) in urea buffer, 50 mM triethylammonium bicarbonate and 500 mM dithiothreitol (DTT).
- Prepare urea buffer (urea 8 M, 100 mM Tris-HCl pH 8): for 10 mL, weigh 4,800 mg of urea and dissolve it in the appropriate amount of HPLC water after adding 1 mL of 1 M Tris pH 8. A volume of 800 µL of urea is needed for each sample.
- Prepare 500 mM dithiothreitol (DTT): dissolve 38.5 mg of DTT in 500 µL of HPLC water. For each sample, 66.7 µL of DTT are needed.
- Prepare 50 mM IAA in Urea Buffer: dissolve 9.25 mg of IAA in 1 mL of urea buffer. For each sample 50 µL of IAA are needed.
- Prepare 50 mM triethylammonium bicarbonate (TEAB). Add 150 µL of 1 M TEAB to 2.85 mL of HPLC water. For each sample 460 µL of TEAB are needed.
- Prepare a solution of trypsin (100 ng/µL) in HPLC water. This solution can be stored at -80 °C and thawed immediately before use. For each sample, 2 µL (200 ng) are needed.
4. Protein digestion by FASP
- Dilute 500 µL of each EPS-urine sample with 66.7 µL of 10% (w/v) sodium dodecyl sulfate (SDS), 66.7 µL of 500 mM DTT and 33.3 µL of 1 M Tris pH 8 to solubilize proteins and to reduce disulphide bonds. Incubate 10 minutes at 95 °C with gentle shaking.
- Add 300 µL of diluted EPS-urine sample on the filter unit (10 kDa) and centrifuge at 14,000 x g for 20 minutes.
- Add 200 µL of urea buffer to the filter and centrifuge at 14,000 x g for 15 minutes. Repeat this step and then discard the flow-through.
- Add 50 µL of IAA solution and centrifuge at 6,000 x g for 25 minutes.
- Add 200 µL of urea buffer and centrifuge at 14,000 x g for 20 minutes. Repeat this step and then discard the flow-through.
- Add 200 µL of 50 mM triethylammonium bicarbonate buffer (TEAB) and centrifuge at 14,000 x g for 20 minutes. Repeat this step.
- Transfer the filter unit into a new collection tube.
- Add 60 µL of 50 mM TEAB buffer and 200 ng of trypsin and incubate the sample in a thermo-mixer at 37 °C overnight.
NOTE: To avoid sample evaporation during overnight incubation, wrap each filter unit with a layer of aluminum foil and then a layer of parafilm. - After trypsin incubation, add 140 µL of water and centrifuge the vial at 14,000 x g for 25 minutes to collect the volume of digest (180-200 µL).
5. Reagent preparation for SCX purification
- Prepare 1 mL of wash 1 (0.5% formic acid (FA) and 20% acetonitrile (ACN)) as follows: add 50 µL of 10% FA and 200 µL of 100% ACN to 750 µL of HLPC water. For each sample, 100 µL of wash 1 are needed.
- Prepare 1.5 mL of wash 2 (0.5% FA and 80% of ACN) as follows: add 75 µL of 10 % FA and 1.2 mL of 100% ACN to 225 µL of HPLC water. For each sample 190 µL of wash 2 are needed.
- Prepare 100 µL of eluting solution (500 mM ammonium acetate (AA) and 20% of ACN): add 25 µL of 2 M AA and 20 µL of 100% ACN to 55 µL of HPLC water.
- Prepare StageTips as follows.
- Pack a 200 µL pipette tip with a layer of SCX resin using a blunt-ended syringe needle (gauge 16). Insert the StageTip into a microcentrifuge lid that has been previously pierced in order to accommodate the microcolumn.
- Assemble the lid onto a 2 mL microcentrifuge from which the original lid was removed. The micro-SPE centrifugal device just assembled should fit into a benchtop centrifuge. Since perforated lids are tedious to prepare, they can be utilized multiple times.
6. SCX purification
- Dilute 30 µL of each sample to 120 µL using wash 2.
- Condition the StageTip by adding 50 µL of wash 1 on top of the extraction resin and centrifuge at 400 x g for 2 minutes. Since flow resistance depends on how strongly the extraction resin was packed into the pipette tip, centrifuge speed may need to be adjusted in order to obtain a flow rate of 20-30 µL/min. Try not to leave the tip dry during sample loading and washing.
- Equilibrate the StageTip with 50 µL of wash 2 and centrifuge at 400 x g for 2 minutes.
- Load the diluted sample (120 µL) using a lower spin speed.
- Wash the StageTip with 50 µL of wash 2 and centrifuge at 400 x g for 2 minutes. Repeat with 50 µL of wash 1.
NOTE: After this wash the resin must be completely dry. - Elute peptide mixture with 7 µL of eluate solution (500 mM ammonium acetate (AA) and 20% of ACN) slowly using a lower spin speed.
- Add 27 µL of 0.1% FA to dilute AA and to obtain a sample for LC-MS/MS preliminary injection. The final dilution to 34 µL will result into a 1:1 volumetric ratio between urine and peptide solution (1 µL of purified digest will correspond to 1 µL of original urine sample).
7. Protein quantification by external standard using DDA analysis (Figure 2)
- Determine protein amount in samples by injecting 2 µL of the resulting peptide digest in LC-MS/MS and interpolating the resulting total peptide area with a calibration curve built as follows:
- Prepare five different HeLa digest solutions (1 ng/µL, 2.5 ng/µL, 7.5 ng/µL, 25 ng/µL, 75 ng/µL) using a HeLa digest stock (100 µg/µL).
- Inject each Hela solution in duplicate (2 µL).
- Set the LC-MS/MS method.
- Inject 2 µL of the five HeLa peptide mixtures and separate peptides through a linear gradient of 75 minutes at a flow rate of 230 nL/minute on a 15 cm, 75 µm i.d column packed with 3 µm C18 silica particles. Use a binary gradient constituted by mobile phase A (0.1% FA, 2% ACN) and mobile phase B (0.1% FA and 80% of ACN).
- Perform gradient elution by increasing mobile phase B content from 6% to 42% in 60 minutes and from 42% to 100% in additional 8 minutes. Maintain for 5 minutes 100% B and then move back to 0% B in two minutes.
- Operate in DDA mode using a top-12 method and set MS full scan range at 350-1800 m/z, resolution 70,000, ACG target 1e6 and maximum injection time 50 ms. Set the mass window for precursor ion isolation at 1.6 m/z, resolution 35,000, ACG target 1e5, maximum injection time 120 ms, HCD fragmentation at of 25 NCE (normalized collision energy) and dynamic exclusion 15 seconds.
- Analyze raw files using Sequest as search engine, and the HUMAN Uniprot Complete protein sequence as the database. In the experiments presented here, the database was downloaded on March 2016 and contained 42.013 sequences.
- Use the following settings: MS tolerance 15 ppm, MS/MS tolerance 0.02 Da, trypsin as enzyme by setting two missed cleavage sites. Set the carbamidomethylation of lysine, serine, threonine, tyrosine (+57.021) and the oxidation of methionines as dynamic modifications and the carbamidomethylation of cysteines (+57.021) as static modifications. Use the cut-off of false discovery rate (FDR) for peptides and proteins identification to 0.01 and filter out by percolator.
- Calculate the peak area for all MS1 peptide signals. Calculate the total area of the peptides.
- Inject 2 µL of peptide mixtures obtained from EPS-urine samples using the same LC-MS/MS method used for the HeLa samples and analyze the raw file using the same parameters used for HeLa data processing.
- Calculate total intensity of peptide signals by summing up area values of all identified peptides and interpolate the external standard curve. This will provide an acceptable estimation of peptide amount present in the EPS-urine protein digest.
8. Reagent preparation for C18 StageTip protocol
- Prepare 500 µL of solution A (0.1% Trifluoroacetic acid (TFA), 50% ACN): add 250 µL of 100% ACN and 10 µL of 5% TFA to 240 µL of HPLC water.
- Prepare 2 mL of solution B (0.1 % TFA): add 40 µL of 5% TFA to 1960 µL of HPLC water.
- Prepare 100 µL of eluting solution (0.1% FA, 50% ACN): add 1 µL of 10% FA and 50 µL of 100% ACN to 49 µL of HPLC water.
- Prepare StageTips as previously described. In this case, C18 extraction disks are used.
9. SCX/C18 StageTip protocol
NOTE: Purify EPS-urine digests by C18 StageTip protocol to remove salts.
- Start from 2 µg of peptides (as quantified by the preliminary injection). Dilute the digest solution 4-fold in wash 2 SCX and proceed as described above (in section "SCX purification"). Elute the peptide mixture with 7 µL of eluate solution (500 mM ammonium acetate (AA) and 20% of ACN) slowly using a lower spin speed (5 µL/min flow rate).
- Acidify the SCX eluate with 150 µL of 0.1% of trifluoroacetic acid (TFA) in order to achieve a pH lower than 3 and a concentration of organic solvent below 5%.
- Condition the C18 StageTip with 50 µL of solution A and centrifuge at 400 x g for 2 minutes. Equilibrate C18 StageTip with 50 µL of solution B and centrifuge at 400 x g for 2 minutes.
- Load the sample on the stage-tip slowly using a lower spin speed.
- Wash C18 StageTip with 50 µL of solution B and centrifuge.
- Slowly elute the peptide mixture with 10 µL of elution solution using a lower spin speed.
- Dry the eluate (10 µL) at 30 °C in in a vacuum centrifuge for 3 minutes. Add 47 µL of 0.1% FA. Expected peptide concentration will be 40 ng/ µL. Analyze by LC-MS/MS in DIA mode.
10. DIA Analysis (Figure 3)
- nanoLC separation: Separate the peptide mixture using a linear gradient of 140 minutes at a flow rate of 230 nL/min on a 15 cm, 75 µm ID column packed with 3 µm C18 silica particles.
- Perform gradient elution at 230 nL/minute flow rate and increase mobile phase B content from 3% to 25% in 90 minutes, then from 25% to 40% in 30 minutes and finally from 40% to 100% in 8 minutes.
- After 10 minutes at 100% B, equilibrate the column with mobile phase A for 15 min.
- Mass spectrometric parameters: perform DIA analysis using a method employing the following scan cycle: (i) a full scan MS event at a resolution of 17,500 (AGC target 1e6, maximum injection time 50 ms) followed by (ii) 20 windows at 20 m/z isolation width, starting from m/z 350, (ii) 5 windows at 50 m/z isolation width and (iv) 1 window at 200 m/z isolation width, ending the scan range of DIA at 1200 m/z. Perform DIA scans at resolution 35,000 (AGC target 5e5, maximum injection time 120 ms and 25 NCE).
11. High- pH reversed phase C18 fractionation for library generation
NOTE: Pool EPS-urine representative samples in an amount of higher than 10 µg in order to build a data-dependent library for DIA analysis by the following procedure:
- Acidify the peptide mixture (11 µg) by 0.2% of TFA and load the resulting solution onto a C18 StageTip packed with two layers of extraction disks to allow higher capacity. We estimated the loading capacity of a single micro disk (from a 16 gauge syringe needle) to be in the 5-10 µg range.
- Condition and equilibrate stationary phase as already described for C18 purification.
- Carry out peptide fractionation by stepwise elution (n=10) from high pH reversed phase C18 using elution solutions containing 0.2% of ammonium hydroxide, 10 mM TEAB, and increasing v/v concentrations of ACN (4%, 8%, 12%, 16%, 20%, 24%, 28%, 32%, 40%, 80%).
- Analyze the 10 fractions using the same chromatographic parameters of the DIA method. Operate the mass spectrometer in DDA mode using the settings previously adopted for the preliminary analysis (see "Protein quantification by external standard using DDA analysis").
12. Data analysis
- Generate the spectral library by importing the protein identification results obtained from the DDA LC-MS/MS experiments of high-reversed phase C18 fractionation in a software dedicated to DIA analysis.
- Set the minimum and maximum number of fragment ions used for identification and quantification to 3 and 6, respectively. Filter the obtained results by a Q-value of 0.01.
- Import DIA raw files and associate to each raw file the same FASTA file used for creating the spectral library.
- Set the minimum and maximum number of unique peptides used for protein quantification to 1 and 10, respectively.
- Calculate the intensity for each protein by summing fragment ion peak area.
- Generate a matrix with the intensity of the quantified proteins in each sample.
Results
This protocol for urinary proteomic analysis includes the following steps: FASP digestion, estimation of protein amount via external standard calibration, double StageTip purification (SCX and C18), and LC-MS/MS analysis in DIA mode.
After protein digestion, preliminary injections are performed after StageTip SCX purification of the resulting peptides. LC-MS/MS raw files are processed to obtain the number of identified peptides, the number of identified proteins and the area of detected peptides. The total area obtained by summing up all identified peptides is used to estimate protein content via interpolation to an external standard: a HeLa protein digest injected at five different amounts (2, 5, 15, 50, 150 ng, respectively). The protein amount for the six samples analyzed in this study varied from sample to sample, showing an average value of 78 ng/µL.
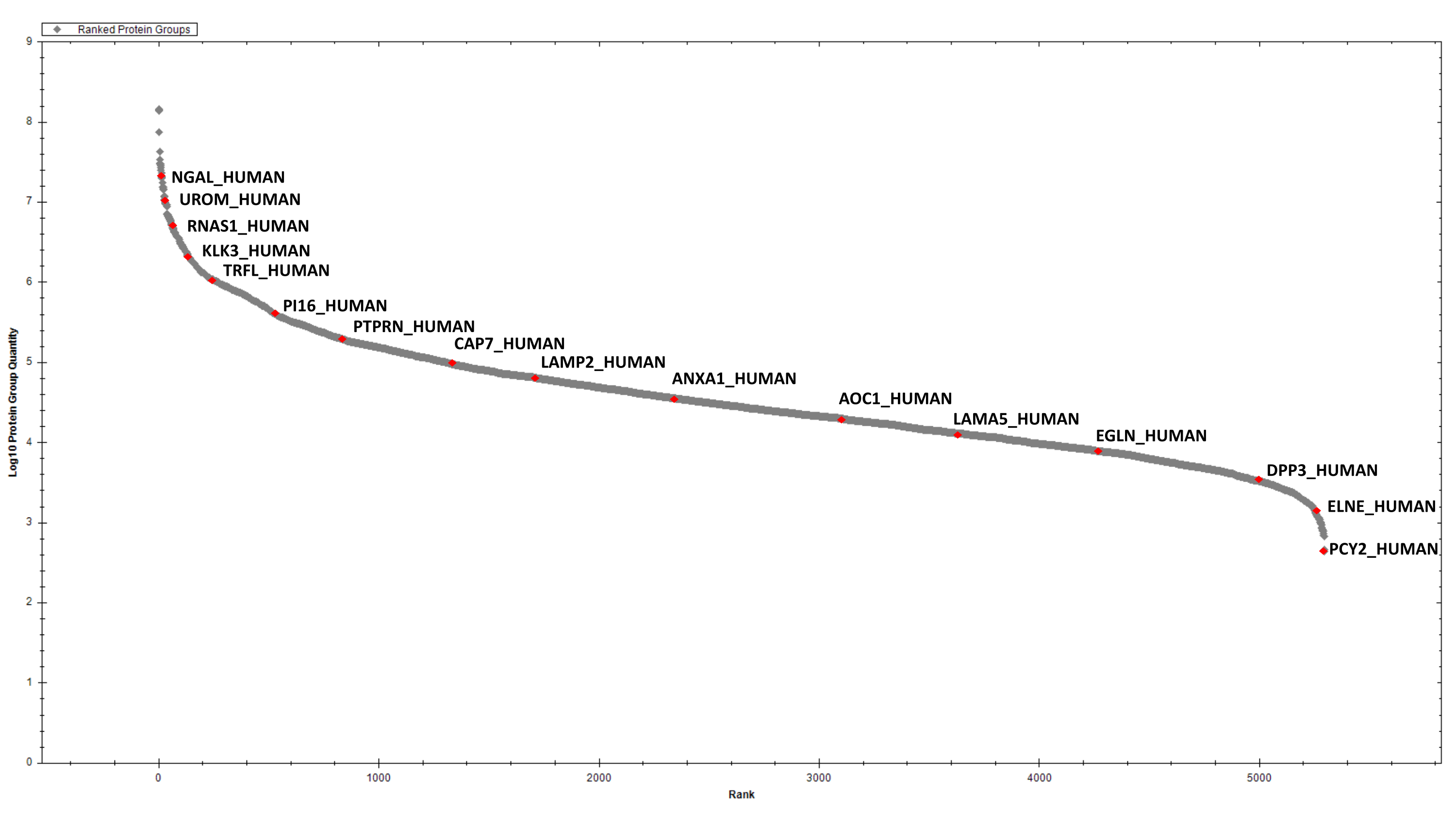
After protein estimation, 2 µg of digested proteins from each sample are purified by sequential SCX and C18 StageTip before DIA analysis. The spectral library for searching DIA data is created after high-pH reversed-phase fractionation and DDA LC-MS/MS analysis of a representative sample (e.g., a sample pool). Using the parameters described above, we have identified and quantified, cumulatively, 2387 protein groups in the six EPS-urine samples under consideration (Supplementary Table 1 and Figure 4).
In order to evaluate from a qualitative point of view the relevance of the list of identified and quantified proteins, the obtained matrix was compared to a list of 624 proteins previously identified in direct- EPS22, a sample collected in a more invasive procedure, which has proven to be a source of interesting candidate prostate cancer biomarkers. In total, 508 out of 624 proteins were successfully detected by our FASP/DIA protocol on EPS-urine.

Figure 1: Proteomic workflow. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representation of our experimental design for protein quantification based on external standard (HeLa digest). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Key steps of DIA analysis: (i) elaboration of the spectral library through high-pH reversed-phase fractionation and DDA analysis, (ii) sample analysis using the DIA approach, (iii) data analysis by Spectronaut. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Ranked plot for the detected EPS-urinary proteins. A few selected hits are labelled. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Table 1: Representative table of quantified proteins in six EPS-urine samples in Spectronaut. Please click here to download this Table.
Discussion
In this work, a strategy for analyzing EPS-urine samples is presented. The FASP protocol is an ideal choice for urinary proteomics because it allows sample concentration before enzymatic digestion. In fact, using this workflow, several hundreds of microliters of urine can be loaded on a single filter and processed. Moreover, on-filter digestion offers relative freedom in the choice of denaturation conditions. In our work, protein denaturation is achieved by diluting urine samples in a buffer containing Tris, SDS and DTT (final concentration: 50 mM Tris, 1% SDS, 50 mM DTT). Proteins are efficiently denatured immediately after thawing the sample, in order to avoid unwanted degradation by active proteases. The original FASP protocol has been improved by increasing the volume of washes from 100 µL to 200 µL. In this way, better removal of residues, especially detergents from the filter, is achieved.
After enzymatic digestion, protein estimation by external standard using a fast LC-MS/MS, data-dependent method is performed before the last steps of the protocol, which comprise double StageTip purification and LC-MS/MS analysis in DIA mode, on equal starting material for all samples (2 µg).
The versatility of FASP is associated with the DIA approach, a sensitive method providing a low number of missing values17. In our work, 2387 proteins were identified and quantified, thus enabling a detailed proteomic profile of EPS-urine to be drawn. The identification and quantification of 2387 proteins was possible through the generation of a rich spectral library, obtained via high-pH reversed fractionation of peptides, and by our DIA method, directed at a wide m/z precursor range. This workflow identified over 80% of proteins previously found in direct-EPS analysis, demonstrating that a considerable fraction of the EPS-urinary proteome is indeed derived from expressed prostatic secretions, thus is a rich source of prostate tissue-specific proteins26.
In conclusion, our experimental design couples the versatility of FASP with the sensitivity of DIA in order to obtain a rich map of the urinary proteome. This strategy is recommended for analyzing EPS-urine samples, but its use can be extended to urinary proteomics in general, or even to other sample types.
Disclosures
All authors declare no conflict of interest.
Acknowledgements
This work was supported by MIUR (Ministero Università Ricerca, PRIN 2017 to MG) and by POR Calabria FESR 2014-2020, action 1.2.2, "INNOPROST".
Materials
| Name | Company | Catalog Number | Comments |
| 1 M Tris HCL pH 8.0 | Lonza | 51238 | |
| 2-iodoacetamide for synthesis | Merck | 8047440100 | |
| Acetonitrile for HPLC LC-MS grade | VWR | 83640.290 | |
| Ammonium acetate | Fluka | 9690 | |
| ammonium hydroxide solution | Sigma | 30501 | |
| ammonium hydroxide volumetric standard, 5N solution in water | Merck | 318612-500ML | |
| Dithiothreitol | Amresco | 0281-25G | |
| Empore Cation 47mm Extraction Disks | Microcolumn | 2251 | |
| Empore Disk C18 | Varian | 12145004 | |
| Formic acid optima | Fisher Scientific | A117-50 | |
| Hela Protein Digest Standard | Fisher Scientific | 88329 | |
| Microcon-10kDa Centrifugal Filter Unit with Ultracel-10 membrane | MERCK | MRCPRT010 | |
| sodium dodecyl sulfate solution | Merck | 71736-500ML | |
| Thiethylammonium bicarbonate buffer | Merck | T7408-100ML | |
| trifluoroacetic acid | Riedel-de Haën | 34957 | |
| Trypsin from porcine pancreas | Merck | T6567-1MG | |
| Urea | Merck | U6504-500G | |
| Water HPLC gradient grade | Fisher Scientific | W/0106/17 | |
| Proteome Discoverer 1,4 | Thermo Fisher Scientific | ||
| Spectronaut 14.0 | Biognosys |
References
- Hüttenhain, R., Soste, M., Selevsek, N., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 10(2013).
- Roobol, M. J., Carlsson, S. V. Risk stratification in prostate cancer screening. Nature Reviews Urology. 10 (1), 38-48 (2013).
- Principe, S., et al. Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. Journal of Proteome Research. 11 (4), 2386-2396 (2012).
- Kim, Y., et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nature Communications. 7, 1-10 (2016).
- Drake, R. R., et al. Clinical collection and protein properties of expressed prostatic secretions as a source for biomarkers of prostatic disease. Journal of Proteomics. 72 (6), 907-917 (2009).
- Macklin, A., Khan, S., Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clinical Proteomics. 17 (1), 1-25 (2020).
- Berger, S. T., et al. MStern blotting-high throughput polyvinylidene fluoride (PVDF) membrane-based proteomic sample preparation for 96-well plates. Molecular and Cellular Proteomics. 14 (10), 2814-2823 (2015).
- Zougman, A., Selby, P. J., Banks, R. E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 14 (9), 1006(2014).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10 (10), 757(2014).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11 (3), 319-324 (2014).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6 (5), 359-362 (2009).
- Ding, H., et al. Urine Proteomics: Evaluation of Different Sample Preparation Workflows for Quantitative, Reproducible, and Improved Depth of Analysis. Journal of Proteome Research. 19 (4), 1857-1862 (2020).
- Zhao, M., et al. A comprehensive analysis and annotation of human normal urinary proteome. Scientific Reports. 7 (1), 1-13 (2017).
- Wiśniewski, J. R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Analytical Chemistry. 88 (10), 5438-5443 (2016).
- Wiśniewski, J. R., Zielinska, D. F., Mann, M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Analytical Biochemistry. 410 (2), 307-309 (2011).
- Yu, Y., et al. Urine sample preparation in 96-well filter plates for quantitative clinical proteomics. Analytical Chemistry. 86 (11), (2014).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Molecular and Cellular Proteomics. 11 (6), 1-17 (2012).
- Liu, Y., Hüttenhain, R., Collins, B., Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Review of Molecular Diagnostics. 13 (8), 811-825 (2013).
- Gallien, S., Duriez, E., Demeure, K., Domon, B. Selectivity of LC-MS/MS analysis: Implication for proteomics experiments. Journal of Proteomics. 81, 148-158 (2013).
- Muntel, J., et al. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. Journal of Proteome Research. 14 (11), 4752-4762 (2015).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Kim, Y., et al. Identification of Differentially Expressed Proteins in Direct Expressed Prostatic Secretions of Men with Organ-confined Versus Extracapsular Prostate Cancer. Molecular & Cellular Proteomics. 11 (12), 1870-1884 (2012).
- Decramer, S., et al. Urine in clinical proteomics. Molecular and Cellular Proteomics. 7 (10), 1850-1862 (2008).
- Thongboonkerd, V. Practical points in urinary proteomics. Journal of Proteome Research. 6 (10), 3881-3890 (2007).
- Konvalinka, A., Scholey, J. W., Diamandis, E. P. Searching for new biomarkers of renal diseases through proteomics. Clinical Chemistry. 58 (2), 353-365 (2012).
- Drake, R. R., et al. In-Depth Proteomic Analyses of Direct Expressed Prostatic Secretions. Journal of Proteome Research. 9 (5), 2109-2116 (2010).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved