
Method Article
Perfil proteômico de EPS-urina através da Digestão FASP e Análise Independente de Dados
Neste Artigo
Resumo
Aqui, apresentamos um protocolo de digestão otimizado no filtro com informações detalhadas sobre o seguinte: digestão de proteínas, purificação de peptídeos e análise de aquisição independente de dados. Esta estratégia é aplicada à análise de amostras de urina de secreções prostáticas expressas e permite alta cobertura proteome e perfil livre de rótulos de baixo valor ausente do proteome urinário.
Resumo
O protocolo de amostra auxiliado por filtro (FASP) é amplamente utilizado para a preparação da amostra de proteômica porque permite concentrar amostras diluídas e é compatível com uma grande variedade de detergentes. Os fluxos de trabalho de proteômica de baixo para cima, como o FASP, dependem cada vez mais dos métodos LC-MS/MS realizados no modo de análise independente de dados (DIA), um método de digitalização que permite uma cobertura proteome profunda e baixa incidência de valores faltantes.
Neste relatório, forneceremos os detalhes de um fluxo de trabalho que combina um protocolo FASP, uma etapa de purificação de Ponta de Estágio duplo e LC-MS/MS no modo DIA para mapeamento de proteome urinário. Como amostra modelo, analisamos secreções prostáticas expressas (EPS)-urina, uma amostra coletada após um exame retal digital (DRE), que é de interesse em estudos de descoberta de biomarcadores de câncer de próstata.
Introdução
A constante evolução das tecnologias proteômicas promete ter um impacto considerável na ajuda ao diagnóstico da doença e na previsão de resposta ao tratamento, fornecendo mapas de alta resolução dos principais efeitos moleculares presentes em uma grande variedade de amostras, como tecidos e biofluidos. Do ponto de vista analítico, a urina oferece diversas vantagens, como facilidade de coleta e maior estabilidade do proteome em relação aos outros biofluidos1. A análise proteômica da urina é de especial interesse em estudos de descoberta de biomarcadores sobre cânceres urológicos, uma vez que permite amostragem não invasiva nas proximidades dos tecidos de interesse2. Em particular, uma amostra que parece ser promissora para o estudo de patologias relacionadas à próstata é a EPS-urina3,4 (ou seja, uma amostra de urina coletada após um exame retal digital (DRE)). Esta última operação antes da coleta amostral enriquece a urina com proteínas específicas da próstata. A ePS-urina é um bom candidato para investigar distúrbios relacionados à próstata5, incluindo câncer de próstata (PCa), uma vez que através de DRE, proteínas secretadas pelo tumor podem ser despejadas na amostra de urina, aumentando a chance de detectar proteínas específicas do tecido do câncer.
Um papel crucial para permitir a detecção e quantificação de biomarcadores potenciais de proteínas é desempenhado pela espectrometria de massa (MS). Nas últimas duas décadas, os protocolos baseados em MS para análise proteômica permitiram que um número cada vez maior de proteínas fosse detectado em uma única execução lc-MS/MS graças a melhorias contínuas na instrumentação de MS e no software de análise de dados6.
A preparação da amostra proteômica baseada em MS geralmente envolve a digestão enzimática da mistura proteica, que pode ser alcançada através de uma ampla variedade de protocolos como: digestão in-solution, MStern blotting7,trapping de suspensão (S-trap)8,preparação de amostra aprimorada em fase sólida (SP3)9, digestão em estágio10 e preparação filtrada de amostra (FASP)11. Todos os protocolos podem ser utilizados para proteômica urinária, embora os resultados possam variar em relação ao número de proteínas e peptídeos identificados e em termos de reprodutibilidade12.
Neste trabalho, nossa atenção foi focada na análise da EPS-urina pelo protocolo FASP. O protocolo FASP foi originalmente projetado para analisar proteínas extraídas de tecidos e culturas celulares, mas seu uso foi então expandido para a análise de outros tipos de amostras, como urina13. Com relação à digestão direta em solução, a FASP é uma abordagem proteômica mais flexível14,uma vez que ao alcançar a remoção efetiva de detergentes e outros contaminantes, como sais da mistura proteica antes da digestão enzimática15,permite a escolha de condições ideais de solubilização de proteínas. Além disso, uma característica adicional da FASP é que ela fornece um meio de concentração amostral. Isso é de particular interesse para a análise proteômica urinária, pois permite partir de volumes amostrais relativamente grandes (centenas de microliters). Diante do potencial do protocolo FASP, diversos estudos têm focado a atenção na automação do fluxo de trabalho, com o objetivo de reduzir a variabilidade experimental e processar um número elevado de amostras emparalelo 16.
Em nosso fluxo de trabalho, a FASP é seguida pela aquisição da LC-MS/MS em análise independente de dados (DIA), que proporciona alta cobertura proteome, boa precisão quantitativa e baixa incidência de valores faltantes. A abordagem DIA é um método sensível onde todos os íons são selecionados para eventos de MS/MS, oposto ao que acontece na análise dependente de dados (DDA) onde apenas íons com maior intensidade são fragmentados. O espectrômetro de massa, operando no modo DIA, realiza ciclos de varredura com largura de isolamento diferente cobrindo toda a faixa precursora m/z. Esta abordagem permite detectar reproduivelmente um alto número de peptídeos por unidade de tempo, fornecendo um instantâneo proteômico da amostra17. Além disso, os dados gerados pelo DIA têm outra característica interessante: a possibilidade de uma análise posterior 18. Os dados do DIA são mais complexos do que os obtidos pelo DDA, pois os espectros de MS/MS no DIA resultam do co-isolamento de vários íons precursores dentro de cada janela m/z 19. O desmembrar o espectro composto de MS/MS em sinais de peptídeos distintos e específicos é alcançado usando dois elementos fundamentais: uma biblioteca espectral e um software dedicado para análise de dados. A biblioteca espectral é gerada por um experimento dependente de dados, geralmente envolvendo fracionamento de peptídeos para maximizar a cobertura proteome, que fornece uma lista de milhares de íons precursores experimentalmente determinados e espectros de peptídeos des detectáveis na amostra em consideração. O software de análise de dados, em vez disso, usa as informações contidas na biblioteca espectral para interpretar os dados do DIA, gerando cromatogramas de íons extraídos específicos que permitem a detecção e quantificação de peptídeos. Embora a análise de dados DIA sem biblioteca seja agora viável, o DIA baseado em bibliotecas ainda fornece melhores resultados em termos de cobertura proteome20.
O protocolo de preparação da amostra aqui descrito (Figura 1) consiste nas seguintes etapas: uma etapa de centrifugação (para remover detritos celulares), digestão FASP, Purificação de Etapa21,quantificação de proteínas e análise dia. Este protocolo foi projetado para a análise da urina EPS no contexto da descoberta do biomarcador do câncer de próstata, mas pode ser aplicado à análise proteômica de qualquer amostra de urina.
Protocolo
O estudo foi aprovado pelo Comitê de Ética Institucional da Universidade Magna Graecia de Catanzaro, RP 41/2018. O consentimento informado por escrito foi obtido de todos os pacientes inscritos no estudo.
1. Preparação da amostra
- Centrifugar amostras de urina EPS dentro de 2h de coleta a 2.100 x g por 10 minutos em temperatura ambiente (RT). Armazene o supernatante a -80 °C até usar.
2. Degelo da amostra
- Transfira a amostra de -80 °C para -20 °C um dia antes da digestão.
- Antes do processamento da amostra, transfira a amostra por 15-20 minutos a 4 °C e, em seguida, na RT.
3. Preparação do reagente para FASP
- No mesmo dia, prepare as seguintes soluções: tampão de ureia, 50 mM iodoacetamida (IAA) no tampão de ureia, 50 mM de trietilamônio bicarbonato e 500 mM dithiothreitol (DTT).
- Prepare o tampão de ureia (ureia 8 M, 100 mM Tris-HCl pH 8): para 10 mL, pese 4.800 mg de ureia e dissolva-o na quantidade apropriada de água HPLC depois de adicionar 1 mL de 1 M Tris pH 8. É necessário um volume de 800 μL de ureia para cada amostra.
- Prepare 500 mM dithiothreitol (DTT): dissolva 38,5 mgs de DTT em 500 μL de água HPLC. Para cada amostra, são necessários 66,7 μL de DTT.
- Prepare 50 mM IAA em Tampão de Ureia: dissolva 9,25 mg de IAA em 1 mL de tampão de ureia. Para cada amostra são necessários 50 μL de IAA.
- Prepare 50 mM trietilamônio bicarbonato (TEAB). Adicione 150 μL de 1 M TEAB a 2,85 mL de água HPLC. Para cada amostra são necessários 460 μL de TEAB.
- Prepare uma solução de trippsina (100 ng/μL) em água HPLC. Esta solução pode ser armazenada a -80 °C e descongelada imediatamente antes de ser usada. Para cada amostra, são necessários 2 μL (200 ng).
4. Digestão de proteínas pela FASP
- Diluir 500 μL de cada amostra de EPS-urina com 66,7 μL de 10% (w/v) sulfato de dodecilo de sódio (SDS), 66,7 μL de 500 mM DTT e 33,3 μL de 1 M Tris pH 8 para solubilizar proteínas e reduzir as uniões de desulphide. Incubar 10 minutos a 95 °C com agitação suave.
- Adicione 300 μL de amostra de EPS-urina diluída na unidade do filtro (10 kDa) e centrífuga a 14.000 x g por 20 minutos.
- Adicione 200 μL de tampão de ureia ao filtro e centrífuga a 14.000 x g por 15 minutos. Repita esta etapa e depois descarte o fluxo.
- Adicione 50 μL de solução IAA e centrífuga a 6.000 x g por 25 minutos.
- Adicione 200 μL de tampão de ureia e centrífuga a 14.000 x g por 20 minutos. Repita esta etapa e depois descarte o fluxo.
- Adicione 200 μL de 50 mM tampão de bicarbonato de trietilamônio (TEAB) e centrífuga a 14.000 x g por 20 minutos. Repita este passo.
- Transfira a unidade do filtro para um novo tubo de coleta.
- Adicione 60 μL de tampão TEAB de 50 mM e 200 ng de trippsina e incubar a amostra em uma termoparla de 37 °C durante a noite.
NOTA: Para evitar a evaporação da amostra durante a incubação durante a noite, enrole cada unidade do filtro com uma camada de papel alumínio e, em seguida, uma camada de parafilm. - Após a incubação da trippsina, adicione 140 μL de água e centrífugue o frasco a 14.000 x g por 25 minutos para coletar o volume de digestão (180-200 μL).
5. Preparação do reagente para purificação de SCX
- Prepare 1 mL de lavagem 1 (0,5% de ácido fórmico (FA) e 20% acetonitrilo (ACN)) da seguinte forma: adicione 50 μL de 10% FA e 200 μL de 100% ACN a 750 μL de água HLPC. Para cada amostra, são necessários 100 μL de lavagem 1.
- Prepare 1,5 mL de lavagem 2 (0,5% FA e 80% de ACN) da seguinte forma: adicione 75 μL de 10 % FA e 1,2 mL de 100% ACN a 225 μL de água HPLC. Para cada amostra são necessários 190 μL de lavagem 2.
- Prepare 100 μL de solução de eluição (acetato de amônio de 500 mM (AA) e 20% de ACN): adicione 25 μL de 2 M AA e 20 μL de 100% ACN a 55 μL de água HPLC.
- Prepare as Dicas de Estágio da seguinte forma.
- Embale uma ponta de pipeta de 200 μL com uma camada de resina SCX usando uma agulha de seringa sem corte (calibre 16). Insira a Ponta de Estágio em uma tampa de microcentrifuge que tenha sido perfurada anteriormente para acomodar a microcolumn.
- Monte a tampa em um microcentrifuuge de 2 mL do qual a tampa original foi removida. O dispositivo centrífuga micro-SPE apenas montado deve caber em uma centrífuga no topo do banco. Uma vez que as tampas perfuradas são tediosas de se preparar, elas podem ser utilizadas várias vezes.
6. Purificação SCX
- Diluir 30 μL de cada amostra para 120 μL usando lavagem 2.
- Condicionar a Ponta de Estágio adicionando 50 μL de lavagem 1 em cima da resina de extração e centrífuga a 400 x g por 2 minutos. Uma vez que a resistência ao fluxo depende da força em que a resina de extração foi embalada na ponta da pipeta, a velocidade da centrífuga pode precisar ser ajustada para obter uma taxa de fluxo de 20-30 μL/min. Tente não deixar a ponta seca durante o carregamento e lavagem da amostra.
- Equilibre a Ponta de Palco com 50 μL de lavagem 2 e centrífuga a 400 x g por 2 minutos.
- Carregue a amostra diluída (120 μL) utilizando uma velocidade de giro mais baixa.
- Lave a Ponta de Palco com 50 μL de lavagem 2 e centrífuga a 400 x g por 2 minutos. Repita com 50 μL de lavagem 1.
NOTA: Após esta lavagem, a resina deve estar completamente seca. - Mistura de peptídeo eluto com 7 μL de solução de eluato (acetato de amônio de 500 mM (AA) e 20% de ACN) lentamente usando uma velocidade de giro mais baixa.
- Adicione 27 μL de FA de 0,1% para diluir AA e obter uma amostra para injeção preliminar de LC-MS/MS. A diluição final para 34 μL resultará em uma razão volumosa de 1:1 entre a solução de urina e peptídeo (1 μL de digestão purificada corresponderá a 1 μL de amostra de urina original).
