
Method Article
Análise de partícula única open-source para Microscopia Super-resolução com VirusMapper
Neste Artigo
Resumo
Este manuscrito usa o open-source VirusMapper pacote de software baseado em Fiji aplicar a análise de uma única partícula de imagens de microscopia de super-resolução, a fim de gerar modelos precisos de estrutura em nanoescala.
Resumo
microscopia de fluorescência de super-resolução está revolucionando investigação em biologia celular. A sua capacidade para romper o limite de resolução de cerca de 300 nm permite a imagiologia rotina de complexos biológicos em escala nano e processos. Este aumento na resolução, também significa que os métodos conhecidos em microscopia electrónica, tal como a análise de uma única partícula, pode ser prontamente aplicada a microscopia de fluorescência de super-resolução. Ao combinar esta abordagem analítica com a imagiologia óptica de super-resolução, torna-se possível aproveitar a capacidade de etiquetagem específica molécula de microscopia de fluorescência para gerar mapas estruturais de elementos moleculares dentro de uma estrutura metastável. Para este fim, desenvolvemos um romance algoritmo - VirusMapper - empacotado como um plugin do ImageJ easy-to-use, de alto desempenho e de alto rendimento. Este artigo apresenta um guia detalhado a este software, mostrando sua capacidade de descobrir novas características estruturais em m biológicacomplexos olecular. Aqui, apresentamos como montar dados compatíveis e fornecer um protocolo passo-a-passo sobre como usar este algoritmo para aplicar a análise de partícula única para imagens de super-resolução.
Introdução
Super-resolução (SR) microscopia teve um grande impacto sobre a biologia celular, fornecendo a capacidade de processos moleculares principais imagem junto com a rotulagem específica molecular crucial para compreendê-los. SR agora permite microscopia de luz para aproximar as resoluções (20-150 nm) anteriormente só alcançáveis com microscopia electrónica (EM), mantendo os benefícios principais de microscopia de luz, tais como o potencial de células vivas da imagem 1, 2. Além disso, a conservação estrutural encontrado em escala nano permite a aplicação da análise de uma única partícula (SPA) de dados SR, um conceito utilizado extensivamente em microscopia electrónica 3. Usando SPA, muitas cópias altamente conservadas de uma estrutura pode ser trabalhada e média em conjunto para melhorar a resolução, precisão, ou o sinal-ruído do objeto visualizado. Quando utilizado em combinação com EL, SPA foi demonstrado ser uma ferramenta poderosa para o alto-pmapeamento recision dos componentes do complexo do poro nuclear, 4, 5, 6, centrossomas e vírus, tais como HIV e HSV-7 1 8.
No entanto, a aplicação combinada de rotina de SR e SPA foi desafiada por uma falta de software disponível. Por este motivo, desenvolvemos VirusMapper, um plugin para o software de processamento de imagem popular ImageJ / Fiji 9. Este é o primeiro pacote de software disponível gratuitamente para SPA generalizada com imagens de fluorescência 10 concebidos para proporcionar rápido user-friendly, média, multi-canal ingênua de estruturas fotografadas com microscopia SR. Embora desenhado para o vírus, pode ser aplicado a qualquer complexo macromolecular em que as espécies moleculares diferentes podem ser visualizados, identificado, e localizada.
VirusMapper pode ser utilizado para produzir produtos de alta precisão molecularmodelos de qualquer estrutura conhecida, permitindo o cálculo de médias dimensões e outros parâmetros. O desenho algoritmo torna-o particularmente útil para separar populações de estruturas, que prevê a determinação de orientações distintas ou diferentes estados morfológicos. Além disso, a imagem latente multicanal pode ser usada para empregar um canal de referência nos casos em que a estrutura subjacente é bem conhecida, permitindo deste modo para a descoberta de estrutura à base de referência. As instruções para baixar e instalar o software são fornecidos no https://bitbucket.org/rhenriqueslab/nanoj-virusmapper . dados de exemplo também pode ser encontrado lá, e os usuários são aconselhados a praticar o uso do software nos dados de exemplo antes de tentar aplicá-la à sua própria.
Aqui, são descritos os passos para usar este plugin para produzir modelos Spa A partir de dados brutos. O software leva imagens cruas contendo o únicor estruturas de multi-rotulado como entrada. Ele retorna, sujeitos a um certo número de parâmetros que são ajustados como o software é executado, modelos SPA que mostram as distribuições médias dos componentes marcados no interior de estruturas de imagens.
