Method Article
Échantillonnage d’une Interface de catalyseur hétérogène eau/métal à l’aide de la théorie fonctionnelle de la densité et de champs de Force dynamique moléculaire multi-échelles
Dans cet article
Résumé
Le protocole présenté ici vise à générer et dégustez les trajectoires des configurations des molécules d’eau liquide autour des espèces catalytiques sur une surface plate métal de transition. Les configurations de l’échantillons peuvent être utilisées comme structures à partir de méthodes basées sur la mécanique quantique.
Résumé
Un nombre important de procédés chimiques catalyse hétérogène se produit dans des conditions liquides, mais simulant le rôle de catalyseur dans de telles conditions est difficile quand il est nécessaire d’inclure les molécules de solvant. La rupture de la liaison et formant des processus modélisés dans ces systèmes nécessitent l’utilisation de méthodes chimiques de quantum. Puisque les molécules en phase liquide sont en constant mouvement thermique, simulations doivent également inclure configurationnelle d’échantillonnage. Cela signifie que les configurations multiples de molécules liquides doivent être simulées pour chaque espèce catalytique d’intérêt. Le protocole présenté ici vise à générer et dégustez les trajectoires des configurations des molécules d’eau liquide autour des espèces catalytiques sur les surfaces plates des métaux de transition d’une façon qui équilibre chimique précision avec dépenses de calcul. Plus précisément, simulations de dynamique moléculaire (FFMD) de champ de force sont utilisées pour générer des configurations des molécules liquides qui peuvent ensuite être utilisés dans des méthodes basées sur la mécanique quantique comme théorie fonctionnelle de la densité ou ab initio moléculaire dynamique. Pour illustrer cela, dans ce manuscrit, le protocole est utilisé pour des intermédiaires catalytiques qui pourraient être impliqués dans la voie de la décomposition du glycérol (C3H8O3). Les structures qui sont générés à l’aide du FFMD sont modélisés dans DFT afin d’estimer les enthalpies de solvatation de l’espèce catalytique et d’identifier comment les molécules de H2O participent à décomposition catalytique.
Introduction
Modélisation des phénomènes moléculaires impliqués dans la catalyse hétérogène dans des conditions de liquides est nécessaire pour la fonction catalytique de compréhension ; Toutefois, cela reste difficile car elle nécessite un équilibre subtil entre la précision chimique et dépenses de calcul. En général, étant donné que la catalyse comporte la rupture et la formation de liaisons chimiques, la mécanique quantique doit être utilisé dans au moins une certaine mesure ; Cependant, des simulations longues sont difficiles en mécanique quantique, car ils nécessitent des ressources système. Puisque les molécules en phase liquide sont en constant mouvement thermique, simulations doivent également inclure l’échantillonnage configurationnelle, c'est-à-dire, qu’ils doivent intégrer les multiples arrangements spatiaux des molécules liquides, comme chaque arrangement spatial différent (c.-à-d., chaque configuration) a une énergie différente. Cela signifie que les configurations multiples de molécules liquides doivent être simulées pour chaque espèce catalytique d’intérêt. Ces besoins – d’utiliser la mécanique quantique et d’effectuer des calculs multiples par espèce catalytique – peuvent rendre la modélisation en catalyse hétérogène sous phase liquide insolubles par le calcul. Le but de la méthode décrite ici est de permettre des simulations tractable par le calcul des phénomènes en catalyse hétérogène sous phase liquide.
Nous sommes particulièrement intéressés par des réactions de catalysées hétérogène qui sont réalisent sous l’eau liquide. Les molécules d’eau ont une influence significative sur les phénomènes catalytiques comme interagissant avec catalytique d’espèces (par exemple, par l’intermédiaire de forces de dispersion et des liaisons hydrogènes)1,2,3,4,5 ,6,7,8,9,10,11,12,13,14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23, participant à des réactions catalytiques1,7,8,9,15,21,22,24 ,25,26,27et influençant les chemins réactionnels et/ou taux catalytique1,11,12,15, 18,23,25,27,28,29,30,31. La modélisation de ces phénomènes a été réalisée à l’aide de QM et/ou ab initio la dynamique moléculaire (DMAI)1,2,6,7,14,22 ,25,27,28,32,33,34, la force dynamique moléculaire champ (FFMD)35 et la mécanique quantique et moléculaire mécanique (QM/MM)10. AIMD et FFMD, les atomes dans le système soient déplacés en application des équations de Newton du mouvement selon les forces qui agissent sur eux. Dans DMAI, les forces et l’énergie du système sont calculées avec la mécanique quantique, alors que dans FFMD, les forces et l’énergie du système sont calculées à l’aide de la force de champs, qui sont des expressions algébriques qui sont paramétrées selon expérimentale ou données QM. En QM/MM, la partie du système où apparaît le lien briser et formant est calculée avec QM, et le reste du système est calculé avec MM, qui emploie des champs de force. Parce qu’ils emploient directement QM, AIMD et QM/MM conviennent mieux pour capturer la rupture de la liaison et formant qui se produit en catalyse hétérogène de phase aqueuse ; Cependant, FFMD est significativement plus mathématiquement tractable et donc mieux adaptée pour générer les configurations des molécules de liquide H2O. La méthode présentée dans le présent protocole soldes précision chimique et dépenses de calcul en utilisant une combinaison des QM et FFMD.
Plus précisément, cette méthode utilise des simulations FFMD pour générer des configurations de liquide H2O et QM pour calculer les énergies du système. FFMD est réalisée à l’aide de LAMMPS. 36 les champs de force utilisés dans FFMD dans ce travail emploient Lennard-Jones + potentiel de Coulomb (LJ + C), où les paramètres LJ ont été tirées de la TIP3P/CHARMM modèle37 H2O, le champ de force universelle38 (UFF) pour le Pt et le Champ de force OPLS-AA39 espèces catalytique ainsi que les paramètres Coulomb sont tirées du modèle TIP3P/CHARMM37 H2O et le champ de force OPLS-AA39 espèces catalytiques. Les paramètres de Coulomb pour Pt atomes ont été mis à 0. Les calculs de QM sont effectués à l’aide de la VASP code40,41,42, qui est un code de théorie de la fonctionnelle (DFT) de densité. Insertions de molécule d’eau sont effectuées avec une code développé interne appelée Monte Carlo plug-in pour les méthodes quantiques (MCPliQ). Conversions de fichier de VASP en LAMMPS dans le présent protocole sont effectuées avec le logiciel de Visual Molecular Dynamics (VMD)43.