7. Quantificação de proteína por padrão externo utilizando análise DDA(Figura 2)
- Determine a quantidade de proteína em amostras injetando 2 μL do digestor de peptídeo resultante em LC-MS/MS e interpolando a área total de peptídeo resultante com uma curva de calibração construída da seguinte forma:
- Prepare cinco soluções diferentes de digestão HeLa (1 ng/μL, 2,5 ng/μL, 7,5 ng/μL, 25 ng/μL, 75 ng/μL) utilizando um estoque de digestão HeLa (100 μg/μL).
- Injete cada solução Hela em duplicata (2 μL).
- Defina o método LC-MS/MS.
- Injete 2 μL das cinco misturas de peptídeos HeLa e peptídeos separados através de um gradiente linear de 75 minutos a uma vazão de 230 nL/minuto em uma coluna de 15 cm, 75 μm i.d embalada com partículas de sílica C18 de 3 μm. Utilize um gradiente binário constituído pela fase móvel A (0,1% FA, 2% ACN) e fase móvel B (0,1% FA e 80% de ACN).
- Realize a elução de gradiente aumentando o conteúdo da fase B móvel de 6% para 42% em 60 minutos e de 42% para 100% em 8 minutos adicionais. Mantenha por 5 minutos 100% B e depois volte para 0% B em dois minutos.
- Opere no modo DDA usando um método top-12 e defina o intervalo total de varredura MS em 350-1800 m/z,resolução 70.000, alvo ACG 1e6 e tempo máximo de injeção de 50 ms. Defina a janela de massa para isolamento de íons precursores em 1,6 m/z,resolução 35.000, alvo ACG 1e5, tempo máximo de injeção de 120 ms, fragmentação de HCD em 25 NCE (energia de colisão normalizada) e exclusão dinâmica de 15 segundos.
- Analise arquivos brutos usando Sequest como mecanismo de busca, e a sequência de proteínas HUMAN Uniprot Complete como banco de dados. Nos experimentos aqui apresentados, o banco de dados foi baixado em março de 2016 e continha 42.013 sequências.
- Use as seguintes configurações: Tolerância a MS 15 ppm, tolerância ms/ms 0.02 Da, trypsin como enzima, definindo dois locais de decote perdidos. Defina a carbamidomeção de linsina, serina, threonina, tyrosina (+57.021) e a oxidação de metioninas como modificações dinâmicas e a carbamidometilação de cisteínas (+57.021) como modificações estáticas. Use a taxa de corte de falsa taxa de descoberta (FDR) para identificação de peptídeos e proteínas para 0,01 e filtrar por percolator.
- Calcule a área de pico para todos os sinais de peptídeos MS1. Calcule a área total dos peptídeos.
- Injete 2 μL de misturas de peptídeos obtidas a partir de amostras de eps-urina usando o mesmo método LC-MS/MS utilizado para as amostras de HeLa e analise o arquivo bruto usando os mesmos parâmetros utilizados para o processamento de dados HeLa.
- Calcule a intensidade total dos sinais de peptídeos somando os valores de área de todos os peptídeos identificados e interpole a curva padrão externa. Isso fornecerá uma estimativa aceitável da quantidade de peptídeo presente no digestor de proteínas de urina EPS.
8. Preparação do reagente para o protocolo C18 StageTip
- Prepare 500 μL de solução A (0,1% ácido trifluoroacético (TFA), 50% ACN): adicione 250 μL de 100% ACN e 10 μL de 5% TFA a 240 μL de água HPLC.
- Prepare 2 mL de solução B (0,1 % TFA): adicione 40 μL de 5% TFA a 1960 μL de água HPLC.
- Prepare 100 μL de solução de eluição (0,1% FA, 50% ACN): adicione 1 μL de FA de 10% e 50 μL de 100% ACN a 49 μL de água HPLC.
- Prepare as Dicas de Estágio como descrito anteriormente. Neste caso, são utilizados discos de extração C18.
9. Protocolo SCX/C18 StageTip
NOTA: Purificar digestores de urina EPS pelo protocolo C18 StageTip para remover sais.
- Comece a partir de 2 μg de peptídeos (quantificados pela injeção preliminar). Diluir a solução de digestão 4 vezes na lavagem 2 SCX e proceder conforme descrito acima (na seção "Purificação SCX"). Elute a mistura de peptídeo com 7 μL de solução de eluato (500 mM acetato de amônio (AA) e 20% de ACN) lentamente usando uma velocidade de giro mais baixa (5 μL/min de fluxo).
- Acidificar o eluato SCX com 150 μL de 0,1% de ácido trifluoroacético (TFA) a fim de obter um pH menor que 3 e uma concentração de solvente orgânico abaixo de 5%.