O objectivo do presente protocolo é a produção de modelos de SPA precisos dando as localizações médios dos componentes dentro de estruturas fotografada de acordo com o oleoduto delineado na Figura 1. Como mostrado na Figura 1, o fluxo de trabalho de software é utilmente dividido em três fases. A primeira etapa é a de segmentos de imagens grandes, resultando em pilhas de partículas para cada canal. Estas partículas são as unidades que será a média para criar modelos e para produzir as sementes para a geração modelo. A segunda etapa é a de gerar imagens de sementes, que são utilizados para registar-se todo o conjunto de partículas na fase final. Isto é feito pela escolha de um canal de referência e manualmente selecção de partículas neste canal que vai contribuir para a seeds. As sementes são escolhidos neste canal de referência, mas pode ser gerada para todos os canais. As partículas são inicialmente realinhados por ajuste de uma Gaussiana 2D neste canal. Todas as partículas que foram seleccionados e realinhados se então a média para produzir uma semente. Para cada estrutura comum visto nos dados que está a ser modelado, as partículas devem ser seleccionado como sementes que clara e precisa que representam a estrutura. A interface nesta fase é também útil para a digitalização dos dados para tais estruturas.
A fase final é gerar modelos utilizando a correspondência de molde. Isto é conseguido através do registo das partículas originalmente extraídos para as imagens de sementes geradas na secção anterior por correlação cruzada. Um subconjunto de partículas registrados é calculada a média em conjunto, e o processo é ainda iterado para reduzir o modelo do erro quadrático médio, se desejado. Este subconjunto é determinado por fixação de uma similaridade mínima contra as sementes que devem ser satisfeitas. Ao criar modelos simultaneamente em múltiplos canais, a semelhança conjunta, ou a média das semelhanças para cada canal, é utilizado. Os modelos resultantes e as partículas registados que contribuíram para eles, em seguida, pode ser ainda analisada.
Protocolo
NOTA: Este protocolo e vídeo complementar o original em papel 10 que descreve o pacote de software em mais detalhes. Os leitores são encorajados a rever isso com cuidado para orientação adicional sobre o uso do software. Existem três fases principais: a extracção de partículas, que segmentos de imagens de grandes dimensões em partículas individuais; selecção de sementes, onde as estruturas comuns são identificados nos dados e alinhadas para produzir sementes, que são utilizados na fase final; e geração de modelos, em que a correspondência modelo com base nestas sementes alinha as partículas e as médias extraídos um subconjunto para produzir os modelos de SPA.
1. Setup antes de executar o pacote de software
- Preparar amostras da estrutura em estudo sobre uma lamela ou nas condições experimentais relevantes.
- Imagem as amostras com microscopia de fluorescência super-resolução, como a microscopia de iluminação estruturada (SIM) 11 ou Stimulaesgotamento ted emissão (STED) 12 microscopia.
NOTA: Os detalhes precisos de como preparar e amostras de imagem depende muito da natureza da estrutura em estudo, de modo que a literatura pertinente deve ser consultado. Como um exemplo, o método preciso para a preparação e imagiologia de amostras de vírus de vaccinia, tais como aqueles usados aqui, está descrito na secção de resultados representativos. - Criar imagens de vários campos de visão mostrando um grande número de cópias separadas da estrutura ou as partículas, de preferência milhares. partículas de imagem que são bem separados um do outro quanto possível e garantir que as imagens são livres de sujeira ou outras estruturas fluorescentes que não são de interesse.
- Abra todas as imagens contendo as partículas em Fiji, arrastando os arquivos para a barra de ferramentas Fiji ou selecionando "Arquivo"> "Abrir".
- Selecione "Imagem"> "pilha"> "Ferramentas"> "Concatene" para concatenar oimagens em uma única pilha. Então, se a imagem resultante é um hyperstack, transformá-lo em uma pilha, selecionando "Imagem"> "Hyperstacks"> "hyperstack Stack".
NOTA: A pilha final deve ter canais intercalados. Se houver dois canais, a primeira fatia na pilha deve ser um canal a partir da primeira imagem, a segunda fatia deve ser o canal correspondente 2, a terceira fatia deve ser um canal a partir da segunda imagem, e assim por diante.
2. Extraia as Partículas
- Escolha a imagem para o segmento e selecione "Extrair Estruturas Virais". Escolha onde deseja salvar as partículas extraídas e ver os "Extrair Estruturas Virais" diálogo (Figura 2).
- Atribuir parâmetros de extração com estimativas iniciais, inserindo-os na caixa de diálogo "Extrair Estruturas Virais" como segue. Ajustar esses parâmetros Depois de visualizar a segmentação de imagens.
- Defina o dormenteer de canais no conjunto de dados como o número de diferentes canais de fluorescência que tem sido representada por imagem (por exemplo, 2).
- Definir o canal de referência a partir do qual as partículas são extraídos por detecção de picos em que canal, introduzindo o número da escolha de canal. Selecione o canal mais consistente; isto é, o canal no qual a maioria das partículas tem a mesma aparência.
NOTA: Se possível, as partículas neste canal terá um máximo central. - Escolher se quer ou não aplicar um borrão Gaussian pré-detecção. Definir o borrão Gaussian pré-detecção de 0 a não aplicar nenhum borrão; Se este valor é aumentado, um filtro de desfocagem gaussiana do raio dado é aplicado antes da detecção de máximos locais. Usar esta característica se o canal de referência não tem um máximo central (por exemplo, forma, anel); indefinição induz o aparecimento de um.
NOTA: Segmentado regiões de interesse (ROI) não têm o Gaussian Blur aplicada a eles, como esse recurso é apenas para usod para posicionar ROIs no canal de referência. - Defina o raio ROI (que será definido em torno de cada máximo local) em pixels. Escolher um valor de tal forma que que os ROIs são ligeiramente maiores do que as partículas maiores, tais como na Figura 3. Por exemplo, se as partículas maiores parecem ter um diâmetro de cerca de 30 pixels (estimados aproximadamente a olho nu), em seguida, definir o raio ROI a 20 pixels.
- Definir o número de ROIs para usar por trama para um valor relativamente pequeno inicial, inferior a 100.
- Definir a sobreposição máxima ROI. Se as partículas são bem separados, manter esta pequena; Se as partículas são aglomeradas, aumentar para permitir que os ROIs a sobrepor-se.
- Selecione "Mostrar previsão".
NOTA: Quando esta opção é seleccionada, os ROIs extraídos para o canal de referência associada com o quadro corrente aparece. - Ajustar o raio ROI, o número de ROIs, e a sobreposição máxima ROI ter ROIs de tamanho apropriado em torno tantas partículas quanto possível, Como na Figura 3.
- Selecione "OK" para executar a segmentação. Feche a imagem eo gerente ROI.
NOTA: Não altere os nomes dos arquivos dos conjuntos de partículas. Estes nomes deverão ser no formato "partículas virais - channelx" para as seções seguintes.
3. Selecione Sementes
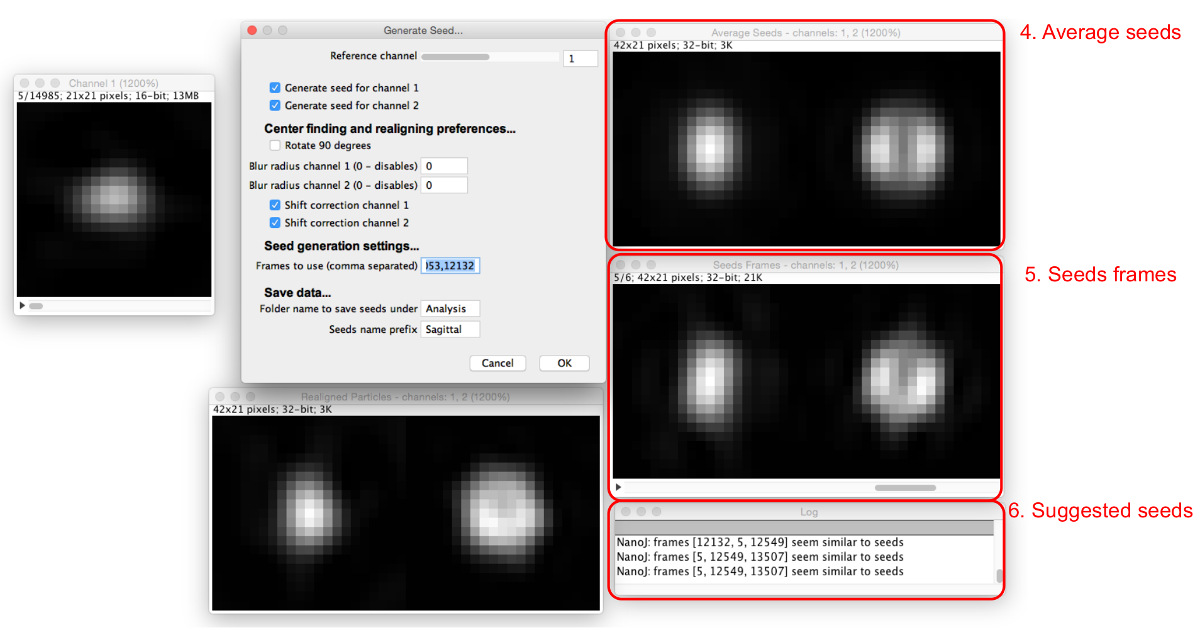
- Selecione "Gerar Sementes", selecione a pasta onde as partículas extraídos são salvos, e ver o diálogo "Gerar Sementes" e janelas (Figura 4).
- Atribuir os parâmetros de selecção de semente iniciais, inserindo-os na caixa de diálogo "Gerar Sementes", como se segue.
- Definir o canal de referência que irão ser montados para alinhar e centro de todos os canais. Escolha de um canal no qual a maioria das partículas tem a mesma aparência e que tem um máximo central, se possível.
- Marque as caixas para todos os canais para que uma semente devem ser gerados.
- Selecione se as sementes devem ser rotated por 90 °. Utilize este recurso para ter o alinhamento consistente com outros modelos
- Escolher se quer ou não aplicar um borrão Gaussian pré-alinhamento. Definir o raio borrão para cada canal de 0 a não aplicar nenhum borrão. Aumentar este valor para aplicar um filtro Gaussian Blur do raio dada antes de realinhamento. Use este recurso se as partículas não têm um máximo central; indefinição induz o aparecimento de um.
NOTA: Sementes não terá o efeito de arrastamento aplicada, como esse recurso é apenas para obter o alinhamento consistente. - Selecione se deseja usar correção mudança para centralizar separadamente sementes para canais não-referência, embora não girá-las, marcando ou desmarcando as caixas de "correção Shift" para cada canal.
NOTA: Use isto se os canais não estão bem alinhados uns com os outros; ele também pode ser útil para o alinhamento de outros canais unicamente de acordo com a referência, sem correcção de deslocamento.
- Escolha partículas para usar como sementes. Busca através de tele partícula sequência de encontrar uma partícula que se assemelha à estrutura que está a ser modelado e registar o número do quadro.
- Digite o número nos "Quadros de usar" caixa, e mais janelas aparecerá (Figura 5). Inserir vários números de quadros separados por vírgulas.
- Ver os quadros de sementes e as sementes médios resultantes nas janelas que aparecem.
NOTA: O log irá sugerir sementes semelhantes à média. - Ajustar o canal de referência, o raio de desfoque de Gauss, e as opções de correcção de mudança para optimizar o processo de selecção das sementes de modo a que um número de quadros de sementes encontradas têm uma aparência semelhante. Continue adicionando sementes até sementes médios são criadas que assemelham-se satisfatoriamente a estrutura visto nos dados que está a ser modelado.
- Nome das sementes e onde eles serão salvos e selecione "OK" para salvar as imagens finais de sementes para uso posterior na geração do modelo.
4. gerar modelos
- Select "Gerar modelos baseados em sementes", seleccionar a pasta, onde as partículas extraídas são guardados, e visualizar o diálogo "gerar modelos" (Figura 6).
- Carregar as sementes para cada canal, utilizando as caixas de verificação.
NOTA: As sementes serão salvos em uma subpasta que foi nomeado na caixa de diálogo "Gerar Seeds". O nome padrão é "Análise". Tome cuidado para selecionar as Médias descendência, não os quadros, que também são salvos para referência. - Atribuir parâmetros iniciais de geração de modelo, inserindo-os na caixa de diálogo "Gerar Models", como se segue. Ajustar esses parâmetros mais tarde.
- Seleccionar se usar um canal de referência para o alinhamento, o qual calcula translações e rotações apenas a partir do canal de referência e aplica-los a todos os canais. Se usando esta opção, seleccionar o canal a ser usado como a referência.
NOTA: Esta opção só deve ser escolhido para usar especificamente um canal como um REFERÊdno, tal como se fazendo descoberta estrutura à base de referência. Caso contrário, ele irá resultar em modelos menos precisos. Os canais devem estar bem alinhados, se usar esta opção. - Selecione se a quadratura intensidades de imagem durante casamento de modelos. Isso vai acentuar pequenas diferenças, então use isso ao criar um modelo que tem características particularmente sutis.
- Escolha uma semelhança mínima contra a semente usando a "Similaridade mínima contra a semente" controle deslizante ou inserindo um número na caixa.
NOTA: Somente as partículas com uma semelhança com as sementes maiores que esse corte será usado. 60-80% é normalmente uma boa escolha para começar. - Definir o número máximo de iterações para 1, optimizar o grau de similaridade contra semente, e aumentá-lo mais tarde, conforme for necessário.
- Marque as caixas para escolher os elementos do processo de geração do modelo que irá aparecer.
NOTA: "Mostrar sementes" vai exibir as sementes que foram carregados com as boxes na parte superior da caixa de diálogo. "Mostrar modelos" irá exibir todas as iterações dos modelos que são criadas a partir de uma média do subconjunto de partículas que se encontra com o grau de similaridade contra semente. "Show MSE" irá mostrar uma imagem do erro quadrático médio (MSE), que destaca as áreas do modelo que são mais variável. "Mostrar partículas" irá exibir o subconjunto de partículas que são usados para criar os modelos, registrados de acordo com o mais alto iteração do modelo que é exibido.
- Seleccionar se usar um canal de referência para o alinhamento, o qual calcula translações e rotações apenas a partir do canal de referência e aplica-los a todos os canais. Se usando esta opção, seleccionar o canal a ser usado como a referência.
- Selecione "Mostrar previsão" para gerar modelos de visualização e ver os resultados.
NOTA: Este é o passo mais intensa computacionalmente do processo. O tempo de execução para um conjunto de alguns milhares de partículas com diâmetros de algumas dezenas de pixels em um PC desktop deve ser em torno de 10 min. Se o tempo de computação é um problema, os usuários devem primeiro tentar o algoritmo em um subconjunto menor dos dados ou usar um raio ROI menor no passo 2.2.4, se possível. - Ver a gerard modelos e optimizar os parâmetros, especialmente o grau de similaridade contra semente. Aumentar o grau de similaridade contra a semente até que só os verdadeiros partículas da morfologia modelado estão incluídas no modelo.
- Aumentar o número máximo de iterações usando o "número máximo de iterações" controle deslizante ou inserindo um número na caixa e permitir que o processo de geração de modelo para percorrer. Use um valor de cerca de 10 para a iteração máxima.
- Nome modelos e selecione "OK" para salvar as pilhas evolução do modelo que contêm todas as iterações do processo final geração do modelo.
NOTA: Se a similaridade mínima contra a semente é tão grande que há partículas têm essa semelhança, nada será atualizado. Se o plugin parece ser congelados, considere a possibilidade de que a semelhança mínimo é muito alto.
Resultados
Aqui, demonstramos o software no modelo de poxvírus, vírus vaccinia. Um dos vírus de mamíferos mais complexos, pacotes de vaccinia cerca de 80 proteínas diferentes dentro de um 350 x 270 x 250 Nm 3 de partículas em forma de tijolo 13, 14. Três sub-estruturas são discerníveis por microscopia electrónica: um núcleo central, que contém o genoma de ADNcd; duas estruturas proteicas, chamados corpos laterais, que flanqueiam o núcleo; e uma única bicamada proteolipídeo envelope 15. O tamanho grande, a estrutura complexa, e receptividade à etiquetagem proteína fluorescente recombinante vaccinia fazer um excelente sistema para demonstrar o fluxo de trabalho VirusMapper.
Utilizando o software conforme descrito aqui, a distribuição de uma variedade de proteínas de virião de vaccinia pode ser modelado. A proteína marcada foi representada por imagem e, possivelmente, em combinação com uma outra proteína de distribuição conhecida como uma referência, e o software foi utilizado como descrito para produzir modelos médios da localização da referida proteína sobre a partícula. Neste exemplo, duas proteínas foram modelados, a proteína L4 de núcleo interior, e o componente principal F17 lateral do corpo.
Um vus vaccinia recombinante que possui F17 marcado com GFP e L4 marcado com mCherry 16 foi utilizada. O vírus purificado foi diluído em Tris 1 mM, pH 9, e obrigado a lamelas lavado, alto desempenho, revestindo-os durante 30 min a temperatura ambiente. As amostras foram em seguida fixadas por aplicação de 4% de formaldeo em PBS durante 20 min. As lamelas foram montadas em lâminas imediatamente em antifade meio de montagem. Imagiologia foi realizada pelo SIM em um microscópio SIM comercial. Um campo de visão foi seleccionada contendo centenas de vírus e as imagens foram obtidas utilizando 5 desvios de fase e 3 rotações de grade com 561 nm (32 ^ m PE ralarRIOD) e 488 nm (32 pm) lasers período de grelha. As imagens foram obtidas utilizando uma câmara scmos e processadas utilizando o software microscópio. Canais estavam alinhados com base em um slide talão multi-colorida fotografada com as mesmas configurações de aquisição de imagem. Depois de imagens e de reconstrução de alinhamento SIM canal foram abertas nas ilhas Fiji e concatenados em uma única pilha de imagens.
As partículas virais foram extraídos a partir das imagens, utilizando o canal de L4 como referência e sem aplicar qualquer borrão de Gauss, como estas partículas têm um máximo central. Cerca de 15.000 partículas foram extraídos nesta experiência.
Devido à geometria da vacínia, os corpos laterais têm uma aparência distintamente diferente, com base na orientação do vírus. Visualizamos duas orientações em que um ou dois corpos laterais poderiam ser distinguidas. Nós nos referimos a essas orientações como frontal e sagital, respeitovamente.
Sementes separadas para as orientações frontal e sagital foram seleccionados através de pesquisa através da lista de partícula na fase "Gerar Sementes" (Figuras 4 e 5); partículas que estavam claramente em uma orientação ou o outro foram escolhidas. O canal L4 foi usada como o canal de refercia para alinhar as sementes com uma outra. Novamente, não Gaussian Blur era necessário. 5 partículas para cada orientação foram selecionados e foram calculadas as médias para produzir as sementes.
Os modelos foram gerados para cada orientação com base nestas sementes. foram utilizados nem uma referência nem canal valores de intensidade ao quadrado. O número máximo de iterações foi definido inicialmente para 1, e a similaridade mínima foi definida para incluir cerca de 1.000 partículas em cada caso, o que deu uma aparência consistente para cada orientação. O número máximo de iterações foi então aumentada parapermitir a convergência do modelo. Os modelos foram assim gerados para as duas orientações dos dois canais (Figura 7).

Figura 1: fluxo de trabalho VirusMapper. O plug-in está organizado em três fases principais. As partículas virais são extraídos a partir de imagens de grandes dimensões, imagens ou sementes de modelo são seleccionados semi-manualmente a partir dos dados, e modelos SPA finais são gerados a partir dos dados, referindo-se as sementes. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: "Extraia Estruturas Virais" diálogo. Ao selecionar "Extrair Viral Structur es", este diálogo irá aparecer. Os parâmetros devem ser preenchidos com as estimativas iniciais para a segmentação ideal. 'Mostrar previsão', então pode ser selecionado, permitindo que as ROIs a ser visualizado e os parâmetros a ser afinado. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: configuração de parâmetros de extracção. Depois de visualizar o ROIs que vai ser extraído, o raio ROI, número de ROIs, e máxima sobreposição ROI são ajustados para atingir uma situação como esta. ROIs são ligeiramente maiores do que as partículas, todas as partículas estão incluídas em uma ROI, e ROIs podem sobrepor-se suficientemente para permitir que as partículas agregadas para ser separado.ank "> Por favor clique aqui para ver uma versão maior desta figura.

Figura 4: Geração de sementes de correspondência de molde. A caixa de diálogo "Gerar Seeds" (1) define os parâmetros a serem atribuídos. A sequência de partículas de referência (2) permite ao utilizador verificar através de partículas no canal de referência. Quando uma partícula é visto na sequência de referência partículas, partículas realinhados para todos os canais podem ser vistos nas pré-visualizações de partículas realinhados (3). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: A adição de imagens de semente. Como as sementes são adicionadas ao "Quadros de usar" caixa, a média de todas as sementes (4) e os quadros envolvidos (5) são exibidos. As partículas que são semelhantes aos atuais sementes médias são sugeridas na caixa de diálogo (6). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: "Gerar Models" diálogo. Ao selecionar "Gerar modelos baseados em sementes," este diálogo irá aparecer. Os parâmetros deve ser preenchido com estimativas iniciais para a geração modelo ideal, e os elementos do processo de geração do modelo a ser mostrado durante o cálculo deve ser seleccionado. "Mostrar previsão", então pode ser selecionado, permitindo que o processo de geração do modelo a ser executado e os parâmetros a ser afinado.ftp_upload / 55471 / 55471fig6large.jpg" target = '_ blank'> Clique aqui para ver uma versão maior desta figura.

Figura 7: Modelos gerados com VirusMapper. viriões de vaccinia com a proteína do núcleo L4 marcado com mCherry e a proteína de corpo laterais F17 marcado com EGFP foram fotografadas usando SIM. Os modelos foram, em seguida, gerado com o software, como descrito no protocolo. Duas orientações, frontal e sagital, distinguem-se pelo aspecto dos corpos laterais. Barra de escala = 100 nm. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Com este método, os pesquisadores estão equipados para combinar o poder do SPA e microscopia SR a fim de gerar de alta precisão, modelos 2D multi-canal da arquitetura proteína de vírus e outros complexos macromoleculares. No entanto, algumas considerações importantes devem ser levados em conta.
As sementes devem ser escolhidos para representar uma estrutura que é consistentemente observado. Assim, os dados em bruto deve ser inspeccionada cuidadosamente antes de as sementes são escolhidos. Isto é importante para a prevenção de modelos enviesados. As escolhas podem ser validados pelo exame dos limiares mínimos de similaridade necessários para incluir um certo número de partículas nos modelos. É evidente que, para uma escolha de semente, quanto maior for este limiar necessita de ser para um determinado número de partículas, o mais que a estrutura é aparente nos dados.
O conceito de modelo de correspondência é particularmente útil quando há heterogeneidade nos dados. Todas as estruturas diferentes que estão viveis devem ser identificados e diferentes modelos criados para cada caso. Ao separar estruturas heterogéneas em um canal, mas simultaneamente a criação de modelos em um segundo canal, padrões podem emergir que não teria sido imediatamente evidente.
Outra consideração para estar ciente de quando se usa este algoritmo é que o método de iteração irá maximizar assimetria estocástica. Por exemplo, quando a modelagem de uma estrutura com dois máximos simétrica, todos os ligeiras assimetrias entre os máximos serão alinhados um com o outro durante a iteração, e o modelo final irá, assim, ser maximamente assimétrica. Se isso não reflete uma simetria conhecido na estrutura que está sendo modelada, então isso deve ser levado em conta. Atualmente, a única maneira de evitar isso maximização é limitar o número de iterações a 1, embora um desenvolvimento potencial seria para VirusMapper incorporar eixos de simetria no processo de geração do modelo. Todas as novas versões do VirusMapper será available no site referenciados (ver Tabela de Materiais). Os usuários também vai encontrar uma FAQ aqui para responder a quaisquer dúvidas comuns.
O software como descrito é aplicável a qualquer estrutura que pode ser trabalhada com resolução suficiente para visualizar as características que o usuário deseja modelar. Embora SPA pode melhorar a resolução, é evidente que não vai melhorar a visibilidade de recursos que de outra forma não são visíveis. Este protocolo não está, por conseguinte, um método para melhorar a qualidade dos dados. Tal como acontece com qualquer técnica, preparação de amostras cuidado e otimização da estratégia de imagem irá fornecer os dados mais limpas e os melhores modelos resultantes.
A escolha da modalidade de imagem EL também é importante e, em geral, irá depender da amostra em questão. VirusMapper foi validado para funcionar bem com SIM e STED 10, e também pode ser usado com dados de microscopia de localização de alta qualidade, mas o cuidado deve ser tomado neste caso,como a rotulagem esparsa pode causar problemas semelhantes aos da maximização assimetria.
Atualmente, VirusMapper é o algoritmo única disponível livremente para a análise de uma única partícula de imagens de fluorescência e de propósito geral única 2D SPA software de média. Outros estudos que fizeram uso dos mesmos princípios 4, 6, 8 têm usado software personalizado especializada para cada estudo particular. Algoritmos de uso geral para a reconstrução de dados 3D foram publicados 5, 18, embora nenhum software foi fornecido.
Quando usado como descrito neste artigo, VirusMapper pode ser usada para produzir modelos precisos, exactos, e robustos da arquitectura proteína macromolecular de vírus e outros complexos. Com estes modelos, os pesquisadores podem fazer medições precisas das dimensões médias do structUres em estudo, potencialmente permitindo-lhes chegar a conclusões biológicas que não teria de outra forma sido possível.
Além disso, com as capacidades multi-canal de esta técnica, é possível mapear um número ilimitado de proteínas e componentes complexos dentro e para descobrir novos organização da proteína. Examinando mudanças na estrutura em nanoescala em diferentes condições biologicamente relevantes, tais como diferentes estágios de um ciclo de vida do vírus, tem o potencial de oferecer informações valiosas sobre biologia.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Nós gostaríamos de agradecer Corina Beerli, Jerzy Samolej, Pedro Matos Pereira, Christopher Bleck, e Kathrin Scherer por suas contribuições para o desenvolvimento original e validação de VirusMapper. Gostaríamos também de agradecer Artur Yakimovich por sua leitura crítica do manuscrito. Este trabalho foi financiado por doações da Biotecnologia e Ciências Biológicas Conselho Investigação (BB / M022374 / 1) (RH); financiamento de base para o MRC Laboratório de Biologia Molecular Cell, University College of London (JM); o Conselho Europeu de Investigação (649101-UbiProPox) (JM); eo Conselho de Pesquisa Médica (RM / K015826 / 1) (RH e JM). RG é financiado pela Engenharia e Ciências Físicas Research Council (EP / M506448 / 1).
Materiais
| Name | Company | Catalog Number | Comments |
| Fiji | Open-source image analysis software | ||
| NanoJ-VirusMapper | developed by the Henriques lab | Open source-Fiji plugin (https://bitbucket.org/rhenriqueslab/nanoj-virusmapper) | |
| VectaShield antifade mounting medium | Vector Labs | H-100 | |
| Elyra PS1 | Zeiss | ||
| ZEN BLACK | Zeiss | Image processing software for SIM | |
| High performance coverslip | Zeiss | 474030-9000-000 | |
| TetraSpeck beads | ThermoFisher | T7279 |
Referências
- Henriques, R., Griffiths, C., Rego, E. H., Mhlanga, M. M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers. 95 (5), 322-331 (2011).
- Gustafsson, N., Culley, S., et al. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 7, 12471 (2016).
- Cheng, Y., Grigorieff, N., Penczek, P. A., Walz, T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 161 (3), 438-449 (2015).
- Szymborska, A., de Marco, A., Daigle, N., Cordes, V. C., Briggs, J. A. G., Ellenberg, J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 341 (6146), 655-658 (2013).
- Broeken, J., Johnson, H., et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl Fluoresc. 3 (1), 14003 (2015).
- Burns, S., Avena, J., Unruh, J., Yu, Z. Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife. , (2015).
- Lelek, M., Di Nunzio, F., Henriques, R., Charneau, P., Arhel, N., Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc Nat Acad Sci U S A. 109 (22), 8564-8569 (2012).
- Laine, R. F., Albecka, A., Svan de Linde, ., Rees, E. J., Crump, C. M., Kaminski, C. F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun. 6, 5980 (2015).
- Schindelin, J., Arganda-Carreras, I., et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9 (7), 676-682 (2012).
- Gray, R. D. M., Beerli, C. VirusMapper: open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci Rep. 6, 29132 (2016).
- Gustafsson, M. G. L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J of Micros. 198 (2), 82-87 (2000).
- Hell, S. W., Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Let. 19 (11), 780 (1994).
- Chung, C. -. S., Chen, C. -. H., Ho, M. -. Y., Huang, C. -. Y., Liao, C. -. L., Chang, W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 80 (5), 2127-2140 (2006).
- Moss, B. Poxviridae: the viruses and their replication. Fields virology. , (2010).
- Condit, R. C., Moussatche, N., Traktman, P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 66 (6), 31-124 (2006).
- Schmidt, F. I., Bleck, C. K. E., et al. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 4 (3), 464-476 (2013).
- . Nanoj-virusmapper Available from: https://bitbucket.org/rhenriqueslab/nanoj-virusmapper (2016)
- Fortun, D., Guichard, P., Unser, M. Reconstruction From Multiple Poses in Fluorescence Imaging: Proof of Concept. IEEE J Sel Topics Signal Process. 10 (1), 61-70 (2016).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados