
Method Article
Haut-débit Gene Tagging dans
Dans cet article
Résumé
Addition of a tag to a protein is a powerful way of gaining insight into its function. Here, we describe a protocol to endogenously tag hundreds of Trypanosoma brucei proteins in parallel such that genome scale tagging is achievable.
Résumé
Improvements in mass spectrometry, sequencing and bioinformatics have generated large datasets of potentially interesting genes. Tagging these proteins can give insights into their function by determining their localization within the cell and enabling interaction partner identification. We recently published a fast and scalable method to generate Trypanosoma brucei cell lines that express a tagged protein from the endogenous locus. The method was based on a plasmid we generated that, when coupled with long primer PCR, can be used to modify a gene to encode a protein tagged at either terminus. This allows the tagging of dozens of trypanosome proteins in parallel, facilitating the large-scale validation of candidate genes of interest. This system can be used to tag proteins for localization (using a fluorescent protein, epitope tag or electron microscopy tag) or biochemistry (using tags for purification, such as the TAP (tandem affinity purification) tag). Here, we describe a protocol to perform the long primer PCR and the electroporation in 96-well plates, with the recovery and selection of transgenic trypanosomes occurring in 24-well plates. With this workflow, hundreds of proteins can be tagged in parallel; this is an order of magnitude improvement to our previous protocol and genome scale tagging is now possible.
Introduction
Trypanosoma brucei est un parasite protozoaire qui provoque trypanosomasis humaine africaine et le nagana chez les bovins. T. brucei est un organisme idéal pour l'analyse de la fonction des protéines en raison de la combinaison d'un génome de haute qualité, de nombreux protéomique et des ensembles de données de transcriptomique et des outils moléculaires bien développés 1-3. Les progrès de la protéomique et de séquençage ont abouti à grands ensembles de données qui mettent en évidence potentiellement gènes intéressants 4-6; Cependant, de nombreux gènes ont un minimum d'information qui leur sont associés dans les bases de données existantes. Il existe donc un besoin d'une méthode à haut débit pour faciliter la caractérisation fonctionnelle des protéines.
L'expression d'une protéine marquée peut donner une pluralité de points de vue sur la fonction d'une protéine. Par exemple, une protéine marquée avec une protéine fluorescente ou un epitope peuvent être localisés par microscopie à fluorescence, ce qui donne des informations sur l'endroit où la protein pourrait exercer son effet biologique. En variante, une protéine marquée avec un TAP 7, 8 ou halotag Son tag peut être purifié pour des analyses biochimiques et l' identification de ses partenaires d'interaction.
Nous avons récemment développé une méthodologie de marquage robuste pour T. 9 brucei. Cette habitude de longue amorce de PCR pour générer l'ADN pour la transfection et a permis le marquage des dizaines de protéines en parallèle - une amélioration importante des protocoles existants. Nous avons maintenant amélioré l'évolutivité de ce protocole dans les formes procycliques par un ordre de grandeur. Ici, nous présentons notre méthode où nous effectuons la PCR et la transfection dans des plaques à 96 puits, avec la récupération et la sélection se produisant dans des plaques à 24 puits. Comme des centaines de protéines peuvent maintenant être étiquetés en parallèle cette méthode fournit une méthode rentable et réalisable pour le marquage du génome du trypanosome entier.
Protocole
1. 96 puits à long PCR Primer
- Pré-gel refroidisseur 96 puits PCR à -80 ° C congélateur.
- Dans un tube de 10 ml, préparer Master Mix A: 2,100 ul ddH 2 O, 105 ul PCR de qualité DMSO, 105 pi 10 mM dNTP, 105 pl pPot gabarit (25 ng / pl) 9, pour un volume total de 2415 pi par plaque .
- Aliquoter 23 ul dans chaque puits d'une plaque à 96 puits PCR.
- Ajouter 2 pi de 100 uM communs amorces à chaque puits chargé à l'aide d'une pipette multicanaux P20 12 canaux. Sceller avec le film, le lieu plaque sur le refroidisseur PCR pré-refroidi et laisser geler pendant un minimum de 20 min à -80 ° C.
- Dans un tube de 10 ml, préparer Master Mix B: 2,048 ul ddH 2 O, 525 ul 10x tampon, 52,5 ul polymérase pour un volume total de 2626 pi par plaque.
- Retirer la plaque et le refroidisseur PCR à partir de -80 ° C congélateur.
- Avec la plaque encore sur le refroidisseur PCR, ajouter 25 pl Master Mix B sur le dessus du figean Master Mix A pour un volume réactionnel total de 50 ul. Ce temps est critique et devrait être achevée avant Mix A peut dégeler. Sceller la plaque avec un film.
- Définir cycleur thermique comme suit: 94 ° C pendant 10 min, puis 30 cycles de 94 ° C pendant 15 s, 65 ° C pendant 30 secondes, 72 ° C pendant 2 minutes suivie d'une période d'extension finale de 72 ° C pendant 7 min .
- Chargez la plaque PCR après le bloc a atteint 94 ° C.
2. Validation de 96 puits à long PCR Primer
- Lancer un grand 1% (p / v) d'un gel d'agarose dans du Tris acétate EDTA (TAE) tampon en cours avec des peignes 4 x 28 puits pour donner un gel avec 4 rangées de puits. Alignez alternés dents du peigne avec les pointes d'une pipette multicanaux 12 canaux. plonger délicatement le gel dans TAE tampon courir après les voies sont chargés.
- Charge 1 kb échelle d' ADN dans le 1 er et le 28 ème puits de chaque rangée.
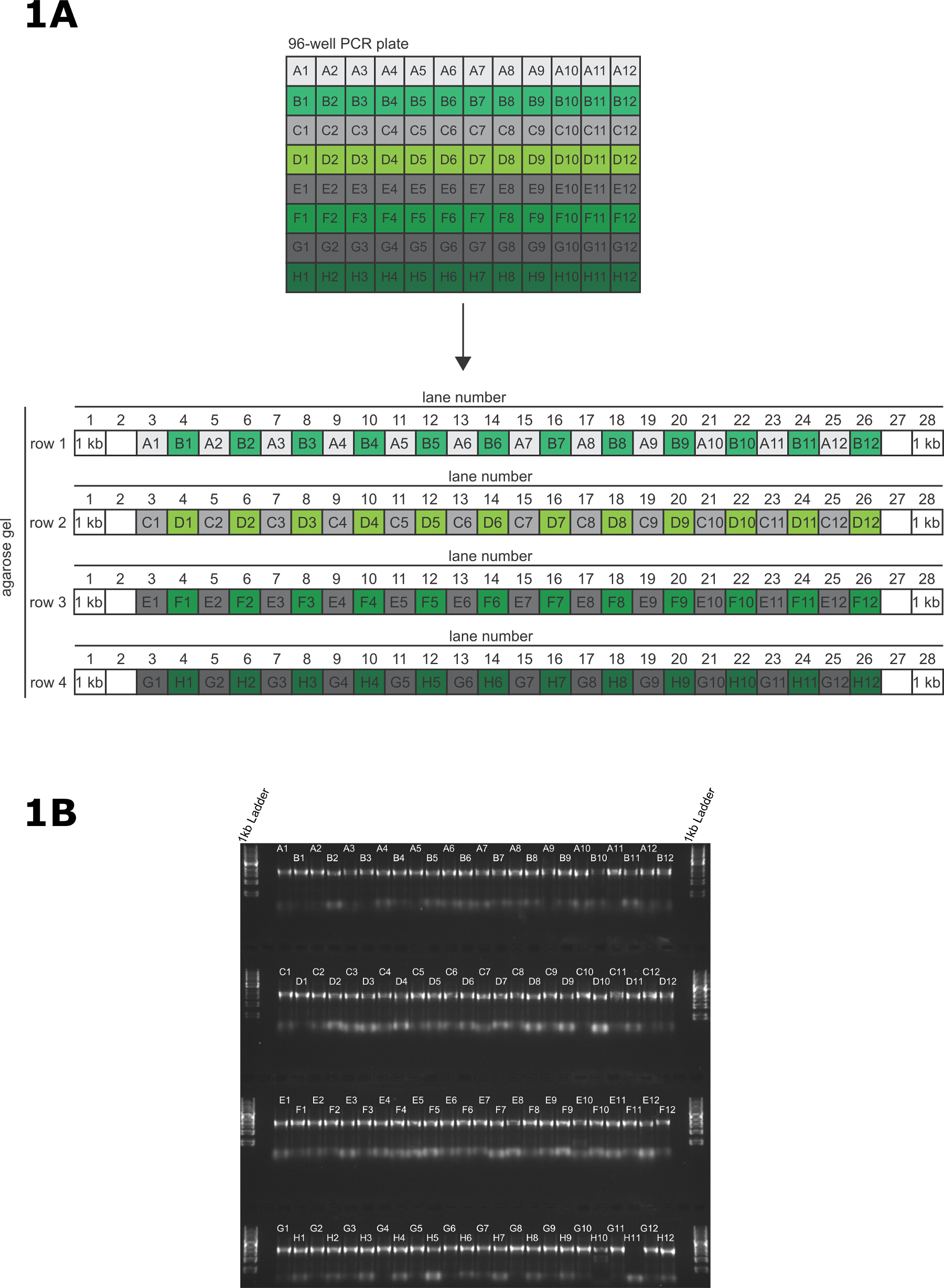
- En utilisant une pipette multicanaux P20 12 canaux, transférer 2 pl de produit PCR directement à chaque piste du gel (figure 1A). Pour ce faire rapidement pour réduire la possibilité de contamination des amplicons.
- Chargez les produits de PCR à partir de la ligne A de la plaque PCR dans les voies impaires de la 1ère rangée du gel (A1 - piste 3, A2 - piste 5 ...... A11 - piste 23, A12 - piste 25) et charger les produits de PCR à partir de la ligne B de la plaque PCR dans les voies paires de la 1 ère ligne du gel (B1 - piste 4, A2 - piste 6 ...... B11 - voie 24, B12 - piste 26).
- Chargez les produits de PCR à partir de la ligne C et D de la plaque PCR en utilisant le même schéma que décrit ci-dessus, avec la ligne C dans l'étrange voies numérotées et rangée D dans les voies paires. Continuer ce modèle de chargement jusqu'à ce que chaque puits de la plaque PCR est chargé sur le gel. tampon ADN de chargement ne soit pas nécessaire de charger ce gel.
- Placer le gel dans le réservoir en cours d'exécution et de remplir le réservoir avec TAE tampon en cours d'exécution jusqu'à ce que le gel est immergé. Exécutez à 100 V pendant 30 min etvisualiser en utilisant un transilluminateur UV. Voir la figure 1B pour un gel d'exemple.
Remarque: Les produits de PCR peuvent être conservés à -20 ° C pendant plusieurs semaines avant la transfection
3. Transfection 96 puits
- Utilisez le T. brucei procyclique ligne sous forme de cellules SMOXP9 10 pour cette procédure et de grandir dans SDM-79 contenant 10% de FCS 10.
- En utilisant 1 x 10 7 cellules par transfection (total de 1,1 x 10 9 cellules) lorsque le marquage sur l'extrémité N de la protéine et 2 x 10 7 cellules par transfection (total de 2,2 x 10 9 cellules) lorsque le marquage sur l' extrémité C - terminale de la protéine. Maintenir les cellules à la mi-log (1,2 x 10 6 -1 x 10 7 cellules / ml) pendant plusieurs jours avant la transfection et récolter les cellules pour la transfection à une densité de 5-8 x 10 6 cellules / ml.
- Laisser les produits congelés de PCR pour la transfection décongeler à la température ambiante.
- Compter les cellules en utilisant un hèmeocytometer ou compteur de cellules automatique.
- Pellet nombre requis de cellules dans plusieurs tubes de 50 ml à 800 x g pendant 10 min.
- Après centrifugation Rejeter le surnageant et remettre en suspension les cellules dans 10 ml CYTOMIX modifiée (0,8 mM d' EGTA, 24 mM de KCl, 0,15 mM de CaCl2, 10 mM de phosphate de potassium pH 7,6 tampon, 25 mM de HEPES-KOH à pH 7,6, 2,6 mM de MgCl2, 0,5% (p / v) de glucose, 100 pg / ml de BSA, 1 mM d'hypoxanthine, 144 mM de saccharose) par tube. Transférer toutes les solutions de cellules à un seul tube et tourner de nouveau à 800 xg pendant 10 min.
- Après centrifugation, éliminer le surnageant et remettre en suspension les cellules dans 23 ml de CYTOMIX modifié pour obtenir une concentration finale de 5 x 10 7 cellules / ml.
- Alors que les cellules tournent, ajouter 1 ml de SDM-79 médias pour chaque puits de 4 x 24 puits des plaques de culture de tissus. Etiqueter les plaques AD et dessiner un anneau autour de puits A1 sur chaque plaque de marqueur permanent pour aider à l'orientation de la plaque.
- Connectez le gestionnaire de la plaque à l'électroporation. Sur la electroporaUnité tor régler la tension de 1500 V, la longueur d'impulsion de 100 microsecondes, et le nombre d'impulsions à 12 et l'intervalle d'impulsion de 500 ms. Sur le manipulateur de plaques, régler le nombre d'impulsions à 1. Ceci assure que cette électroporation appliquera une impulsion unique à chacune des 12 colonnes de la plaque d'électroporation.
- L'utilisation d'un P200 12 canaux pipette multicanaux, transférer les réactions de PCR de la plaque PCR sur les plaques d'électroporation 96 puits jetables, écart de 4 mm.
- Lorsque les cellules ont été filé et sont remises en suspension à la concentration finale de 5 x 10 7 cellules / ml (étape 3.7), le transfert vers un réservoir de réactif. Pipette 200 pi de la solution de cellules dans chaque puits de la plaque d'électroporation en utilisant un canal pipette multicanaux P200 12, mélanger avec le produit PCR par pipetage.
- En utilisant un tissu enlever les gouttelettes de la partie supérieure de la plaque pour éviter les courts-circuits.
- Appliquer le film d'étanchéité prévue dans l'emballage de plaque à la partie supérieure de la plaque d'électroporation, posil itioning de façon à laisser des trous pour les électrodes à découvert en haut et en bas de chaque colonne. Évitez de couvrir les points soulevés utilisés pour guider le film.
- Chargez la plaque d'électroporation dans le manipulateur de plaques et fermer le couvercle. Appuyez sur 'Pulse' sur l'unité de electroporator.
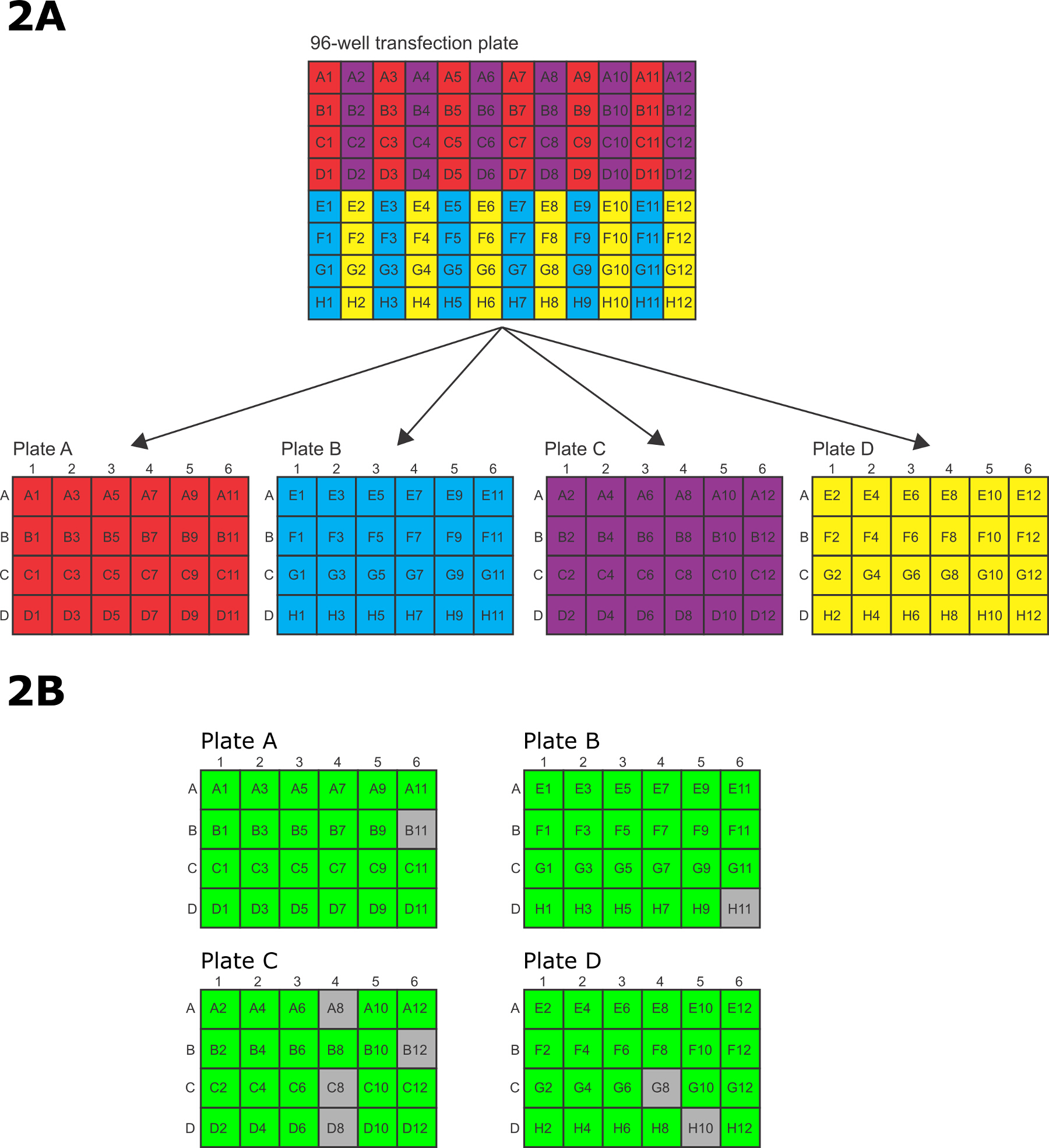
- Après l'électroporation, le transfert rapide des cellules du 96 puits électroporation plaque à 4 x 24 puits des plaques de culture tissulaire. Pour transférer les cellules, utilisez un P200 12 canaux pipette multicanaux avec chaque autre bout manquant de telle sorte que les 6 conseils restants alignés avec les 6 rangs sur une plaque de culture tissulaire de 24 puits. Transférer les cellules électroporées dans les 96 puits de plaques à électroporations motif prédéfini pour les 24 puits des plaques de culture tissulaire (figure 2A).
- Préparer les embouts de pipette P200 - pour chaque transfection de 96 puits préparer 2 boîtes de 96 pointes avec chaque autre colonne supprimée.
- Transfert puits A1 / A3 / A5 / A7 / A9 / A11 sur la plaque d'électroporation 96 puits àplaque A la ligne A des plaques de culture de tissus à 24 puits, B1 / B3 / B5 / B7 / B9 / B11 dans la rangée B, C1 / C3 / C5 / C7 / C9 / C11 dans la ligne C et D1 / D3 / D5 / D7 / D9 / D11 en ligne D. Après le transfert de chaque ensemble de 6 puits de la plaque de 96 puits à la plaque de 24 puits, les puits de la plaque de 96 puits sont ensuite lavés avec les médias des puits correspondants dans le 24- bien plaque et le lavage est ensuite transféré vers les puits dans la plaque de 24 puits.
- puits de transfert E1 / E3 / E5 / E7 / E9 / E11 sur la plaque d'électroporation 96 puits à 24 puits plaque B rangée A, F1 / F3 / F5 / F7 / F9 / F11 dans la rangée B, G1 / G3 / G5 / G7 / G9 / G11 dans la ligne C et H1 / H3 / H5 / H7 / H9 / H11 en ligne D. Après le transfert de chaque ensemble de 6 puits de la plaque de 96 puits à la plaque de 24 puits, les puits dans le 96 plaque -Bien sont ensuite lavées avec les médias des puits correspondants de la plaque de 24 puits et le lavage est ensuite transféré dans les puits dans la plaque de 24 puits.
- Transfert puits A2 / A4 / A6 / A8 / A10 / A12 sur la plaque d'électroporation 96 puits à 24 puits plaque C rangée A, B2 / B4 / B6 / B8 / B10 / B12 dans la rangée B, C2 / C4 / C6 / C8 / C10 / C12 dans la ligne C et D2 / D4 / D6 / D8 / D10 / D12 en ligne D. Après le transfert de chaque ensemble de 6 puits de la 96 plaque eh bien à la plaque de 24 puits, les puits de la plaque de 96 puits sont ensuite lavées avec les médias des puits correspondants de la plaque de 24 puits et le lavage est ensuite transféré dans les puits dans la plaque de 24 puits.
- puits de transfert E2 / E4 / E6 / E8 / E10 / E12 sur la plaque d'électroporation 96 puits à 24 puits plaque D rangée A, F2 / F4 / F6 / F8 / F10 / F12 dans la rangée B, G2 / G4 / G6 / G8 / G10 / G12 dans la ligne C et H2 / H4 / H6 / H8 / H10 / H12 en ligne D. Après le transfert de chaque ensemble de 6 puits de la plaque de 96 puits à la plaque de 24 puits, les puits dans le 96 plaque -Bien sont ensuite lavées avec les médias des puits correspondants de la plaque de 24 puits et le lavage est ensuite transféré dans les puits dans la plaque de 24 puits.
- En utilisant un microscope à contraste de phase inversée, examiner 1 puits de chaque colonne pour vérifier les cellules. Il y aura un mélange de cellules vivantes et mortes avecquantité importante de débris cellulaires. Les cellules vivantes peuvent être distinguées des cellules mortes parce qu'ils seront brillants au microscope à contraste de phase et seront en mouvement.
- Placez les plaques à 24 puits dans une boîte en plastique propre avec un couvercle qui forme un joint étanche. Mettez la boîte dans un incubateur à 28 °.
- Entre 6-8 heures plus tard, ajouter 1 ml de SDM-79 avec 10% de FCS contenant un antibiotique sélectif 2x à chaque puits. Dans notre expérience, les lignées cellulaires où les gènes sont marqués sur l'extrémité N sont résistantes à des concentrations plus élevées de médicaments sélectifs. Par conséquent, en utilisant la blasticidine, par exemple, le marquage à l'extrémité N-terminale nécessite une concentration finale de 20 pg / ml de blasticidine et le marquage sur l'extrémité C-terminale nécessite une concentration finale de 10 pg / ml de blasticidine.
Note: les lignées cellulaires transgéniques deviendront visibles 9-10 jours après la transfection. - Passage au moins deux fois dans la drogue nouvelle et les médias avant l'analyse. La figure 2B montre les résultats d'un transfe 96 puits typiquection pour le marquage 96 protéines différentes sur la terminaison N. Évaluer chaque puits par microscopie à contraste de phase afin de déterminer si elle contient des cellules saines, résistantes aux médicaments. Un puits sain contient principalement (<95%) des cellules très actives qui sont lumineuses en microscopie contrat de phase. Une analyse subséquente par microscopie épifluorescence ou western blot est nécessaire pour déterminer si la protéine a été identifiée avec succès.
Résultats
Dans cette transfection représentative, les amorces ont été conçues en utilisant le script TagIt perl 9 et synthétisés dans le commerce. Le 96 puits PCR a été réalisée et validée comme décrit (figure 1A); dans cet exemple, ont réussi 95/96 PCR (figure 1B). Dans notre expérience, en répétant les réactions ont échoué soit avec les amorces originales ou des amorces de re-synthèse ne se traduit pas dans une PCR réussie.
Les amplicons ont été transférées dans des plaques de 96 puits électroporation pour l' électroporation (figure 2A), puis transférées à 24 puits des plaques de culture de récupération et de sélection. Après un temps suffisant pour permettre la sélection de se produire, les puits ont été notés pour la survie avant l'analyse ultérieure. Dans cet exemple, 88/96 puits (92%) étaient positifs après la sélection de 15 jours.
Figure 1: Validation de longue amorce de PCR (A) Motif de transfert de produits de PCR provenant de plaque à 96 puits par PCR sur le gel d' agarose pour la validation de la PCR.. (B) A 96 puits PCR gel de validation représentant pour le marquage 95 gènes sur la terminaison N et 1 amplicon ne contenant pas 5 'homologie de ciblage pour agir en tant que témoin négatif dans la transfection. Les échantillons sont chargés sur un 1% (p / v) gel d'agarose et résolues à 100 V pendant 30 min. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 2: (A) Motif de transfert de cellules électroporées 96 puits électroporation plat à 96 puits électroporation et sélection.e à 24 puits des plaques de culture tissulaire. (B) montrant un schéma d' un pointage en direct / mort typique après 15 jours de sélection dans des plaques à 24 puits. Le vert représente une transfection réussie, gris représente un puits avec seulement les cellules mortes. Dans cet exemple, 88/96 (92%) des puits contenaient des parasites transfectées avec succès. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Discussion
Des progrès spectaculaires dans la sensibilité de la protéomique et les méthodes de transcriptomique dans les 5-10 dernières années a fourni des données précieuses sur des milliers de gènes et de leurs produits. Cependant, les outils pour traiter la fonction de ces protéines n'a pas suivi le rythme.
Tagging une protéine facilite de nombreuses expériences pour déterminer sa fonction. Par exemple, une protéine peut être fusionnée à une protéine fluorescente dans une variété de différentes couleurs pour faciliter les études de localisation et de co-localisation. Mots - développés pour la microscopie électronique, tels que APEX2 ou miniSOG 11,12, permettent la localisation ultrastructurale de la protéine marquée. Mots - clefs pour la biochimie, comme la balise TAP et ProtC-TEV-ProtA (PTP) tag 7,13 permettent la purification des complexes associés à la protéine pour l' identification de partenaires de liaison ou dans des dosages biochimiques in vitro.
Les étapes spécifiques qui sont essentiels à la réussite de la protocol sont les suivantes: l'incorporation de DMSO dans le maître-PCR Mix 1, la congélation de la PCR Master Mix 1 avant l'addition du mélange maître 2, l'utilisation de la polymerase commerciale Master Mix 2 et la modification du tampon CYTOMIX électroporation. Dans notre expérience, il est nécessaire d'utiliser le double du nombre de cellules pour C transfections terminaux de marquage que pour N terminal de marquage transfections afin d'atteindre une proportion similaire de puits positifs. Par conséquent, toutes les mesures doivent être effectuées comme décrit.
Cette technique est seulement susceptible d'être couronnée de succès lorsque transfecter l'insecte forme procyclique trypanosome. Formes sanguines ont une efficacité de transfection inférieure 14, de plus ils sont susceptibles de mourir lors de la sélection en raison de la toxicité dépendante de la densité qui est sans rapport avec la drogue sélective. Par conséquent, notre protocole précédent représente la meilleure technologie actuelle pour le marquage de forme sanguine trypanosomes 9. Il est également probable que le transefficacité fection variera dépendant de la trypanosome isolat spécifique. Ce protocole a été optimisé à l'aide de 927 SMOX formes procycliques - d'autres souches peuvent nécessiter une optimisation supplémentaire. Les mesures qui peuvent augmenter la probabilité de succès comprennent: l'augmentation de la quantité de amplicon PCR, ce qui augmente le nombre de cellules incluses dans la transfection.
Nous présentons une méthode où des centaines de protéines peuvent être étiquetés en parallèle. Cela facilitera études à grande échelle sur la localisation et de l'interaction, en complément de grands ensembles de données existants et de fournir des informations précieuses à la communauté. modèles Primer sont disponibles sur demande et une liste de plasmides modèles est disponible à partir de: http://www.sdeanresearch.com/
Déclarations de divulgation
The authors have nothing to disclose.
Remerciements
SD was supported by a Sir Henry Wellcome Fellowship [092201/Z/10/Z]. This work was funded by Wellcome Trust grants [WT066839MA][104627/Z/14/Z][108445/Z/15/Z] to Professor Keith Gull. We would like to thank Professor Keith Gull for helpful conversations and insights.
matériels
| Name | Company | Catalog Number | Comments |
| Oligonucleotide | Life technologies | na | desalt only (do not use additional purification) |
| DMSO | Roche | Included with the Expand HiFi pack | Must be PCR grade |
| dNTP | any reputable | ||
| Expand HiFi polymerase | Roche | 11759078001 | |
| BTX ECM830 | Harvard Apparatus | Electroporator | |
| HT-200 plate handler | Harvard Apparatus | HT-200 | Handles the 96-well eletroplates |
| MOS 96 ELECTROPLATE 4 mm | Harvard Apparatus | 45-0452 | Electroplate for the 96-well electroporation |
| Blasticidin S Hydrochloride | Melford | 3513-03-9 | |
| Hygromycin b Gold | Invivogen | ant-hg-1 | |
| Phleomycin | Melford | P0187 | |
| 225 cm TC Flask, canted neck, phenolic cap | Appletonwoods | BC006 | |
| 24 well culture plates | Appletonwoods | BC017 | |
| Eppendorf® PCR Cooler, iceless cold storage system for 96 well plates and PCR tubes | Sigma Aldrich | Z606634-1EA | |
| 96-well Multiply® PCR plate with lateral skirt | Sarstedt | 72.1979.203 |
Références
- Kelly, S., Reed, J., et al. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol Biochem Parasitol. 154 (1), 103-109 (2007).
- Wirtz, L. E., Leal, S., Ochatt, C., Cross, G. A. M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 99 (1), 89-101 (1999).
- Shi, H., Djikeng, A., Mark, T., Wirtz, L. E., Tschudi, C., Ullu, E. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA. 6 (7), 1069-1076 (2000).
- Alsford, S., Turner, D. J., et al. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21 (6), 915-924 (2011).
- Alsford, S., Eckert, S., et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 482 (7384), 232-236 (2012).
- Mony, B. M., MacGregor, P., et al. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature. 505 (7485), 681-685 (2014).
- Rigaut, G., Shevchenko, A., Rutz, B., Wilm, M., Mann, M., Séraphin, B. A generic protein purification method for protein complex characterization and proteome exploration : Abstract Nature Biotechnology. Nature Biotechnol. 17 (10), 1030-1032 (1999).
- Los, G. V., Encell, L. P., et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem Biol. 3 (6), 373-382 (2008).
- Dean, S., Sunter, J. D., Wheeler, R. J., Hodkinson, I., Gluenz, E., Gull, K. A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids. Open biol. 5 (1), 140197 (2015).
- Poon, S. K., Peacock, L., Gibson, W., Gull, K., Kelly, S. A modular and optimized single marker system for generating Trypanosoma brucei cell lines expressing T7 RNA polymerase and the tetracycline repressor. Open biol. 2 (2), 110037 (2012).
- Lam, S. S., Martell, J. D., et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nature meth. 12 (1), 51-54 (2014).
- Shu, X., Lev-Ram, V., Deerinck, T. J., Qi, Y., Ramko, E. B. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS biology. , (2011).
- Schimanski, B., Nguyen, T. N., Günzl, A. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryotic Cell. , (2005).
- Burkard, G., Fragoso, C. M., Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol. 153 (2), 220-223 (2007).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.