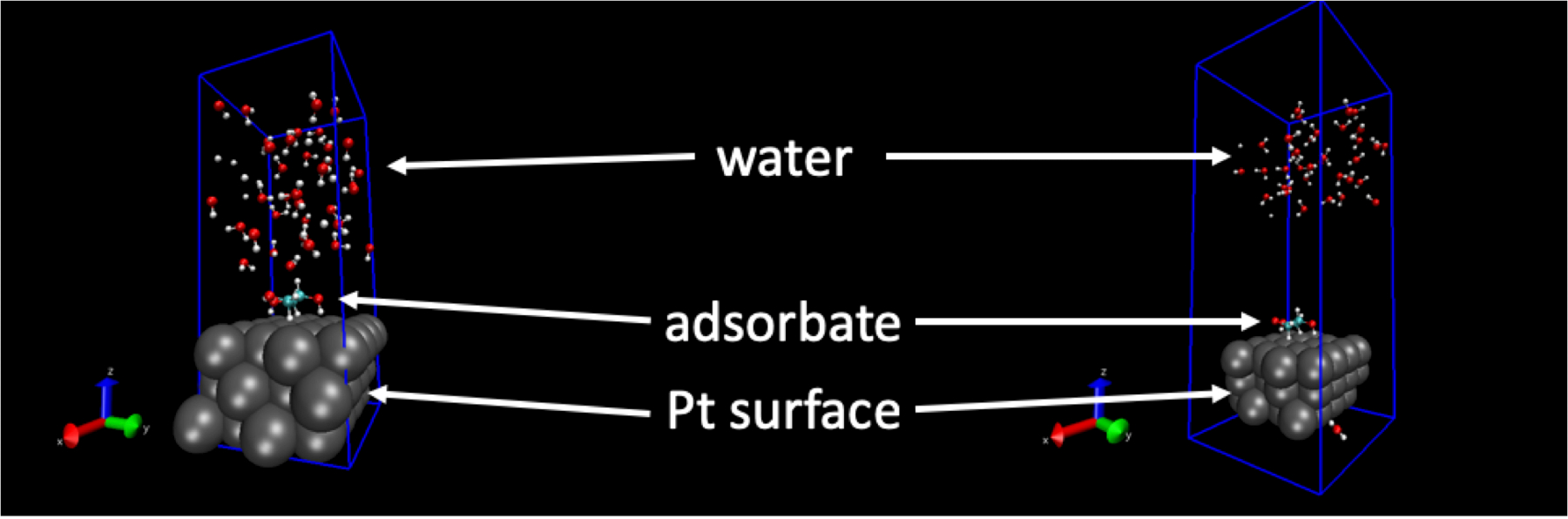
Le protocole vise à générer des configurations des molécules d’eau liquide autour des espèces catalytiques sur les surfaces plates des métaux de transition à faible couverture. La couverture est notée θ et défini comme le nombre des adsorbats par atome de métal surface (c.-à-d., le nombre des adsorbats surfaces normalisée par le nombre d’atomes métalliques dans la couche superficielle de la dalle du métal dans le modèle de catalyseur). Dans ce manuscrit, faible taux de couverture est définie comme θ ≤ 1/9 monocouche (ML), où 1 ML signifie une espèce catalytique par atome de métal surface. Les modèles de catalyseur doivent être placés dans des boîtes de simulation périodique. Les boîtes de simulation n’ont pas à être des cubes. Ce manuscrit illustre l’utilisation du protocole pour générer des configurations de liquide H2O qui peut être utilisé pour calculer les quantités d’intérêt en catalyse hétérogène de phase aqueuse.
Ce protocole requiert que l’utilisateur a accès à des versions du logiciel VASP, MCPliQ, LAMMPS et VMD installées et fonctionnel. Plus d’informations sur VASP (https://www.vasp.at/), LAMMPS (https://Lammps.sandia.gov/) et VMD (https://www.ks.uiuc.edu/Research/vmd/) sont disponibles sur leurs sites Web respectifs. Le logiciel MCPliQ est documenté à https://github.com/getman-research-group/JoVE_article, ainsi que tous les fichiers d’entrée et des scripts Python mentionnés dans le présent protocole. Ce protocole suppose que les fichiers exécutables et les scripts mentionnés dans s’exécutera sur un ordinateur de recherche performant et sont installés dans un répertoire qui se trouve dans la variable $PATH de l’utilisateur. Si un fichier exécutable ou un script est placé dans un endroit qui n’est pas à l’utilisateur de $PATH, puis le chemin vers l’exécutable doit être inclus pour l’exécuter. Exécutables et les scripts sont exécutés en étapes 2.1.2, 2.2.1, 2.2.8, 3.1, 4.2, 5.2 et 6.1.2. Par exemple, pour exécuter le code de MCPliQ à l’étape 2.1.2 provenant d’un répertoire qui n’est pas à l’utilisateur de $PATH, l’utilisateur devez taper $PATHTOMCPLIQ/mcpliq à l’interface de ligne de commande au lieu de mcpliq, où $PATHTOMCPLIQ est l’emplacement où le mcpliq fichier exécutable a été stocké (par exemple, $PATHTOMCPLIQ pourrait être ~ / bin). Avant de commencer ce protocole, tous les scripts et exécutables doivent recevoir des autorisations exécutables (par exemple, sous Linux, cela pourrait se faire en tapant chmod + x mcpliq à l’interface de ligne de commande à partir du répertoire où l’exécutable mcpliq est stockée). En outre, tous les modules requis par les logiciels ou les scripts doivent être chargés (ces dépendances seront spécifiques à des installations individuelles des différents logiciels et l’ordinateur où s’exécuteront les simulations).