- Condiça a Ponta de Estágio C18 com 50 μL de solução A e centrífuga a 400 x g por 2 minutos. Equilibre c18 StageTip com 50 μL de solução B e centrífuga a 400 x g por 2 minutos.
- Carregue a amostra na ponta do palco lentamente usando uma velocidade de giro mais baixa.
- Lave C18 StageTip com 50 μL de solução B e centrífuga.
- Lentamente elute a mistura de peptídeo com 10 μL de solução de eluição usando uma velocidade de giro mais baixa.
- Seque o elunato (10 μL) a 30 °C em uma centrífuga de vácuo por 3 minutos. Adicionar 47 μL de 0,1% FA. A concentração de peptídeos esperados será de 40 ng/ μL. Analisar por LC-MS/MS no modo DIA.
10. Análise DIA (Figura 3)
- separação nanoLC: Separe a mistura de peptídeos usando um gradiente linear de 140 minutos a uma taxa de fluxo de 230 nL/min em uma coluna de 15 cm, 75 μm ID embalada com partículas de sílica C18 de 3 μm.
- Realize a elução gradiente a 230 nL/minuto de fluxo e aumente o conteúdo da fase B móvel de 3% para 25% em 90 minutos, depois de 25% a 40% em 30 minutos e, finalmente, de 40% para 100% em 8 minutos.
- Após 10 minutos a 100% B, equilibre a coluna com a fase móvel A por 15 minutos.
- Parâmetros espectrométricos em massa: realize a análise dia usando um método que emprega o seguinte ciclo de varredura: (i) um evento ms de varredura completa com uma resolução de 17.500 (alvo AGC 1e6, tempo máximo de injeção de 50 ms) seguido de (ii) 20 janelas a 20 m/z de largura de isolamento, começando a partir de m/z 350, (ii) 5 janelas a 50 m/z de largura de isolamento e (iv) 1 janela a 200 m/z de largura de isolamento, terminando a faixa de varredura de DIA a 1200 m/z. Realize as varreduras DIA na resolução 35.000 (alvo AGC 5e5, tempo máximo de injeção de 120 ms e 25 NCE).
11. Fracionamento de fase C18 invertido de alta pH para geração de biblioteca
NOTA: Pool EPS-urine representative amostras em uma quantidade superior a 10 μg, a fim de construir uma biblioteca dependente de dados para análise dia pelo seguinte procedimento:
- Acidificar a mistura de peptídeos (11 μg) por 0,2% de TFA e carregar a solução resultante em uma C18 StageTip embalada com duas camadas de discos de extração para permitir maior capacidade. Estimamos que a capacidade de carregamento de um único micro disco (de uma agulha de seringa de 16 calibres) esteja na faixa de 5-10 μg.
- Condição e fase estacionária de equilíbrio, conforme já descrito para purificação de C18.
- Realizar fracionamento de peptídeos por eluição stepwise (n=10) a partir da fase C18 invertida de pH alto usando soluções de elução contendo 0,2% de hidróxido de amônio, 10 mM TEAB, e aumento das concentrações v/v de ACN (4%, 8%, 12%, 16%, 20%, 24%, 28%, 32%, 40%, 80%).
- Analise as 10 frações utilizando os mesmos parâmetros cromatográficos do método DIA. Opere o espectrômetro de massa no modo DDA usando as configurações anteriormente adotadas para a análise preliminar (ver "Quantificação de proteína por padrão externo usando análise DDA").
12. Análise de dados
- Gere a biblioteca espectral importando os resultados de identificação de proteínas obtidos a partir dos experimentos DDA LC-MS/MS de fracionamento C18 de fase ultraversa em um software dedicado à análise do DIA.
- Fixar o número mínimo e máximo de íons de fragmento utilizados para identificação e quantificação para 3 e 6, respectivamente. Filtre os resultados obtidos por um valor Q de 0,01.
- Importe arquivos brutos DIA e associe a cada arquivo bruto o mesmo arquivo FASTA usado para criar a biblioteca espectral.
- Defina o número mínimo e máximo de peptídeos únicos utilizados para quantificação proteica para 1 e 10, respectivamente.
- Calcule a intensidade de cada proteína somando a área de pico de íons fragmentos.
- Gerar uma matriz com a intensidade das proteínas quantificadas em cada amostra.
Resultados
Este protocolo de análise proteômica urinária inclui as seguintes etapas: digestão fasp, estimativa da quantidade de proteína via calibração padrão externa, purificação de duplo Estágio (SCX e C18),e análise LC-MS/MS no modo DIA.
Após a digestão da proteína, as injeções preliminares são realizadas após a purificação do StageTip SCX dos peptídeos resultantes. Os arquivos brutos LC-MS/MS são processados para obter o número de peptídeos identificados, o número de proteínas identificadas e a área de peptídeos detectados. A área total obtida somando todos os peptídeos identificados é usada para estimar o conteúdo proteico via interpolação a um padrão externo: um digestor de proteína HeLa injetado em cinco quantidades diferentes (2, 5, 15, 50, 150 ng, respectivamente). A quantidade de proteína para as seis amostras analisadas neste estudo variou de amostra para amostra, mostrando um valor médio de 78 ng/μL.
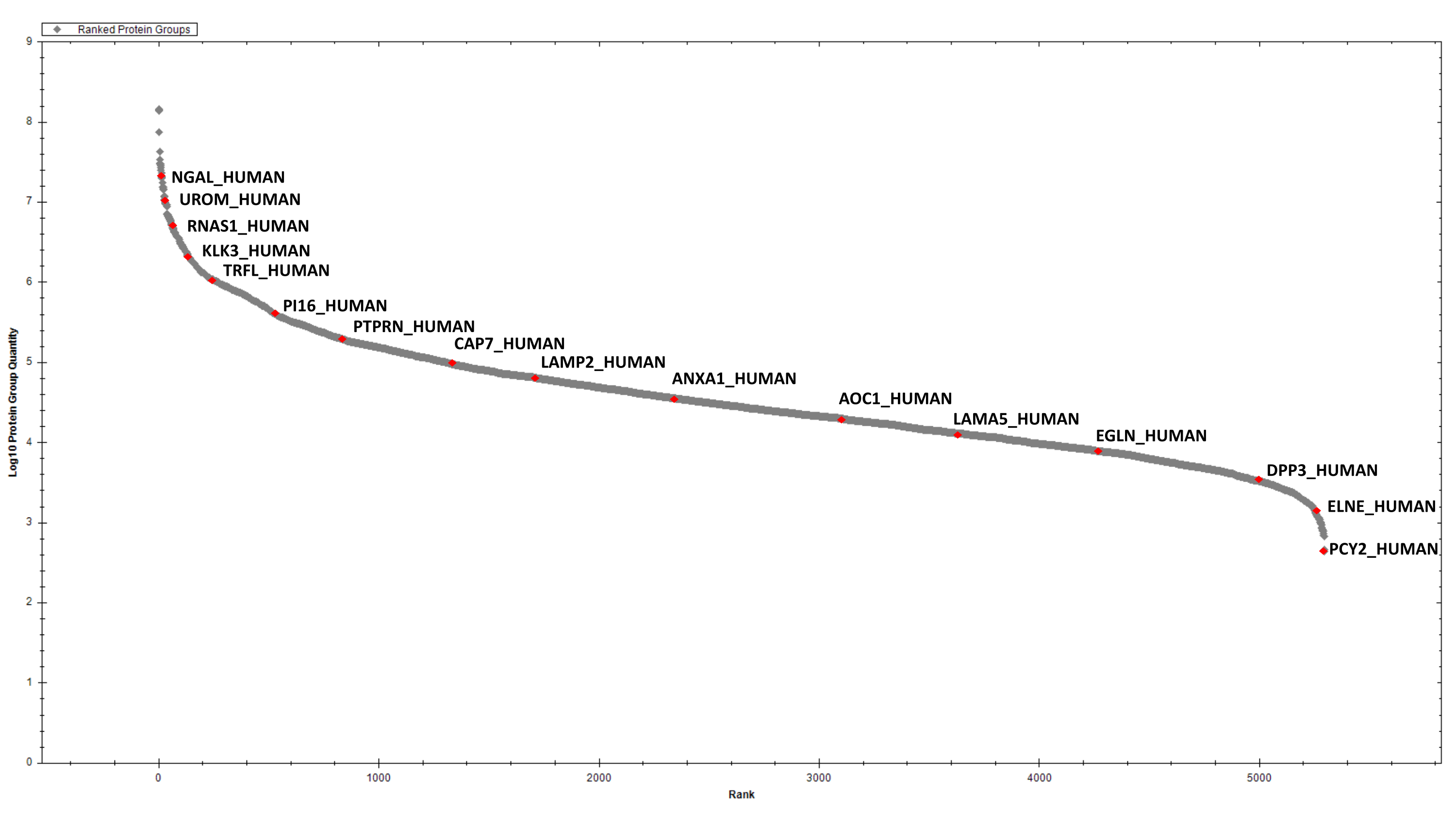
Após a estimativa da proteína, 2 μg de proteínas digeridas de cada amostra são purificadas por SCX sequencial e C18 StageTip antes da análise dia. A biblioteca espectral para pesquisa de dados DIA é criada após fracionamento de fase invertida de alto pH e análise de DDA LC-MS/MS de uma amostra representativa (por exemplo, um pool de amostras). Utilizando os parâmetros descritos acima, identificamos e quantificamos, cumulativamente, 2387 grupos proteicos nas seis amostras de EPS-urina em consideração(Tabela Suplementar 1 e Figura 4).
Para avaliar do ponto de vista qualitativo a relevância da lista de proteínas identificadas e quantificadas, a matriz obtida foi comparada a uma lista de 624 proteínas previamente identificadas em EPS22direto, amostra coletada em um procedimento mais invasivo, que provou ser fonte de biomarcadores de câncer de próstata candidatos interessantes. No total, 508 das 624 proteínas foram detectadas com sucesso pelo nosso protocolo FASP/DIA sobre eps-urina.

Figura 1: Fluxo de trabalho proteômico. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Representação do nosso design experimental para quantificação de proteínas com base no padrão externo (HeLa digest). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Etapas-chave da análise do DIA: (i) elaboração da biblioteca espectral através de fracionamento de fase invertida de alto pH e análise de DDA, (ii) análise amostral utilizando a abordagem DIA, (iii) análise de dados pelo Spectronaut. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Gráfico classificado para as proteínas eps-urinárias detectadas. Alguns hits selecionados são rotulados. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela suplementar 1: Tabela representativa de proteínas quantificadas em seis amostras de EPS-urina em Spectronaut. Clique aqui para baixar esta Tabela.
Discussão
Neste trabalho, é apresentada uma estratégia de análise de amostras de EPS-urina. O protocolo FASP é uma escolha ideal para proteômica urinária porque permite concentração de amostras antes da digestão enzimática. De fato, usando este fluxo de trabalho, várias centenas de microliters de urina podem ser carregados em um único filtro e processados. Além disso, a digestão no filtro oferece relativa liberdade na escolha das condições de desnaturação. Em nosso trabalho, a desnaturação de proteínas é obtida pela diluição de amostras de urina em um tampão contendo Tris, SDS e DTT (concentração final: 50 mM Tris, 1% SDS, 50 mM DTT). As proteínas são eficientemente desnaturadas imediatamente após o descongelamento da amostra, a fim de evitar a degradação indesejada por proteases ativas. O protocolo FASP original foi melhorado aumentando o volume de lavagens de 100 μL para 200 μL. Dessa forma, é possível uma melhor remoção de resíduos, especialmente detergentes do filtro.
Após a digestão enzimática, a estimativa de proteína por padrão externo utilizando um método rápido de LC-MS/MS, dependente de dados é realizado antes das últimas etapas do protocolo, que compreendem a purificação dupla de StageTip e a análise de LC-MS/MS no modo DIA, em material inicial igual para todas as amostras (2 μg).
A versatilidade do FASP está associada à abordagem DIA, um método sensível que proporciona um baixo número de valores faltantes17. Em nosso trabalho, 2387 proteínas foram identificadas e quantificadas, permitindo a elaboração de um perfil proteômico detalhado da urina EPS. A identificação e quantificação de 2387 proteínas foi possível através da geração de uma rica biblioteca espectral, obtida através de fracionamento reverso de peptídeos de alto pH, e pelo nosso método DIA, direcionado a uma ampla gama precursora de m/z. Este fluxo de trabalho identificou mais de 80% das proteínas encontradas anteriormente na análise direct-EPS, demonstrando que uma fração considerável do proteome EPS-urinário é de fato derivada de secreções prostáticas expressas, sendo assim uma rica fonte de proteínas específicas do tecido da próstata26.
Em conclusão, nosso design experimental acomente a versatilidade da FASP com a sensibilidade do DIA para obter um rico mapa do proteome urinário. Esta estratégia é recomendada para analisar amostras de EPS-urina, mas seu uso pode ser estendido para proteômica urinária em geral, ou mesmo para outros tipos de amostra.
Divulgações
Todos os autores não declaram conflito de interesses.
Agradecimentos
Este trabalho contou com o apoio da MIUR (Ministero Università Ricerca, PRIN 2017 a MG) e da POR Calabria FESR 2014-2020, ação 1.2.2, "INNOPROST".
Materiais
| Name | Company | Catalog Number | Comments |
| 1 M Tris HCL pH 8.0 | Lonza | 51238 | |
| 2-iodoacetamide for synthesis | Merck | 8047440100 | |
| Acetonitrile for HPLC LC-MS grade | VWR | 83640.290 | |
| Ammonium acetate | Fluka | 9690 | |
| ammonium hydroxide solution | Sigma | 30501 | |
| ammonium hydroxide volumetric standard, 5N solution in water | Merck | 318612-500ML | |
| Dithiothreitol | Amresco | 0281-25G | |
| Empore Cation 47mm Extraction Disks | Microcolumn | 2251 | |
| Empore Disk C18 | Varian | 12145004 | |
| Formic acid optima | Fisher Scientific | A117-50 | |
| Hela Protein Digest Standard | Fisher Scientific | 88329 | |
| Microcon-10kDa Centrifugal Filter Unit with Ultracel-10 membrane | MERCK | MRCPRT010 | |
| sodium dodecyl sulfate solution | Merck | 71736-500ML | |
| Thiethylammonium bicarbonate buffer | Merck | T7408-100ML | |
| trifluoroacetic acid | Riedel-de Haën | 34957 | |
| Trypsin from porcine pancreas | Merck | T6567-1MG | |
| Urea | Merck | U6504-500G | |
| Water HPLC gradient grade | Fisher Scientific | W/0106/17 | |
| Proteome Discoverer 1,4 | Thermo Fisher Scientific | ||
| Spectronaut 14.0 | Biognosys |
Referências
- Hüttenhain, R., Soste, M., Selevsek, N., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 4 (142), 10(2013).
- Roobol, M. J., Carlsson, S. V. Risk stratification in prostate cancer screening. Nature Reviews Urology. 10 (1), 38-48 (2013).
- Principe, S., et al. Identification of prostate-enriched proteins by in-depth proteomic analyses of expressed prostatic secretions in urine. Journal of Proteome Research. 11 (4), 2386-2396 (2012).
- Kim, Y., et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nature Communications. 7, 1-10 (2016).
- Drake, R. R., et al. Clinical collection and protein properties of expressed prostatic secretions as a source for biomarkers of prostatic disease. Journal of Proteomics. 72 (6), 907-917 (2009).
- Macklin, A., Khan, S., Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clinical Proteomics. 17 (1), 1-25 (2020).
- Berger, S. T., et al. MStern blotting-high throughput polyvinylidene fluoride (PVDF) membrane-based proteomic sample preparation for 96-well plates. Molecular and Cellular Proteomics. 14 (10), 2814-2823 (2015).
- Zougman, A., Selby, P. J., Banks, R. E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics. 14 (9), 1006(2014).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10 (10), 757(2014).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11 (3), 319-324 (2014).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6 (5), 359-362 (2009).
- Ding, H., et al. Urine Proteomics: Evaluation of Different Sample Preparation Workflows for Quantitative, Reproducible, and Improved Depth of Analysis. Journal of Proteome Research. 19 (4), 1857-1862 (2020).
- Zhao, M., et al. A comprehensive analysis and annotation of human normal urinary proteome. Scientific Reports. 7 (1), 1-13 (2017).
- Wiśniewski, J. R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Analytical Chemistry. 88 (10), 5438-5443 (2016).
- Wiśniewski, J. R., Zielinska, D. F., Mann, M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Analytical Biochemistry. 410 (2), 307-309 (2011).
- Yu, Y., et al. Urine sample preparation in 96-well filter plates for quantitative clinical proteomics. Analytical Chemistry. 86 (11), (2014).
- Gillet, L. C., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Molecular and Cellular Proteomics. 11 (6), 1-17 (2012).
- Liu, Y., Hüttenhain, R., Collins, B., Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Review of Molecular Diagnostics. 13 (8), 811-825 (2013).
- Gallien, S., Duriez, E., Demeure, K., Domon, B. Selectivity of LC-MS/MS analysis: Implication for proteomics experiments. Journal of Proteomics. 81, 148-158 (2013).
- Muntel, J., et al. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. Journal of Proteome Research. 14 (11), 4752-4762 (2015).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Kim, Y., et al. Identification of Differentially Expressed Proteins in Direct Expressed Prostatic Secretions of Men with Organ-confined Versus Extracapsular Prostate Cancer. Molecular & Cellular Proteomics. 11 (12), 1870-1884 (2012).
- Decramer, S., et al. Urine in clinical proteomics. Molecular and Cellular Proteomics. 7 (10), 1850-1862 (2008).
- Thongboonkerd, V. Practical points in urinary proteomics. Journal of Proteome Research. 6 (10), 3881-3890 (2007).
- Konvalinka, A., Scholey, J. W., Diamandis, E. P. Searching for new biomarkers of renal diseases through proteomics. Clinical Chemistry. 58 (2), 353-365 (2012).
- Drake, R. R., et al. In-Depth Proteomic Analyses of Direct Expressed Prostatic Secretions. Journal of Proteome Research. 9 (5), 2109-2116 (2010).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados