
Method Article
Open Source Analyse unique pour particules super-résolution Microscopie avec VirusMapper
Dans cet article
Résumé
Ce manuscrit utilise le logiciel open-source basée à Fidji VirusMapper d'appliquer une analyse unique particule aux images de microscopie de super-résolution afin de générer des modèles précis de la structure à l'échelle nanométrique.
Résumé
La microscopie de fluorescence super-résolution est en train de révolutionner la recherche en biologie cellulaire. Sa capacité à briser la limite de résolution de l'ordre de 300 nm permet l'imagerie de routine des complexes biologiques à l'échelle nanométrique et des processus. Cette augmentation de la résolution signifie également que les méthodes populaires en microscopie électronique, telles que l'analyse seule particule, peuvent facilement être appliquées à la microscopie de fluorescence super-résolution. En combinant cette approche analytique avec l'imagerie optique à super-résolution, il devient possible de tirer parti de la capacité d'étiquetage spécifique à la molécule de la microscopie à fluorescence pour générer des cartes d'éléments de structure moléculaire dans une structure métastable. À cette fin, nous avons développé un nouvel algorithme - VirusMapper - emballé comme un outil facile à utiliser, de haute performance et plugin ImageJ haut débit. Cet article présente un guide détaillé à ce logiciel, mettant en valeur sa capacité à découvrir de nouvelles caractéristiques structurelles biologiques mcomplexes. Molecular Nous présentons ici comment assembler des données compatibles et de fournir un protocole étape par étape sur la façon d'utiliser cet algorithme pour appliquer une analyse unique particule images à haute résolution.
Introduction
Super-résolution (SR) La microscopie a eu un impact majeur sur la biologie cellulaire en offrant la possibilité d'images clés processus moléculaires ainsi que le marquage spécifique moléculaire crucial pour les comprendre. SR permet maintenant la microscopie optique à approcher les résolutions (20-150 nm) jusqu'ici réalisables avec la microscopie électronique (EM) tout en conservant les principaux avantages de la microscopie optique, tels que le potentiel de l' image des cellules vivantes 1, 2. En outre, la conservation de la structure trouvée à l'échelle nanométrique permet l'application d' une analyse unique de particules (SPA) à des données de RS, un concept largement utilisé en microscopie électronique 3. En utilisant SPA, de copies hautement conservées d'une structure peut être imagée et moyenne ensemble pour améliorer la résolution, la précision ou signal bruit de l'objet visualisé. Lorsqu'il est utilisé en combinaison avec SR, SPA a été démontré être un outil puissant pour le haut-pla cartographie recision des composants du complexe du pore nucléaire 4, 5, 6, centrosomes et des virus tels que le VIH 7 et 8 HSV-1.
Cependant, l'application combinée de routine SR et SPA a été contestée par un manque de logiciels disponibles. Pour cette raison, nous avons développé VirusMapper, un plug - in pour le logiciel de traitement d'image populaire ImageJ / Fidji 9. Ceci est le premier logiciel librement disponible pour SPA généralisée avec des images de fluorescence 10 conçues pour fournir une moyenne rapide, naïve, multi-canal convivial des structures imagées par microscopie SR. Bien que conçu pour les virus, il peut être appliqué à tout complexe macromoléculaire dans lequel les différentes espèces moléculaires peuvent être imagés, identifiés et localisés.
VirusMapper peut être utilisé pour produire moléculaire de haute précisionles modèles de toute structure connue, permettant le calcul des dimensions moyennes et d'autres paramètres. La conception de l'algorithme rend particulièrement utile pour séparer les populations de structures, prévoyant la détermination des orientations distinctes ou différents états morphologiques. En outre, l'imagerie multi-canaux peut être utilisé pour utiliser un canal de référence dans le cas où la structure sous-jacente est bien connu, permettant ainsi la découverte de la structure à base de référence. Les instructions pour le téléchargement et l' installation du logiciel sont fournis sur https://bitbucket.org/rhenriqueslab/nanoj-virusmapper . Exemple de données peuvent également être trouvées ici, et les utilisateurs sont invités à pratiquer l'utilisation du logiciel sur l'exemple des données avant de tenter de l'appliquer à eux-mêmes.
Ici, les étapes pour utiliser ce plugin pour produire des modèles de SPA à partir de données brutes sont décrites. Le logiciel prend des images brutes contenant o uniquer structures à étiquettes multiples en entrée. Il revient, sous réserve d'un certain nombre de paramètres sont ajustés comme le logiciel est exécuté, les modèles SPA montrant les distributions moyennes des composants marqués dans les structures imagées.
Le but de ce protocole est de produire des modèles de SPA précises donnant les localisations moyennes de composants au sein des structures imagées selon l'pipeline présenté sur la figure 1. Comme le montre la Figure 1, le flux de travail de logiciel est utilement divisé en trois étapes. La première étape consiste à segmenter des images de taille, ce qui entraîne des piles de particules pour chaque canal. Ces particules sont les unités qui seront moyennées pour créer des modèles et de produire des semences pour la production de modèles. La deuxième étape consiste à générer des images de semences, qui sont utilisés pour enregistrer l'ensemble des particules dans la phase finale. Cela se fait en choisissant un canal de référence et la sélection manuelle des particules dans ce canal qui contribuera à la seeds. Les graines sont choisies dans ce canal de référence, mais peuvent être générés pour tous les canaux. Les particules sont initialement réalignés en ajustant une gaussienne 2D dans ce canal. Toutes les particules qui ont été sélectionnés et réalignés sont ensuite moyennées pour produire une graine. Pour chaque structure commune vu dans les données à modéliser, les particules doivent être choisies comme des graines qui représentent de façon claire et précise que la structure. L'interface à ce stade est également utile pour la numérisation des données pour de telles structures.
La dernière étape consiste à générer des modèles en utilisant la correspondance de modèle. Ceci est obtenu grâce à l'enregistrement des particules extraites à l'origine pour les images de semences produites dans la section précédente par corrélation croisée. Un sous-ensemble de particules moyenne est enregistré en même temps, et le procédé est en outre itérée afin de réduire l'erreur quadratique moyenne modèle, si on le souhaite. Ce sous-ensemble est déterminée en définissant une similitude minimale contre la graine qui doit être satisfaite. Lors de la création modèles simultanément dans plusieurs canaux, la similitude commune, ou la moyenne des similarités pour chaque canal, est utilisé. Les modèles résultants et les particules enregistrées qui les ont contribué à peuvent ensuite être analysées.
Protocole
NOTE: Ce protocole vidéo et compléter le document original 10 décrivant le logiciel plus en détail. Les lecteurs sont invités à examiner attentivement pour des indications supplémentaires en ce qui concerne l'utilisation du logiciel. Il existe trois grandes étapes: l'extraction des particules, lequel des segments de grandes images en particules individuelles; la sélection des semences, où les structures communs sont identifiés dans les données et alignées pour produire des graines, qui sont utilisés dans l'étape finale; et la génération de modèle, où concordance de modèles à partir de ces graines aligne les particules extraites et un sous-ensemble des moyennes pour produire les modèles de SPA.
1. Configuration Avant l'exécution du package logiciel
- Préparer les échantillons de la structure à l'étude sur une lamelle couvre-objet ou dans les conditions expérimentales appropriées.
- Imager les échantillons avec la microscopie de fluorescence super-résolution, comme la microscopie d'éclairage structuré (SIM) 11 ou stimulated depletion d'émission (STED) 12 microscopie.
REMARQUE: Les détails précis de la façon de préparer et d'échantillons d'image dépend fortement de la nature de la structure à l'étude, devrait donc être consulté la documentation pertinente. À titre d'exemple, la méthode précise pour la préparation et des échantillons d'imagerie du virus de la vaccine, tels que ceux utilisés ici, est décrite dans la section des résultats représentatifs. - Créer des images de plusieurs champs de vue montrant un grand nombre de copies séparées de la structure ou de particules, de préférence des milliers. des particules de l'image qui sont ainsi séparées les unes des autres que possible et faire en sorte que les images sont exempts de saleté ou d'autres structures fluorescentes qui ne sont pas d'intérêt.
- Ouvrez toutes les images contenant les particules dans les îles Fidji en faisant glisser les fichiers dans la barre d'outils Fidji ou en sélectionnant « Fichier »> « Ouvrir ».
- Sélectionnez « Image »> « Stack »> « Outils »> « concaténer » pour concaténer lales images en une seule pile. Ensuite, si l'image résultante est un HyperStack, la transformer en une pile en sélectionnant « Image »> « Hyperstacks »> « HyperStack à pile ».
REMARQUE: La pile finale doit avoir des canaux intercalés. S'il y a deux canaux, le premier tranche de la pile doit être le canal 1 de la première image, la deuxième tranche devrait être le canal correspondant 2, la troisième tranche devrait être le canal 1 de la deuxième image, et ainsi de suite.
2. Extraire les particules
- Choisissez l'image à segmenter et sélectionnez « Extraire structures virales ». Choisissez où enregistrer les particules extraites et voir « l'extrait » Structures virales boîte de dialogue (figure 2).
- Affecter des paramètres d'extraction avec des estimations initiales en les entrant dans la boîte de dialogue « Extraire structures virales » comme suit. Peaufinez ces paramètres après la prévisualisation de la segmentation d'images.
- Réglez le engourdiser de canaux dans l'ensemble de données que le nombre des différents canaux de fluorescence qui ont été imagé (par exemple, 2).
- Régler le canal de référence à partir de laquelle les particules sont extraites par la détection de pics dans ce canal en saisissant le numéro du choix du canal. Sélectionnez le canal le plus cohérent; autrement dit, le canal dans lequel la plupart des particules ont la même apparence.
REMARQUE: Si possible, les particules dans ce canal aura un maximum central. - Choisissez ou non d'appliquer un flou gaussien pré-détection. Réglez le flou gaussien pré-détection à 0 pour ne pas appliquer le flou; si cette valeur est augmentée, un filtre de flou gaussien du rayon donné est appliqué avant la détection de maxima locaux. Utiliser cette fonctionnalité si le canal de référence ne possède pas un maximum central (par exemple, la forme de l' anneau); flou induit l'apparition d'un.
NOTE: régions segmentées d'intérêt (ROIs) ne sont pas le flou gaussien de les appliquer, car cette fonctionnalité est utilisé uniquementd pour positionner ROI dans le canal de référence. - Régler le rayon de la ROI (qui sera fixé autour de chaque maximum local) en pixels. Choisir une valeur telle que ce que les régions d' intérêt sont légèrement plus grandes que les plus grosses particules, par exemple dans la figure 3. Par exemple, si les plus grosses particules semblent avoir un diamètre d'environ 30 pixels (estimées à peu près à l'oeil nu), puis régler le rayon de la ROI à 20 pixels.
- Définissez le nombre de régions d'intérêt à utiliser par image à une première valeur relativement faible inférieure à 100.
- Réglez le recouvrement de retour sur investissement maximal. Si les particules sont bien séparées, garder ce petit; si les particules sont groupées, augmenter ce pour permettre aux régions d'intérêt à se chevaucher.
- Sélectionnez "preview show".
REMARQUE: Lorsque cette option est sélectionnée, les extraits pour ROIs le canal de référence associé à l'image actuelle apparaît. - Régler le rayon de la ROI, le nombre de régions d'intérêt, et le chevauchement de retour sur investissement maximal d'avoir des ROI de taille appropriée autour autant que possible des particules, Comme dans la figure 3.
- Sélectionnez « OK » pour lancer la segmentation. Fermez l'image et le gestionnaire de retour sur investissement.
REMARQUE: Ne modifiez pas les noms des fichiers des ensembles de particules. Ces noms doivent être au format « particules virales - ChannelX » pour les sections suivantes.
3. Sélectionnez Graines
- Sélectionnez « Générer » Graines, sélectionnez le dossier dans lequel les particules extraites sont sauvegardées et afficher la boîte de dialogue « Générer les semences » et les fenêtres (Figure 4).
- Paramétrez les semences de sélection initiale en les entrant dans la boîte de dialogue « Générer les semences », comme suit.
- Réglez le canal de référence qui équipera aligner et centrer tous les canaux. Choisissez un canal dans lequel la plupart des particules ont la même apparence et qui a un maximum central, si possible.
- Cochez les cases pour tous les canaux pour lesquels une graine doit être générée.
- Sélectionnez si les graines doivent être RoTated de 90 °. Utilisez cette fonction pour avoir l'alignement compatible avec les autres modèles
- Choisissez ou non d'appliquer un flou gaussien pré-alignement. Réglez le rayon de flou pour chaque canal à 0 pour ne pas appliquer le flou. Augmenter cette valeur pour appliquer un filtre de flou gaussien du rayon donné avant réalignement. Utilisez cette fonction si les particules ne sont pas un maximum central; flou induit l'apparition d'un.
NOTE: Les graines ont pas le flou appliqué, car cette fonction est uniquement pour obtenir un alignement constant. - Indiquez si vous souhaitez utiliser la correction de décalage pour centrer séparément les graines pour les canaux non-référence, mais pas les faire pivoter, en cochant ou décochant les cases « de correction de décalage » pour chaque canal.
REMARQUE: Utilisez cette option si les canaux ne sont pas bien alignés les uns aux autres; il peut aussi être utile pour aligner d'autres canaux uniquement en fonction de la référence, sans correction de décalage.
- Choisissez des particules à utiliser comme graines. Recherche par til séquence de particules pour trouver une particule qui ressemble à la structure qui est à modéliser et d'enregistrer le numéro de l'image.
- Entrez le numéro dans les « cadres à utiliser » boîte, et plus de fenêtres apparaîtront (Figure 5). Entrer plusieurs numéros de trames séparées par des virgules.
- Voir les images de semences et les graines moyennes obtenues dans les fenêtres qui apparaissent.
REMARQUE: Le journal proposera des graines semblables à la moyenne. - Régler le canal de référence, le rayon de flou gaussien, et les options de correction de décalage pour optimiser le processus de sélection des semences de telle sorte qu'un nombre de trames de semences trouvés ont une apparence similaire. Continuez d'ajouter des graines jusqu'à ce que les graines moyennes sont créées qui ressemblent de manière satisfaisante la structure vu dans les données à modéliser.
- Nommez les graines et où ils seront sauvegardés et sélectionnez « OK » pour enregistrer les images de semences finales pour une utilisation ultérieure dans la génération de modèle.
4. Générer modèles
- SeLect dialogue « Générer des modèles basés sur les semences », sélectionnez le dossier dans lequel les particules extraites sont enregistrées, et voir la « Générer des modèles » (Figure 6).
- Chargez les graines pour chaque canal en utilisant les cases à cocher.
NOTE: Les graines seront enregistrées dans un sous-dossier qui a été nommé dans la boîte de dialogue « Générer les semences ». Le nom par défaut est « Analyse ». Prenez soin de sélectionner les graines moyennes, pas les cadres, qui sont également enregistrés pour référence. - Assigner des paramètres initiaux de génération de modèle en les entrant dans la boîte de dialogue « Générer des modèles », comme suit. Peaufinez ces paramètres plus tard.
- Indiquez si vous souhaitez utiliser un canal de référence pour l'alignement, qui calcule les traductions et les rotations qu'à partir du canal de référence et les applique à tous les canaux. Si vous utilisez cette option, sélectionnez le canal à utiliser comme référence.
REMARQUE: Cette option ne doit être choisi d'utiliser spécifiquement un canal comme reference, comme si faire la découverte de la structure à base de référence. Dans le cas contraire, elle se traduira par des modèles moins précis. Les canaux doivent être bien alignées si vous utilisez cette option. - Indiquez si vous souhaitez carré intensités d'image lors de l'appariement de modèle. Cela va accentuer les petites différences, donc utiliser lors de la création d'un modèle qui a des caractéristiques particulièrement subtiles.
- Choisissez une similitude minimale contre les graines en utilisant le curseur « similitude minimale contre les graines » ou en entrant un numéro dans la zone.
NOTE: Seules les particules avec une similitude avec les graines supérieures à ce seuil sera utilisé. 60-80% est généralement un bon choix pour commencer. - Définissez le nombre maximum d'itérations à 1, optimiser la similitude minimale contre les semences et l'augmenter par la suite, au besoin.
- Cochez les cases pour choisir les éléments du processus de génération de modèle qui apparaîtra.
NOTE: « Afficher les graines » affiche les graines qui ont été chargés de la boxes en haut de la boîte de dialogue. « Afficher les modèles » affichera toutes les itérations des modèles créés à partir de la moyenne du sous-ensemble de particules qui répond à la similitude minimale contre les semences. « Afficher MSE » affiche une erreur quadratique moyenne (MSE) image qui met en évidence les zones du modèle qui sont les plus variables. « Afficher les particules » afficheront le sous-ensemble de particules qui sont utilisées pour créer les modèles, enregistré selon la plus haute itération du modèle qui est affiché.
- Indiquez si vous souhaitez utiliser un canal de référence pour l'alignement, qui calcule les traductions et les rotations qu'à partir du canal de référence et les applique à tous les canaux. Si vous utilisez cette option, sélectionnez le canal à utiliser comme référence.
- Sélectionnez « Afficher preview » pour générer des modèles de prévisualisation et afficher les résultats.
NOTE: Ceci est l'étape la plus intensive informatiquement du processus. Le temps d'exécution pour un ensemble de quelques milliers de particules dont le diamètre est de quelques dizaines de pixels sur un PC de bureau devrait être d'environ 10 min. Si le temps de calcul est un problème, les utilisateurs doivent d'abord essayer l'algorithme sur un plus petit sous-ensemble des données ou utiliser un plus petit rayon de retour sur investissement à l'étape 2.2.4, si possible. - Voir les générerd modèles et d'optimiser les paramètres, en particulier la similitude minimale contre les semences. Augmenter la similitude minimale contre les graines jusqu'à ce que seules vraies particules de la morphologie modélisé sont incluses dans le modèle.
- Augmenter le nombre maximum d'itérations en utilisant le « nombre maximal d'itérations » curseur ou en entrant un numéro dans la boîte et permettre au processus de génération de modèle pour itérer. Utilisez une valeur d'environ 10 pour l'itération maximale.
- Nom modèles et sélectionnez « OK » pour enregistrer les piles de modèle d'évolution qui contiennent toutes les itérations du processus final de génération de modèle.
NOTE: Si la similitude minimale contre les graines est si élevé qu'il n'y a pas de particules ont cette similitude, rien ne sera mis à jour. Si le plug-in semble être gelé, envisager la possibilité que la similitude minimale est trop élevée.
Résultats
Ici, nous démontrons le logiciel sur le modèle poxvirus, le virus de la vaccine. L' un des plus complexes virus de mammifères, les paquets de la vaccine environ 80 protéines différentes dans un 350 x 270 x 250 nm 3 particule en forme de brique 13, 14. Trois structures sont discernables au microscope électronique: un noyau central, qui contient le génome ADNdb; deux structures protéiques, appelés corps latéraux, qui flanquent le noyau; et une seule double couche proteolipid enveloppe 15. La grande taille, structure complexe, et à l'étiquetage des amenability protéine fluorescente recombinant font la vaccine un excellent système pour démontrer le flux de travail VirusMapper.
En utilisant le logiciel comme décrit ici, la distribution d'une variété de protéines sur le virion de la vaccine peut être modélisé. Une protéine a été marquée et imagée, éventuellement en combination avec une autre protéine de distribution connue comme référence, et le logiciel a été utilisé comme décrit pour produire des modèles moyens de la localisation de cette protéine sur la particule. Dans cet exemple, deux protéines ont été modélisées, la protéine de noyau interne L4, et le composant principal corps latéral F17.
Un virus de la vaccine recombinant qui a marqué F17 avec GFP et L4 marqué avec mCherry 16 a été utilisé. Le virus purifié a été dilué dans 1 mM de Tris, pH 9, et lié à laver, des lamelles de haute performance en les revêtant pendant 30 min à température ambiante. Les échantillons ont ensuite été fixées en appliquant 4% de formaldehyde dans du PBS pendant 20 min. Les lames ont immédiatement montées sur des lames à antifade moyen de montage. L'imagerie a été réalisée par SIM sur un microscope SIM commerciale. Un champ de vue a été sélectionné contenant des centaines de virus et les images ont été acquises à l'aide des décalages de phase 5 et 3 rotations de la grille avec grille de 561 nm (32 um periode) et les lasers 488 nm (32 um de période de réseau). Les images ont été acquises à l'aide d'une caméra sCMOS et traitées à l'aide du logiciel de microscope. Les canaux ont été alignés sur la base d'une glissière à billes multicolores en image avec les mêmes paramètres d'acquisition d'image. Après les images de reconstruction SIM et d'alignement de canal ont été ouverts en Fiji et concaténées en une seule pile d'images.
Les particules virales ont été extraites des images en utilisant le canal L4 comme référence et sans appliquer de flou gaussien, étant donné que ces particules ont un maximum central. Environ 15 000 particules ont été extraites dans cette expérience.
En raison de la géométrie de la vaccine, les corps latéraux ont un aspect nettement différent en fonction de l'orientation du virus. Nous visualisé deux orientations dans lesquelles un ou deux corps latéraux pourraient être distingués. Nous avons fait référence à ces orientations que, le respect frontal et sagittalivement.
Graines séparées pour les orientations frontales et sagittales ont été sélectionnées en effectuant une recherche dans la liste des particules au stade « Générer Seeds » (figures 4 et 5); les particules qui ont été clairement dans une orientation ou l'autre été choisis. Le canal L4 a été utilisé comme canal de référence pour aligner les uns avec les autres graines. Encore une fois, pas de flou gaussien était nécessaire. 5 particules pour chaque orientation ont été sélectionnées et ont été moyennées pour produire des graines.
Les modèles ont été générés pour chaque orientation à partir de ces graines. Ni un canal de référence, ni les valeurs d'intensité au carré ont été utilisés. Le nombre maximum d'itérations a été initialement fixé à 1, et la similitude minimale a été fixée à inclure environ 1 000 particules dans chaque cas, ce qui a donné une apparence uniforme pour chaque orientation. Le nombre maximum d'itérations a été portée àpermettre la convergence du modèle. Les modèles ont ainsi été générés pour les deux orientations dans les deux canaux (figure 7).

Figure 1: flux de VirusMapper. Le plug-in est organisé en trois étapes principales. Les particules virales sont extraits de grandes images, des images ou des graines modèle sont sélectionnées semi-manuellement à partir des données et des modèles de SPA finales sont générées à partir des données en se référant aux graines. S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2: Boîte de dialogue "Extraire structures virales". Lors de la sélection « Extrait virale structur es », cette boîte de dialogue apparaît. Les paramètres doivent être remplis avec des estimations initiales pour la segmentation optimale. « Prévisualisation » peut être sélectionné, ce qui permet aux régions d' intérêt à être prévisualisé et les paramètres à être peaufiné. S'il vous plaît cliquer ici pour voir une plus grande version de ce chiffre.
{kind=link}

Figure 3: Réglage des paramètres d'extraction. Après avoir prévisualisé ROIs qui sera extrait, le rayon de retour sur investissement, le nombre de régions d'intérêt, et le chevauchement de retour sur investissement maximal sont ajustés pour parvenir à une telle situation. ROI sont légèrement plus grandes que les particules, les particules sont comprises dans une ROI et ROI peuvent se chevaucher suffisamment pour permettre aux particules en grappes à séparer.ank "> S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.

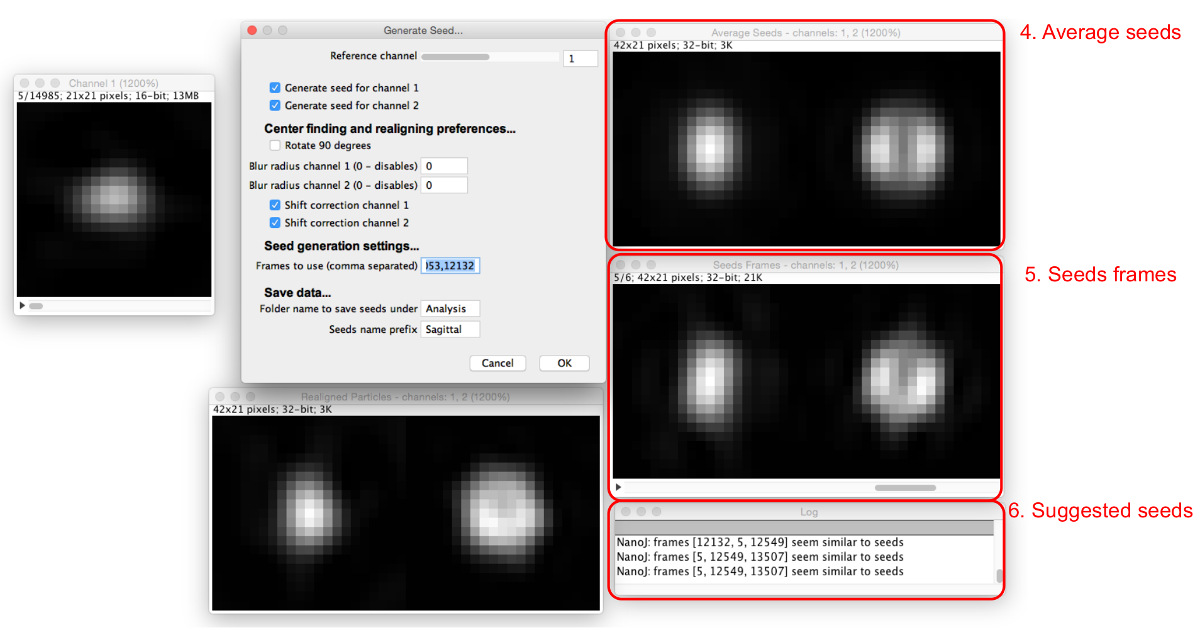
Figure 4: Génération graines de correspondance de modèle. La boîte de dialogue « Générer les semences » (1) définit les paramètres à affecter. La séquence des particules de référence (2) permet à l'utilisateur de balayer à travers les particules dans le canal de référence. Lorsqu'une particule est considérée dans la séquence des particules de référence, les particules réalignées pour tous les canaux peuvent être consultés dans les extraits de particules réalignés (3). S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5: Ajout d' images de semences. Comme les graines sont ajoutées à la « Cadres utiliser » boîte, la moyenne de toutes les semences (4) et les cadres concernés (5) sont affichés. Les particules qui sont semblables aux graines moyennes actuelles sont proposées dans la boîte de dialogue (6). S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6: Boîte de dialogue "Générer des modèles". Lors de la sélection « Générer des modèles basés sur les semences », cette boîte de dialogue apparaîtra. Les paramètres doivent être remplis avec des estimations initiales pour la génération de modèle optimal, et les éléments de la procédure de génération de modèle pour être vues pendant le calcul doit être sélectionné. « Afficher l'aperçu » peut être sélectionné, ce qui permet le processus de génération de modèle pour exécuter et les paramètres à peaufiné.ftp_upload / 55471 / 55471fig6large.jpg » target = "_ blank"> S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.

Figure 7: Les modèles générés avec VirusMapper. virions de la vaccine avec la protéine de noyau L4 étiqueté avec mCherry et la protéine de corps latéral F17 étiqueté avec EGFP ont été imagées en utilisant la carte SIM. Les modèles ont ensuite été générés avec le logiciel, tel que décrit dans le protocole. Deux orientations, par l'apparition des corps latéraux frontal et sagittal, se distinguent. Barre d'échelle = 100 nm. S'il vous plaît cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Avec cette méthode, les chercheurs disposent de combiner la puissance de la microscopie SPA et SR afin de générer de haute précision, les modèles 2D multi-canal de l'architecture des protéines de virus et d'autres complexes macromoléculaires. Cependant, il faut prendre quelques considérations importantes en compte.
Les graines doivent être choisis pour représenter une structure qui est toujours vu. Ainsi, les données brutes doivent être soigneusement inspectés avant que les graines sont choisies. Ceci est important pour la prévention des modèles biaisées. Les choix peuvent être validés par l'examen des seuils de similarité minimale requise pour inclure un certain nombre de particules dans les modèles. Il est clair que, pour un choix de semences, plus ce seuil doit être pour un nombre donné de particules, plus cette structure est apparente dans les données.
Le concept de correspondance de modèle est particulièrement utile quand il existe une hétérogénéité dans les données. Toutes les différentes structures qui sont vible doivent être identifiés et différents modèles créés pour chaque cas. En séparant les structures hétérogènes dans un canal, mais en créant simultanément des modèles dans un second canal, les modèles peuvent émerger qui n'auraient pas été immédiatement évidente.
Une autre considération à prendre en compte lors de l'utilisation de cet algorithme est que la procédure d'itération permettra de maximiser l'asymétrie stochastique. Par exemple, lors de la modélisation d'une structure avec deux maxima symétrique, toutes les légères dissymétries entre les maxima sont alignés les uns avec les autres lors de l'itération, et le modèle final seront donc au maximum asymétrique. Si cela ne reflète pas une symétrie connue dans la structure en cours de modélisation, alors cela devrait être pris en compte. À l'heure actuelle, la seule façon d'éviter cette maximisation est de limiter le nombre d'itérations à 1, bien qu'un potentiel de développement serait pour VirusMapper d'incorporer des axes de symétrie dans le processus de génération de modèle. Toutes les nouvelles versions de VirusMapper seront avaiD spon sur le site Web référencé (voir tableau des matériaux). Les utilisateurs trouveront également une FAQ ici pour répondre à toutes les questions communes.
Le logiciel tel que décrit est applicable à toute structure qui peut être imagée avec une résolution suffisante pour visualiser les caractéristiques que l'utilisateur souhaite modéliser. Bien que SPA peut améliorer la résolution, il ne sera pas clairement améliorer la visibilité des caractéristiques qui sont par ailleurs pas visibles. Ce protocole est donc pas une méthode pour améliorer la qualité des données. Comme toute technique, une préparation minutieuse de l'échantillon et l'optimisation de la stratégie d'imagerie fournira les plus propres données et les meilleurs modèles résultants.
Le choix de la modalité d'imagerie SR est également important et, en général, dépendra de l'échantillon à portée de main. VirusMapper a été validé pour fonctionner avec la carte SIM et STED 10, et il peut également être utilisé avec des données de microscopie de localisation de haute qualité, mais il faut prendre soin , dans ce cas,que l'étiquetage clairsemés pourrait causer des problèmes similaires à ceux de la maximisation de l'asymétrie.
À l'heure actuelle, VirusMapper est l'algorithme que librement disponible pour l'analyse unique particule d'images de fluorescence et le seul usage général logiciel de calcul de la moyenne 2D SPA. D' autres études qui ont fait usage des mêmes principes 4, 6, 8 ont utilisé un logiciel personnalisé spécialisé pour chaque étude. Algorithmes à usage général pour la reconstruction des données 3D ont été publiées 5, 18, même si aucun logiciel n'a été fourni.
Lorsqu'il est utilisé comme décrit dans cet article, VirusMapper peut être utilisé pour produire des modèles précis, précis et robuste de l'architecture des protéines macromoléculaire de virus et d'autres complexes. Avec ces modèles, les chercheurs peuvent prendre des mesures précises des dimensions moyennes des structUres à l'étude, ce qui pourrait leur permettre d'atteindre des conclusions biologiques qui n'auraient pas été autrement possible.
En outre, les capacités multi-canaux de cette technique, il est possible de mapper un nombre illimité de protéines et de composants au sein de complexes et de découvrir l'organisation de nouvelles protéines. Examiner les changements dans la structure nanométrique dans différentes conditions biologiquement pertinentes, telles que différentes étapes d'un cycle de vie du virus, a le potentiel d'offrir des indications précieuses sur la biologie.
Déclarations de divulgation
Les auteurs n'ont rien à dévoiler.
Remerciements
Nous tenons à remercier Corina Beerli, Jerzy Samolej, Pedro Pereira Matos, Christopher Bleck, et Kathrin Scherer pour leur contribution au développement initial et la validation des VirusMapper. Nous tenons également à remercier Artur Yakimovich pour sa lecture critique du manuscrit. Ce travail a été financé par des subventions de la biotechnologie et du Conseil de recherches en sciences biologiques (BB / M022374 / 1) (RH); financement de base au Laboratoire MRC de biologie cellulaire et moléculaire, University College London (JM); le Conseil européen de la recherche (649101-UbiProPox) (JM); et le Conseil de recherches médicales (MR / K015826 / 1) (RH et JM). RG est financé par le Conseil ingénierie et sciences physiques de la recherche (EP / M506448 / 1).
matériels
| Name | Company | Catalog Number | Comments |
| Fiji | Open-source image analysis software | ||
| NanoJ-VirusMapper | developed by the Henriques lab | Open source-Fiji plugin (https://bitbucket.org/rhenriqueslab/nanoj-virusmapper) | |
| VectaShield antifade mounting medium | Vector Labs | H-100 | |
| Elyra PS1 | Zeiss | ||
| ZEN BLACK | Zeiss | Image processing software for SIM | |
| High performance coverslip | Zeiss | 474030-9000-000 | |
| TetraSpeck beads | ThermoFisher | T7279 |
Références
- Henriques, R., Griffiths, C., Rego, E. H., Mhlanga, M. M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers. 95 (5), 322-331 (2011).
- Gustafsson, N., Culley, S., et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 7, 12471 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 161 (3), 438-449 (2015).
- Szymborska, A., de Marco, A., Daigle, N., Cordes, V. C., Briggs, J. A. G., Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341 (6146), 655-658 (2013).
- Broeken, J., Johnson, H., et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl Fluoresc. 3 (1), 14003 (2015).
- Burns, S., Avena, J., Unruh, J., Yu, Z. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. , (2015).
- Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Nat Acad Sci U S A. 109 (22), 8564-8569 (2012).
- Laine, R. F., Albecka, A., Svan de Linde, ., Rees, E. J., Crump, C. M., Kaminski, C. F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun. 6, 5980 (2015).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Gray, R. D. M., Beerli, C. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci Rep. 6, 29132 (2016).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J of Micros. 198 (2), 82-87 (2000).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Let. 19 (11), 780 (1994).
- Chung, C. -. S., Chen, C. -. H., Ho, M. -. Y., Huang, C. -. Y., Liao, C. -. L., Chang, W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 80 (5), 2127-2140 (2006).
- Moss, B. Poxviridae: the viruses and their replication. Fields virology. , (2010).
- Condit, R. C., Moussatche, N., Traktman, P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 66 (6), 31-124 (2006).
- Schmidt, F. I., Bleck, C. K. E., et al. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 4 (3), 464-476 (2013).
- . Nanoj-virusmapper Available from: https://bitbucket.org/rhenriqueslab/nanoj-virusmapper (2016)
- Fortun, D., Guichard, P., Unser, M. Reconstruction From Multiple Poses in Fluorescence Imaging: Proof of Concept. IEEE J Sel Topics Signal Process. 10 (1), 61-70 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.