Protocole
1. générer la structure de l’adsorbat
- Créez un fichier de VASP POSCAR comprenant un orage supercellulaire avec conditions aux limites périodiques, comme vous le feriez si vous exécutiez simulations des adsorbats sur des surfaces métalliques sous vide. L’orage supercellulaire devrait inclure une estimation de la structure de l’adsorbat et la surface métallique ainsi que l’espace vide au-dessus de l’adsorbat pour l’ajout de molécules de H2O. Détails sur la façon de le faire sont fournis dans les précédents travaux35,44.
Remarque : Il est important que la hauteur de l’espace vide doit être au moins 12 Å au-dessus de l’adsorbat. - Détendre la structure et de minimiser son énergie en utilisant le code VASP. Détails sur la façon de le faire sont fournis dans les précédents travaux35,44. Cela produira un fichier appelé CONTCAR, qui sera utilisé dans la section suivante.
2. ajouter des molécules de2O H explicites
- Ajouter N molécules d’eau à l’espace vide dans le CONTCAR créé à l’étape 1.2 en utilisant le code de MCPliQ, où N = ρV, ρ est la masse volumique de l’eau, et V est le volume de l’espace vide situé au-dessus de l’adsorbat.
: Ρ il convient noter que la densité de l’eau, tel que déterminé par le modèle TIP3P/CHARMM de l’eau à la température de la simulation. V va être affinée à l’étape suivante.- Spécifiez les informations suivantes dans le fichier de master_input.txt de MCPliQ : le nombre de molécules2O H à ajouter (N) en changeant le premier argument de la ligne 28, le chemin vers le fichier water.txt en changeant le second argument dans la ligne 28 et le minimum et hauteur maximale de la supercellule qui peut être occupé par les molécules d’eau en changeant la coordonnée z Minimum en ligne 11 et la coordonnée z Maximum en ligne 12.
- Exécuter le code de MCPliQ en tapant mcpliq partir de l’interface de ligne de commande pour insérer des molécules d’eau dans le fichier CONTCAR. Le code va afficher un ou plusieurs fichiers avec l’extension de fichier. POSCAR.
Remarque : Si plusieurs fichiers de POSCAR sont produits, ils seront appellera POSCAR_n.POSCAR. Sélectionnez le fichier où n est plus grand.
- Générer les fichiers d’entrée LAMMPS pour une simulation du TNP et équilibrer le volume cellulaire utilisant FFMD dans la troupe du TNP dans LAMMPS.
- Exécutez le script lmps_bond_angle.py sur le. Fichier POSCAR généré à l’étape 2.1.2 en tapant lmps_bond_angle.py $filename. POSCAR à l’interface de ligne de commande, où $filename est le nom de la. Fichier POSCAR généré à l’étape 2.1.2. Ce script crée un fichier nommé $filename. POSCAR.bond_angle_info.txt, qui énumère les obligations et les angles qui seront utilisés dans le fichier de données LAMMPS.
- Ouvrez VMD et sélectionnez fichier > Nouvelle molécule dans la fenêtre principale pour ouvrir la fenêtre de l’Explorateur de fichiers de molécule . Sélectionnez VASP_POSCAR dans la liste déroulante Type de fichier déterminer . Cliquez sur parcourir et accédez à la $filename. Fichier POSCAR. Cliquez sur Load pour ouvrir le $filename. Fichier POSCAR.
- Ouvrez la Console de savoirs traditionnels au sein de VMD en sélectionnant Extensions > Tk Console à partir de la fenêtre principale de VMD.
- Exécutez la commande suivante dans la console de Tk : topo writelammpsdata $WDPATH/data.myadsorbate complet, où $WDPATH est le répertoire sur l’ordinateur où VMD va écrire le fichier de données LAMMPS et data.myadsorbate est le nom du fichier de données LAMMPS.
- Supprimez la section liaisons et les Angles au bas du fichier data.myadsorbate. Ensuite, ajoutez les listes bond et angle dans le fichier $filename. POSCAR.bond_angle_info.text en data.myadsorbate.
NOTE : Les indices pour le type de liaison O-H et le type d’angle H-O-H pour les molécules d’eau dans le $filename. Fichier de POSCAR.bond_angle_info.txt ont la valeur 1. Ainsi, types bond et angle des adsorbats devraient commencer à compter à 2. - Modifiez le fichier data.myadsorbate en ajoutant les paramètres de Lennard-Jones à la section de la Paire Coeffs et les paramètres de Coulomb à la section des atomes . Lennard-Jones et paramètres Coulomb pour les molécules de H2O, atomes adsorbée et des atomes de surface métalliques doivent être ajoutées.
Remarque : Les paramètres de Lennard-Jones pour Pt atomes, des molécules d’eau et atomes d’adsorbat dans le présent protocole sont obtenus à partir de l’UFF38, TIP3P/CHARMM37et OPLS-AA39 champs de force, respectivement. Paramètres de Coulomb pour les atomes des molécules et des adsorbats eau proviennent de la TIP3P/CHARMM37 et OPLS-AA39 champs de force, respectivement. Paramètres de Coulomb pour Pt atomes sont définis à 0 dans le présent protocole. Alternativement, calculées charges partielles pourraient être utilisées pour les paramètres Coulomb pour adsorbat atomes et Pt. - Copiez le LAMMPS fichier d’entrée input.equil dans le répertoire $WDPATH. Modifier la variable de groupe en ligne 34 pour indiquer l’index de type atom pour l’oxygène de l’eau et les atomes d’hydrogène de l’eau et la variable de groupe en ligne 35 pour indiquer l’index de type atom pour les atomes Pt et d’adsorbat.
- Exécuter le logiciel LAMMPS en tapant mpiexec np - XX lmp_mpi < input.equil à l’interface de ligne de commande, où XX correspond au nombre de cœurs de processeurs à utiliser, et lmp_mpi est le nom de l’exécutable LAMMPS. Cela se déroulera une minimisation d’énergie pour affiner la configuration2O H, suivie d’une simulation de FFMD effectuée au nombre constant de molécules de H2O (N), volume (V) et la température (T) d’apporter la l’eau à la température de la simulation, suivie par une simulation du FFMD exécuter à la constante N, (P), la pression et la température (T) pour déterminer la hauteur physiquement correcte de la boîte de simulation. Les fichiers de sortie qui seront utilisées à l’article 3 sont appelés data.myadsorbate_npt et log.myadsorbate.
Remarque : La durée de la simulation du TNP doit être longtemps assez pour former un « équilibrage » exécuté, où le volume de la supercellule vient à l’état stationnaire et un « tirage », qui est utilisé pour l’échantillon, que la moyenne de l’ensemble (ici, la hauteur de la supercellule). Pendant le passage de l’équilibration, le volume de la supercellule lorsque comploté contre temps doit se stabiliser à une valeur d’état d’équilibre. Une fois que cela se produit, la simulation du TNP peut être considérée comme dans son cycle de production. Vérifiez l’équilibration de la simulation du TNP en veillant à ce que les fluctuations de la hauteur de la supercellule (lz) sont minimes ou ont convergé vers une valeur constante. En cas de fluctuations importantes, puis re-générer une configuration de2O H en diminuant la timestep en ligne 92 dans le fichier input.equil et répétition étape 2.2.8 ou recommencer à l’étape 2.1.1.
3. extraire la hauteur correcte de la supercellule
- Exécutez le script get_npt_lz.py sur le fichier log.myadsorbate en tapant get_npt_lz.py log.myadsorbate à l’interface de ligne de commande. Ce script affiche la hauteur moyenne supercell de la partie cycle de « production » de la simulation du TNP dans le fichier avg_lz.txt.
Remarque : Le script de get_npt_lz.py suppose que LAMMPS écrit la longueur de la cellule z-dimension (lz) dans le fichier log.myadsorbate chaque 1000 fs (personnalisable à la ligne 20 du script get_npt_lz.py), qui est la valeur par défaut dans le fichier d’entrée fourni input.equil LAMMPS. Le script get_npt_lz.py détecte et élimine les 2 premières ns (personnalisable à la ligne 19 du script get_npt_lz.py) une valeur de lz valeurs dans le fichier log.myadsorbate, car ils représentent la portion de l’équilibration de la simulation, alors que les 3 autres ns comprennent le " partie de la production » et sont donc utilisés par le script get_npt_lz.py pour calculer la longueur de z-dimension moyenne. En plus du fichier avg_lz.txt, le script get_npt_lz.py génère un fichier nommé npt_data.txt, qui fournit les valeurs de lz en fonction de l’instant, mais aussi un fichier appelé npt_plot.png, qui trace les mêmes données. L’intrigue peut être utilisé pour vérifier l’équilibration de la simulation du TNP. - Reconstruire la supercellule à l’aide de la hauteur moyenne déterminée au TNP.
- Copiez le fichier data.myadsorbate_npt dans un nouveau répertoire, désigné ici par $WD2PATH et renommez-le data.myadsorbate.
- Modifiez le nouveau fichier data.myadsorbate afin que la hauteur de lz est égale à la valeur moyenne de sortie du script get_npt_lz.py en modifiant les arguments zlo et zhi dans le fichier data.myadsorbate tel que zlo est 0,0 et zhi est la valeur de lz provient du fichier avg_lz.txt en s TEP 3.1.
4. générer des configurations de molécules de H2O
- Copiez le LAMMPS fichier d’entrée input.prod dans $WD2PATH. Modifier la variable de groupe en ligne 32 pour indiquer l’index de type atom pour l’oxygène de l’eau et les atomes d’hydrogène de l’eau et la variable de groupe en ligne 33 pour indiquer l’index de type atom pour les atomes Pt et d’adsorbat.
- Exécuter le logiciel LAMMPS en tapant mpiexec np - XX lmp_mpi < input.prod dans l’interface de ligne de commande, où XX correspond au nombre de cœurs de processeurs à utiliser, et lmp_mpi est le nom de l’exécutable LAMMPS. Cela fonctionnera une simulation NVT constante sur les molécules de2O H. Le principal extrant de cette simulation est le fichier dump.myadsorbate.lammpstrj.
Remarque : La durée de la simulation NVT doit être longtemps assez pour former une équilibration exécuté, où l’énergie du système est de stabiliser l’état, et une cycle, dont l’ensemble est en moyenne de production (ici, la position spatiale des molécules d’eau) sont échantillonnés. Pendant le passage de l’équilibration, l’énergie du système lorsque comploté contre temps doit se stabiliser à une valeur d’état d’équilibre. Une fois que cela se produit, la simulation NVT peut être considérée comme dans son cycle de production.
5. déterminer la durée de vie de liaisons hydrogènes pour l’échantillonnage de temps propre
- Modifiez le script hb_lifetime_dist.py pour spécifier : l’instant de la première image du fichier dump.myadsorbate.lammpstrj en changeant la variable actualStart sur la ligne 22, combien de cadres sont écrits dans le fichier de trajectoire LAMMPS en changeant la variable timestep sur ligne 23, le premier et le dernier pas de temps horaire le script devrait envisager (c.-à-d. la partie de la production de la trajectoire) en changeant les variables N_first et N_last sur les lignes 24 et 25, que ce soit images consécutives sont considérés ou cadres sont ignorés en changeant le nevery variable sur la ligne 26 et le nombre de lignes par section de trame du fichier trajectoire en changeant la variable frameLine à la ligne 27. En outre, modifiez les lignes 31 à 35 pour spécifier les types d’atomes dans le fichier de data.myadsorbate appartenant à l’adsorbat et types d’atomes appartenant aux molécules H2O.
Remarque : Le script hb_lifetime_dist.py analyse les configurations de2O H dans le terme de production et détermine si les molécules d’O2H sont les hydrogènes de l’adsorbat. Il puis les chefs d’accusation le temps de simulation qui chaque hydrogène demeure intacte et rapporte cette information comme une distribution de vies de liaison hydrogène dans les unités du ps. La version spécifique du script qui est fourni avec ce protocole suppose que LAMMPS écrit la configuration des molécules de H2O dans le fichier dump.myadsorbate.lammpstrj chaque fs 1000, qui est la valeur par défaut dans le fichier d’entrée fourni input.prod LAMMPS. Il détecte et élimine les 2 premières worth ns de configurations dans le le dump.myadsorbate.lammpstrj du fichier, car ils représentent la portion de l’équilibration de la simulation et utilise les 3 autres ns pour calculer les durées de vie de liaison hydrogène. - Exécutez le script hb_lifetime_dist.py sur le fichier dump.myadsorbate.lammpstrj en tapant hb_lifetime_dist.py à l’interface de ligne de commande. Cela produira un fichier appelé distribution_HB_lifetime.dat.
- Tracer les données dans le fichier distribution_HB_lifetime.dat pour voir la répartition des durées de vie de liaison hydrogène qui ont eu lieu au cours de la simulation NVT.
- Déterminer l’incrément de temps à utiliser pour l’intervalle d’échantillonnage basé sur les durées de vie calculée de liaison hydrogène. Le meilleur choix est la durée de vie maximale de liaison hydrogène ; Sinon, une valeur qui saisirait l’intervalle de confiance de 95 % peut être utilisée.
6. configurations d’échantillon de liquides molécules de H2O
- Déterminer le nombre de configurations de la série de production de la trajectoire NVT FFMD pour plus de calculs. Le nombre de configurations doit être choisi de sorte que le temps minimum entre les configurations est égale ou supérieure à l’intervalle d’échantillonnage indiquée dans la section 5.
- Modifiez la valeur par défaut de la variable num_frames sur la ligne 21 du script lammps_frames.py pour spécifier le nombre de configurations d’extraire.
- Exécutez le script lammps_frames.py sur le dump.myadsorbate.lammpstrj fichier en tapant lammps_frames.py à l’interface de ligne de commande. Cela affichera une liste de simulation temps correspondant aux configurations qui doivent être extraites à partir du fichier dump.myadsorbate.lammpstrj. Ces configurations peuvent servir à partir de structures simulations AIMD ou QM.
NOTE : 1) le script lammps_frames.py automatiquement détecte des fichiers de vidage et le journal LAMMPS ainsi que la partie de la production de la trajectoire dans le fichier de vidage et divise le nombre de configurations dans le fichier de vidage en 10 groupes. Par ailleurs, l’utilisateur peut spécifier le fichier journal, le fichier de vidage et le nombre de configurations de l’interface de ligne de commande à l’aide de la -l, -d, - options et n, respectivement. Pour ce faire, l’utilisateur doit taper lammps_frames.py - n XX -l $logfilename -d $dumpfilename à l’interface de ligne de commande, où XX est le nombre désiré de configurations, $logfilename est le nom du fichier journal LAMMPS et $dumpfilename est le nom de la Fichier LAMMPS de trajectoire (dump). Les temps de simulation qui sont produits se référer aux fois médians dans chaque groupe. 2) si les configurations seront calculées en VASP avec l’indicateur LDIPOLE allumé, une petite couche d’espace vide s’ajouteront à la partie supérieure de l’orage supercellulaire au-dessus de la couche d’eau ; Cela facilitera la convergence de la structure électronique dans le calcul de la VASP. Ajout d’un Å 3 supplémentaires d’espace vide au-dessus de molécules H2O a réussi dans les simulations ci-dessous.
Résultats
Une utilisation de ce protocole consiste à calculer les énergies d’interaction entre l’eau liquide et catalytique, c'est-à-dire, ΔEint35:
∆Eint=Eespèce catalytique + H2O+Esurface du catalyseur propre-Eespèce catalytique-Ecatalyseur propre surface + H2O
où Eespèce catalytique + H2Oest l’énergie d’une configuration de molécules2O H autour d’une espèce catalytique sur une surface métallique, lasurface du catalyseur propre de E est l’énergie de la surface du catalyseur propre dans le vide, E Catalytiques espèces est l’énergie de l’espèce catalytique sur une surface métallique dans le vide, et Enettoyer catalyseur surface + H2O est l’énergie de la configuration de H2O sur la surface du catalyseur avec l’espèce catalytique supprimé. Les positions des molécules H2O permettant de calculer Eespèce catalytique + H2O et Enettoyer catalyseur surface + H2O doivent être identiques. Toutes les valeurs de E sont calculés en utilisant le code VASP. La quantité ΔEint comprend toutes les interactions physiques et chimiques entre toutes les molécules dans la structure de l’eau liquide et l’espèce catalytique et donne une estimation raisonnable de l’enthalpie de solvatation du catalytique espèces, ce qui sont nécessaire pour calculer son énergie libre de l’énergie libre de solvatation et de total. Le tableau 1 fournit des valeurs pour ΔEint calculées pour les espèces sur un catalyseur de Pt(111) de surface avec des formules chimiques égales à CxHyOz en unités de eV (1 eV = 96.485 kJ/mol). Les valeurs ont été calculées couvertures ≤1/9 ML.35,46 les valeurs indiquées sont les moyennes repris 10 configurations de liquide H2O, et les incertitudes sont signalées comme des écarts-types. Toutes les valeurs sont négatives, indiquant des interactions favorables avec de l’eau.
Une autre application de ce protocole est de générer des structures de départ pour DMAI. Film 1 est un film d’une trajectoire AIMD qui a été démarrée à partir d’une configuration générée par le présent protocole. Au début de ce film, un adsorbant COH est indiquée sur une surface de Pt(111) sous une structure de liquide H2O. Molécule d’un H2O est soulignée, qui forme une liaison hydrogène avec COH. Au cours du film, cette molécule de2O H abstracts le proton de l’adsorbat COH et dépose un second atome d’hydrogène à la surface de Pt(111). La molécule de H2O contribue ainsi à catalyser la réaction COH * + * → CO * + H *, où le * s indiquent les sites catalytiques. Cette simulation met en évidence la force principale et l’objectif principal de la méthode d’échantillonnage multi-échelles décrite ci-après. De nombreuses configurations de molécules de H2O sont générées avec FFMD, en raison de sa force en traçabilité informatique. Toutefois, une limitation du FFMD est qu’il ne peut pas capturer rupture de liaison et formant à moins qu’un champ de force réactif est mis en œuvre. AIMD utilise la mécanique quantique pour calculer les énergies et ainsi peut capturer bond cassant et formant. Cependant, AIMD réclame par trop le calcul générer toutes les configurations possibles des molécules de2O H nécessaires pour échantillonnage suffisant a été atteint. Par conséquent, ce protocole combine les deux méthodes.
Les structures du liquide H2O molécules générées par cette procédure dépendent de paramètres d’entrée. Définir incorrectement peut avoir des influences involontaires sur les structures de l’eau. Par exemple, lorsque les distances intermoléculaires devient trop petites ou que les autres paramètres dans les fichiers d’entrée dynamique moléculaire sont définies incorrectement ou prennent des valeurs non-physiques, la structure de l’eau peut devenir déraisonnable. Dans ces conditions, la structure de l’eau va « sauter » involontairement pendant la trajectoire FFMD. La figure 1 montre un exemple de cela. La capture instantanée sur le côté gauche est la structure de départ pour une course FFMD et la capture instantanée sur la partie droite est un instantané pris au sein de ps 1 de démarrage de la simulation. Comme peut être vu, les molécules d’O2H sont passés loin de la surface. Ceci est causé par certains paramètres apportées dans les fichiers d’entrée de simulation et n’est pas une structure qui est susceptible de se produire dans la réalité.

Figure 1 : Exemple d’un résultat négatif. La simulation de dynamique moléculaire de champ de force « explose » en raison d’une valeur ou le paramètre non-physiques. Image de gauche : la départ de la géométrie de la surface Pt(111), adsorbée et la structure de l’eau liquide. Image de droite : la géométrie de la surface Pt(111), adsorbée et eau liquide structure inférieure à 1 ch plus tard. Dans l’image de droite, les molécules de2O H ont séparé de la surface en raison d’unphysically de grandes forces. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Film 1 : Ab initio (DMAI) simulation de dynamique moléculaire effectuée à partir d’une configuration générée dans multi-échelles échantillonnage. Une molécule H2O d’origine de hydrogène avec un adsorbat COH sur une surface de Pt(111) abstracts le proton de COH et dépose une deuxième hydrogène à la surface de Pt(111). Ce lien briser et formant des événements peut être capturé par DMAI, mais pas avec le champ de force dynamique moléculaire (FFMD) sauf si un champ de force réactif est utilisé. La configuration initiale des molécules de2O H utilisée dans cette simulation AIMD a été générée à l’aide FFMD tel que décrit dans ce manuscrit. S’il vous plaît cliquez ici pour voir cette vidéo. (Clic droit pour télécharger.)
| Espèces catalytiques | ∆Eint (eV) |
| COH | -0,70 ± 0,07 |
| CO | -0,03 ± 0,03 |
| CH2OH | -0.64 ± 0,12 |
| CHO-CHOH-CH2OH | -0,93 ± 0,22 |
| COH-COH-CH2OH | -0,87 ± 0,23 |
| COH-CHOH-COH | -1.72 ± 0,26 |
| CHOH-COH-CO | -1.57 ± 0,25 |
| CHO-CO-CO | -0.31 ± 0,19 |
Tableau 1 : Eau catalytique espèces interaction énergie résultant. Des énergies d’interaction en eV calculé pour huit CxHyOz adsorbats sur Pt(111). Valeurs rapportées les moyennes sont pris en charge plusieurs configurations du liquide H2O. Les incertitudes sont les écarts types des moyennes. 1 eV = 96.485 kJ/mol.
Discussion
La méthode présentée a été choisie pour sa facilité de mise en oeuvre, mais on pouvaient faire plusieurs personnalisations. D’une part, les champs de force utilisés dans les simulations FFMD peuvent être modifiés. Modifier les paramètres de champ de force ou potentiels peut être fait en éditant les fichiers de données et l’entrée LAMMPS. De même, les solvants autres que H2O pourraient être employés. Pour effectuer cette modification, la molécule de solvant désirée devront être insérées à partir d’étape 2.1.1 et les fichiers d’entrée LAMMPS devra être modifié afin d’incorporer les possibilités appropriées et les paramètres. Insertion de la nouvelle molécule de solvant nécessiterait également fournissant les coordonnées internes de la molécule de solvant dans un fichier .txt analogue au fichier water.txt.
Une autre modification qui pourrait être faite consiste à modifier la zone de la dalle de surface. Les résultats discutés dans ce manuscrit employé 3 Pt x 3 Pt ou Pt 4 x 4 Pt dalles de surface, qui ont des zones de surface inférieure à 120 Å2. Augmentation de la surface de la dalle, les dépenses de calcul augmente également. Dépenses de calcul a le plus d’impact sur l’article 5 du présent protocole. Si les étapes de traitement des données à l’article 5 deviennent prohibitifs par le calcul, données volumineuses post traitement des stratégies telles que celles mentionnées dans Li et coll. 201845 peuvent être employées.
Les sources possibles d’incertitude pour cette procédure sont le champ de force utilisée, la méthode d’échantillonnage et la fréquence d’échantillonnage. La structure de l’eau est déterminée par le champ de force qui est utilisé, ce qui signifie que le choix du champ de force pourrait influencer les configurations spécifiques de molécules de H2O. Notre groupe a évalué comment les choix du champ de force pour H2O molécules et atomes Pt influencent les énergies d’interaction calculé en FFMD et trouvé que le choix du champ de force contribue à moins de 0,1 eV à cette énergie d’interaction. Une autre source d’incertitude est la méthode d’échantillonnage, ce qui influe sur les configurations spécifiques qui sont utilisées pour calculer une quantité d’intérêt. Notre groupe a comparé les performances de la méthode de « temps d’échantillonnage » présentée dans le présent protocole avec une méthode « échantillonnage d’énergie », qui est biaisée à des configurations d’énergie inférieures de molécules de H2O, sur l’interaction énergie calculée dans la DFT et trouvé à la fois de ces méthodes d’échantillonnage statistiquement égaux de donner des valeurs35,46. La fréquence d’échantillonnage peut également influencer les résultats. Nous avons évalué comment augmenter le nombre de configurations de 10 à 30 000 influe sur les énergies d’interaction moyenne calculées en FFMD pour 40 différents C3HxO3 adsorbats et trouvé que la fréquence d’échantillonnage contribue moins que 0,1 eV l’interaction moyenne énergie44.
La principale limitation de cette méthode est que les adsorbats sont approximées par des structures sous vide pendant les simulations FFMD. En réalité, les adsorbats exposera les changements conformationnels (bond s’étire, coudes d’angle, des mouvements de torsion, etc.) en raison des mouvements normaux thermiques, y compris les interactions avec les molécules de solvant. Tentatives d’intégrer des changements de conformation des adsorbats dans les simulations FFMD exigerait développement détaillé des champs de force pour adsorbats surfaces catalytiques, c'est-à-dire qui comportent des termes qui décrivent les obligations s’étend, coudes d’angle et termes de torsion, parmi d’autres. Comme une orientation future de ce protocole, nous développons ces champs de force pour les adsorbats à des surfaces solides, qui nous permettront de déterminer l’étendue à laquelle l’utilisation des adsorbats rigides influe sur les résultats.
Déclarations de divulgation
Les auteurs ne divulguer aucun conflit d’intérêt.
Remerciements
Cette recherche a été financée par la National Science Foundation, par le biais d’attribution numéro CBET-1438325. Support de bourse pour CJB grâce à la NASA formation Grant NX14AN43H tient à reconnaître. Simulations ont été effectuées sur le Cluster de supercalculateur Palmetto, qui est maintenue par la Cyberinfrastructure Technology Group à l’Université Clemson. Nous remercions le docteur Paul J. Meza-Morales pour le protocole d’essai.
matériels
| Name | Company | Catalog Number | Comments |
| VASP software | Computational Materials Physics, Dept. of Physics, University of Vienna | vasp.5.4.4 | Standard parallel VASP executable in the newest version. |
| LAMMPS software | Sandia National Laboratory | 31Mar17-dp | Double-precision, parallel LAMMPS executable from 31 March 2017. |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | 1.9.3 | Standard VMD executable in the newest version. |
| MCPliQ software | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Executable and input files for the MCPliQ software availabe from the Getman Research Group GitHub page. | |
| JoVE article scripts | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Python scripts for this JoVE manuscript available from the Getman Research Group GitHub page. | |
| H2O PDB file | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University or RCSB Protein Data Bank | PDB file for a water molecule, available from the Getman Research Group GitHub page or at http://www.rcsb.org/ligand/HOH. |
Références
- Liu, J. L., Cao, X. M., Hu, P. Density functional theory study on the activation of molecular oxygen on a stepped gold surface in an aqueous environment: A new approach for simulating reactions in solution. Physical Chemistry Chemical Physics. 16 (9), 4176-4185 (2014).
- Okamoto, Y., Sugino, O., Mochizuki, Y., Ikeshoji, T., Morikawa, Y. Comparative study of dehydrogenation at Pt(111)/water and Pt(111)/vacuum of methanol interfaces. Chemical Physics Letters. 377 (1-2), 236-242 (2003).
- Santana, J. A., Cabrera, C. R., Ishikawa, Y. A density-functional theory study of electrochemical adsorption of sulfuric acid anions on Pt(111). Physical Chemistry Chemical Physics. 12 (32), 9526-9534 (2010).
- Artrith, N., Kolpak, A. M. Understanding the composition and activity of electrocatalytic nanoalloys in aqueous solvents: A combination of DFT and accurate neural network potentials. Nano Letters. 14 (5), 2670-2676 (2014).
- Jinnouchi, R., Kodama, K., Morimoto, Y. DFT calculations on H, OH and O adsorbate formations on Pt(111) and Pt(332) electrodes. Journal of Electroanalytical Chemistry. 716, 31-44 (2014).
- Yoon, Y., Rousseau, R., Weber, R. S., Mei, D. H., Lercher, J. A. First-principles study of phenol hydrogenation on Pt and Ni catalysts in aqueous phase. Journal of the American Chemical Society. 136 (29), 10287-10298 (2014).
- Desai, S. K., Pallassana, V., Neurock, M. A periodic density functional theory analysis of the effect of water molecules on deprotonation of acetic acid over Pd(III). Journal of Physical Chemistry B. 105 (38), 9171-9182 (2001).
- Huang, Z. Q., Long, B., Chang, C. R. A theoretical study on the catalytic role of water in methanol steam reforming on PdZn(111). Catalysis Science & Technology. 5 (5), 2935-2944 (2015).
- Chang, C. R., Huang, Z. Q., Li, J. Hydrogenation of molecular oxygen to hydroperoxyl: An alternative pathway for O2 activation on nanogold catalysts. Nano Research. 8 (11), 3737-3748 (2015).
- Faheem, M., Heyden, A. hybrid quantum mechanics/molecular mechanics solvation scheme for computing free energies of reactions at metal-water interfaces. Journal of Chemical Theory and Computation. 10 (8), 3354-3368 (2014).
- Behtash, S., et al. Solvation effects in the hydrodeoxygenation of propanoic acid over a model Pd(211) catalyst. Journal of Physical Chemistry C. 120 (5), 2724-2736 (2016).
- Behtash, S., Lu, J. M., Walker, E., Mamun, O., Heyden, A. Solvent effects in the liquid phase hydrodeoxygenation of methyl propionate over a Pd(111) catalyst model. Journal of Catalysis. 333, 171-183 (2016).
- Norskov, J. K., et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. Journal of Physical Chemistry B. 108 (46), 17886-17892 (2004).
- Skachkov, D., Rao, C. V., Ishikawa, Y. Combined first-principles molecular dynamics/density functional theory study of ammonia electrooxidation on Pt(100) electrode. Journal of Physical Chemistry C. 117 (48), 25451-25466 (2013).
- Hibbitts, D. D., Loveless, B. T., Neurock, M., Iglesia, E. Mechanistic role of water on the rate and selectivity of Fischer-Tropsch synthesis on ruthenium catalysts. Angewandte Chemie International Edition. 52 (47), 12273-12278 (2013).
- Abdelrahman, O. A., Heyden, A., Bond, J. Q. Analysis of kinetics and reaction pathways in the aqueous-phase hydrogenation of levulinic acid to form γ-valerolactone over Ru/C. ACS Catalysis. 4 (4), 1171-1181 (2014).
- Wang, H. F., Liu, Z. P. Formic acid xxidation at Pt/H2O interface from periodic DFT calculations integratd with a continuum solvation model. Journal of Physical Chemistry C. 113, 17502-17508 (2009).
- Behtash, S., Lu, J., Faheem, M., Heyden, A. Solvent effects on the hydrodeoxygenation of propanoic acid over Pd(111) model surfaces. Green Chemistry. 16, 605-616 (2014).
- Montemore, M. M., Andreussi, O., Medlin, J. W. Hydrocarbon adsorption in an aqueous environment: A computational study of alkyls on Cu(111). The Journal of Chemical Physics. 145, 074702 (2016).
- Hartnig, C., Grimminger, J., Spohr, E. Adsorption of formic acid on Pt(111) in the presence of water. Journal of Electroanalytical Chemistry. 607, 133-139 (2007).
- Hartnig, C., Grimminger, J., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (211). Electrochimica Acta. 52 (6), 2236-2243 (2007).
- Hartnig, C., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (111). Chemical Physics. 319, 185-191 (2005).
- Michel, C., et al. Role of water in metal catalyst performance for ketone hydrogenation: A joint experimental and theoretical study on levulinic acid conversion into gamma-valerolactone. Chemical Communications. 50 (83), 12450-12453 (2014).
- Zope, B. N., Hibbitts, D. D., Neurock, M., Davis, R. J. Reactivity of the gold/water interface during selective oxidation catalysis. Science. 330 (6000), 74-78 (2010).
- Pavlova, A., Meijer, E. J. Understanding the role of water in aqueous ruthenium-catalyzed transfer hydrogenation of ketones. ChemPhysChem. 13 (15), 3492-3496 (2012).
- Saavedra, J., Doan, H. A., Pursell, C. J., Grabow, L. C., Chandler, B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science. 345 (6204), 1599-1602 (2014).
- Desai, S., Neurock, M. A first principles analysis of CO oxidation over Pt and Pt66.7%Ru33.3%(111) surfaces. Electrochimica Acta. 48 (25-26), 3759-3773 (2003).
- Gohda, Y., Schnur, S., Gross, A. Influence of water on elementary reaction steps in electrocatalysis. Faraday Discussions. 140, 233-244 (2008).
- Nie, X. W., Luo, W. J., Janik, M. J., Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. Journal of Catalysis. 312, 108-122 (2014).
- Michel, C., Auneau, F., Delbecq, F., Sautet, P. C-H versus O-H bond dissociation for alcohols on a Rh(111) surface: A strong assistance from hydrogen bonded neighbors. ACS Catalysis. 1 (10), 1430-1440 (2011).
- Neurock, M., Wasileski, S. A., Mei, D. From first principles to catalytic performance: tracking molecular transformations. Chemical Engineering Science. 59 (22-23), 4703-4714 (2004).
- Camellone, M. F., Marx, D. On the impact of solvation on a Au/TiO2 nanocatalyst in contact with water. Journal of Physical Chemistry Letters. 4 (3), 514-518 (2013).
- Santana, J. A., Mateo, J. J., Ishikawa, Y. Electrochemical hydrogen oxidation on Pt(110): A combined direct molecular dynamics/density functional theory study. Journal of Physical Chemistry C. 114 (11), 4995-5002 (2010).
- Santana, J. A., Saavedra-Arias, J. J., Ishikawa, Y. Electrochemical hydrogen xxidation on Pt(100): A combined direct molecular dynamics/density functional theory study. Electrocatalysis-US. 6 (6), 534-543 (2015).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 119 (24), 13642-13651 (2015).
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics. 117, 1-19 (1995).
- MacKerell, A. D., et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 102, 3586-3616 (1998).
- Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A., Skiff, W. M. UFF, A full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society. 114, 10024-10035 (1992).
- Kahn, K., Bruice, T. C. Parameterization of OPLS-AA force field for the conformational analysis of macrocyclic polyketides. Journal of Computational Chemistry. 23, 977-996 (2002).
- Kresse, G., Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science. 6, 15-50 (1996).
- Kresse, G., Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B. 54, 11169-11186 (1996).
- Kresse, G., Hafner, J. Ab initio molecular dynamics for liquid metals. Physical Review B. 47, 558-561 (1993).
- Humphrey, W., Dalke, A., Schulten, K. VMD - visual molecular dynamics. Journal of Molecular Graphics and Modelling. 14, 33-38 (1996).
- Xie, T., Sarupria, S., Getman, R. B. A DFT and MD study of aqueous phase dehydrogenation of glycerol on Pt(111): Comparing chemical accuracy versus computational expense in different methods for calculating aqueous phase system energies. Molecular Simulation. 43, 370-378 (2017).
- Li, Y., Zhang, X., Srinath, A., Getman, R. B., Ngo, L. B. Combining HPC and big data infrastructures in large-scale post-processing of simulation data: A case3 study in PEARC ’18. Proceedings of the Practice and Experience on Advanced Research Computing. , (2018).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Correction to "Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 120, 801 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.