
Method Article
Мультимасштабный выборки гетерогенных воды/металла катализатора интерфейса с использованием теории функционала плотности и Force-Field молекулярной динамики
В этой статье
Резюме
Цель Протокола, представленные здесь заключается в генерации и образцы траекторий конфигураций молекул жидкой воды вокруг каталитического видов на поверхности плоских переходных металлов. Выбранные конфигурации может использоваться как начиная структур квантовой механики на основе методов.
Аннотация
Значительное количество гетерогенно катализируемой химические процессы происходят в условиях жидкой, но имитируя функции катализатора в таких условиях является сложной задачей, когда это необходимо для включения молекул растворителя. Разорвать связь и формирования процессов, моделируется в этих системах требуют применения химических методов квантовой. Поскольку под постоянным теплового движения молекул в жидкой фазе, моделирования должна также включать конфигурационное выборки. Это означает, что несколько конфигураций жидких молекул должны имитировать для каждого вида каталитической интерес. Цель Протокола, представленные здесь будет генерировать и образцы траекторий конфигураций молекул жидкой воды вокруг каталитического видов на поверхности плоских переходных металлов в пути, что остатки химическая точность с вычислительных затрат. В частности силовое поле моделирования молекулярной динамики (FFMD) используются для создания конфигурации жидкого молекул, которые впоследствии могут быть использованы в методы, основанные на квантовой механике, теории функционала плотности или ab initio молекулярной динамика. Чтобы проиллюстрировать это, в этой рукописи, протокол используется для каталитического промежуточных продуктов, которые могут быть вовлечены в путь для разложения глицерина (C3H8O3). Структуры, которые создаются с помощью FFMD моделируются в DFT с целью оценить энтальпиями сольватации каталитического видов и определить, как молекулы H2O участвовать в каталитического разложения.
Введение
Моделирования молекулярных явлений участвующих в несродное катализирование жидкого условиях является необходимым для понимания катализатора; Однако это остается сложным, потому что он требует тонкий баланс между химическая точность и вычислительных затрат. В целом поскольку катализ предполагает нарушение и формирование химических связей, квантовой механики должны использоваться для по крайней мере некоторой степени; Однако долго моделирования сложны в квантовой механике, как они требуют значительных вычислительных ресурсов. Поскольку под постоянным теплового движения молекул в жидкой фазе, моделирования должна также включать конфигурационное выборки, т.е., они должны включать несколько пространственных механизмов жидкого молекул, как каждый другой пространственное расположение (то есть, каждый Конфигурация) имеет разную энергию. Это означает, что несколько конфигураций жидких молекул должны имитировать для каждого вида каталитической интерес. Эти потребности – использовать квантовой механики и выполнить несколько вычислений на каталитические виды – может оказать моделирования в несродное катализирование под жидкой фазы вычислительно неразрешимыми. Цель метода, описанного здесь является возможность вычислительно шансов справиться с возникающими моделирования явлений в несродное катализирование в жидкой фазе.
Мы особенно заинтересованы в гетерогенно катализируемой реакции, которые проводятся под жидкой воды. Молекулы воды имеют значительное влияние на каталитической явления, такие как взаимодействие с каталитического видов (например, с помощью распыления сил и водорода склеивание)1,2,3,4,5 ,6,,78,9,10,11,12,13,14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23, участвуя в каталитических реакций1,,78,9,15,21,22,24 ,25,26,27и влияющие на пути реакции и/или каталитической ставки1,11,12,15, 18,23,25,27,28,,2930,31. Моделирование этих явлений была выполнена с использованием QM или ab initio молекулярной динамики (AIMD)1,2,6,7,14,22 25, ,27,28,,3233,34, сила поля молекулярной динамики (FFMD)35 и квантовой механике/молекулярной механики (QM/мм)10. В AIMD и FFMD атомы в системе перемещаются во исполнение Ньютона уравнений движения согласно сил, действующих на них. В AIMD системы энергии и силы рассчитываются с квантовой механикой, тогда как в FFMD, системы энергии и силы рассчитываются с использованием силы, что поля, которые являются алгебраические выражения, которые параметризуются на основе экспериментальных или данных QM. В QM/мм часть системы, где происходит Бонд разрыва и формирования вычисляется с QM, и остальная часть системы является с мм, которая использует силу поля. Потому, что они непосредственно используют QM, AIMD и QM/мм лучше подходят для захвата разорвать связь и формирования, происходит в водной фазе гетерогенного катализа; Однако FFMD, значительно более вычислительно шансов справиться с возникающими и таким образом лучше подходит для создания конфигураций жидкости H2O молекул. Метод, представленный в настоящем Протоколе остатков химических точность и вычислительные затраты, используя сочетание QM и FFMD.
В частности этот метод использует FFMD моделирования для создания конфигураций жидкости H2O и QM для расчета энергии системы. FFMD осуществляется с помощью будущее. 36 силовые поля, используемые в FFMD в этой работе используют Леннард-Джонса + потенциал кулона (LJ + C), где ЖЖ параметры были взяты из TIP3P/CHARMM модель37 H2O, универсальная области силы38 (ФФУ) для Pt и OPLS-AA силовое поле39 для каталитического видов и кулона параметры были взяты из модель37 H2O TIP3P/CHARMM и OPLS-AA силовое поле39 для каталитического видов. Кулоновская параметры для Pt атомов было равным 0. QM вычисления выполняются с помощью VASP код40,,41,42, который плотность теории функционала (DFT) код. Молекулы воды вставки выполняются с кода, разработанного собственными силами под названием Монте-Карло плагин для квантовых методов (MCPliQ). Файл преобразования VASP в будущее в настоящем Протоколе выполняются с программного обеспечения визуального молекулярной динамики (VMD)43.
Протокол предназначен для создания конфигурации молекул жидкой воды вокруг каталитического видов на поверхности плоских переходных металлов в низкой степени охвата. Охват обозначается θ и определяется как количество адсорбатов на поверхности атома металла (то есть, количество поверхности адсорбатов нормализуется на количество атомов металла в верхнем слое металлические плита в модели катализатора). В этой рукописи, низкий охват определяется как θ ≤ 1/9 монослоя (мл), где 1 мл означает один каталитического видов на поверхности атома металла. Катализатора модели должны быть помещены в периодических моделирования коробки. Моделирование коробки не нужно быть кубов. Эта рукопись демонстрирует использование протокола для создания конфигурации жидкого H2O который может использоваться для расчета количества интерес в водной фазе несродное катализирование.
Этот протокол требует, что пользователь имеет доступ к установлен и рабочих версий программного обеспечения VASP, MCPliQ, будущее и ВМД. Дополнительные сведения о VASP (https://www.vasp.at/), будущее (https://Lammps.sandia.gov/) и VMD (https://www.ks.uiuc.edu/Research/vmd/) доступны на их соответствующих веб-сайтах. Программное обеспечение MCPliQ описана в https://github.com/getman-research-group/JoVE_article, наряду с все входные файлы и скрипты Python, упомянутых в настоящем Протоколе. Этот протокол предполагает, что исполняемые файлы и сценарии, упомянутых в будет выполняться на компьютере с высокой производительности исследований и устанавливаются в каталог, находящийся в переменной $PATH пользователя. Если в месте, которое не помещается, исполняемый файл или сценарий пользователя $PATH, то путь к исполняемому файлу должны быть включены для его выполнения. Исполняемые файлы и скрипты выполняются в шагах 2.1.2, 2.2.1, 2.2.8, 3.1, 4.2, 5.2 и 6.1.2. Например, чтобы выполнить код MCPliQ в шаге 2.1.2 из каталога, который не является пользователем в $PATH, пользователь будет вводить $PATHTOMCPLIQ/mcpliq в интерфейсе командной строки вместо mcpliq, где $PATHTOMCPLIQ это место где mcpliq хранится исполняемый файл (например, $PATHTOMCPLIQ может быть ~ / bin). Перед началом этого протокола, все исполняемые файлы и скрипты следует исполняемый разрешения (например, в Linux, это может быть сделано в интерфейсе командной строки из каталога, где хранится исполняемый файл mcpliq, введя chmod + x mcpliq ). Кроме того, должны быть загружены все модули, необходимые для программного обеспечения или скриптов (эти зависимости будет характерных для индивидуальных установок различных программного обеспечения и компьютер, где будет выполняться расчеты).
протокол
1. Сгенерируйте структуру адсорбата
- Создайте файл VASP POSCAR, включающий суперячеек с периодические граничные условия, как вы бы, если выполнялось моделирование адсорбатов на металлических поверхностях под вакуумом. Суперячеек должна включать первоначальное предположение адсорбата структуры и поверхности металла, а также вакуума пространство выше для добавления H2O молекул адсорбата. В предыдущей работе35,44предоставляются сведения о том, как это сделать.
Примечание: Важно, что высота вакуумного пространства быть по меньшей мере 12 Е выше верхней части адсорбата. - Расслабьтесь структуры и свести к минимуму свою энергию с помощью кода VASP. В предыдущей работе35,44предоставляются сведения о том, как это сделать. Это позволит производить файл с названием CONTCAR, который будет использоваться в следующем разделе.
2. Добавьте явные H2O молекул
- Добавить N молекул воды в вакуумном пространстве в CONTCAR, созданный на шаге 1.2 с использованием кода MCPliQ, где N = ρV, ρ — плотность воды, и V является объем вакуума пространство над адсорбата.
Примечание: ρ должны приниматься как плотность воды, как определено в модели TIP3P/CHARMM воды при температуре моделирования. V будут уточнены в следующем шаге.- Укажите следующую информацию в файле master_input.txt MCPliQ: количество молекул H2O для добавления (N), изменив первый аргумент в линии 28, путь к файлу water.txt, изменив второй аргумент в линии 28 и минимального и Максимальная высота суперячеек, которая может быть занята молекул воды, изменив минимум z координаты в строке 11 и Максимальная координата z в строке 12.
- Выполните код, MCPliQ, набрав mcpliq из интерфейса командной строки для вставки молекул воды в файле CONTCAR. Код будет выводить один или несколько файлов с расширением файла. POSCAR.
Примечание: Если более чем один POSCAR файлы создаются, они будут называться POSCAR_n.POSCAR. Выберите файл, где n является крупнейшим.
- Создавать будущее входных файлов для моделирования ДНЯО и сбалансировать объем ячейки, используя FFMD в ансамбле ДНЯО в будущее.
- На выполнение скрипта lmps_bond_angle.py. POSCAR файл, созданный на шаге 2.1.2, набрав lmps_bond_angle.py $filename. POSCAR в интерфейс командной строки, где $filename это имя. Файл POSCAR, созданный в шаге 2.1.2. Этот сценарий создает файл с именем $filename. POSCAR.bond_angle_info.txt, в котором перечислены облигаций и углы, которые будут использоваться в файле данных будущее.
- VMD открыть и выберите файл > Новые молекулы в главном окне, чтобы открыть окно Браузера файлов молекулы . Выберите VASP_POSCAR из раскрывающегося меню Определяют тип файла . Нажмите кнопку Обзор и перейдите к $filename. Файл POSCAR. Нажмите кнопку загрузки для открытия $filename. Файл POSCAR.
- Откройте консоль Tk в VMD, выбрав расширения > Tk консоли от VMD главного окна.
- Выполните следующую команду в консоли ТЗ: топо writelammpsdata $WDPATH/data.myadsorbate полный, где $WDPATH — это каталог на компьютере, где VMD будет писать в файл данных будущее и data.myadsorbate — это имя файла данных, будущее.
- Удалите раздел облигаций и углы в нижней части файла data.myadsorbate. Затем добавьте списки облигаций и угол в файл $filename. POSCAR.bond_angle_info.text в data.myadsorbate.
Примечание: Индексы для типа связи O-H и H-O-H угловой для молекул воды в $filename. Оба файла POSCAR.bond_angle_info.txt равным 1. Таким образом типы Бонд и угол для адсорбатов должна начать подсчет на 2. - Отредактируйте файл data.myadsorbate путем добавления параметров Леннард-Джонса в секцию Пара Coeffs и кулона параметры в разделе атомов . Леннард-Джонса и кулона параметры для H2O молекулы, атомы адсорбата и поверхностные атомы металла должны быть добавлены.
Примечание: Параметры Леннард-Джонса Pt атомов, молекул воды и адсорбата атомов в этом протоколе получаются от ФФУ38, TIP3P/CHARMM37и39 OPLS-AA силовые поля, соответственно. Кулоновская параметры для атомов молекул и адсорбатов воды получаются из37 TIP3P/CHARMM и OPLS-AA39 силовые поля, соответственно. Кулоновская параметров для Pt атомы имеют значение 0 в этом протоколе. Кроме того вычисляемые частичной обвинения могут использоваться для кулона параметры адсорбата атомов и Pt атомов. - Скопируйте будущее входной файл input.equil в каталог $WDPATH. Измените переменную группы на линии 34 для обозначения атома типа индексов для воды кислородом и атомы водорода воды и групповой переменной в строке 35 для обозначения атома типа индексов для Pt и адсорбата атомов.
- Выполнение программного обеспечения будущее, набрав mpiexec - np XX lmp_mpi < input.equil в интерфейс командной строки, где XX — это число ядер процессоров для использования, и lmp_mpi — это имя исполняемого файла будущее. Это будет выполняться Минимизация энергии для уточнения H2O конфигурации, следуют FFMD моделирования, выступал на постоянное количество молекул H2O (N), объем (V) и температура (T), чтобы принести воду до температуры моделирования, а затем FFMD моделирования в постоянном N, давление (P) и температура (T), чтобы определить физически правильную высоту окна моделирования. Выходные файлы, которые будут использоваться в разделе 3, называются data.myadsorbate_npt и log.myadsorbate.
Примечание: Продолжительность моделирования ДНЯО должно быть достаточно, чтобы включать «уравновешивания» перспективе, где объем суперячеек доходит до стабильного состояния и «производство» запуска, которая используется для выборки, которые ансамбля усредняет (здесь, высота суперячеек). Во время выполнения уравновешивания объем суперячеек когда заговор против времени должны выравниваться в значение устойчивого состояния. Как только это происходит, моделирование ДНЯО можно сказать в его производства. Проверьте уравновешивания моделирования ДНЯО путем обеспечения того, что колебания в высоту суперячеек (lz) являются минимальными или сошлись в постоянное значение. Если большие колебания происходят, затем заново сгенерировать конфигурацию H2O уменьшается скорость на линии 92 в файле input.equil и повторяющиеся шаг 2.2.8 или снова начиная с шага 2.1.1.
3. Извлеките надлежащей высоты суперячеек
- Выполните сценарий get_npt_lz.py на файл log.myadsorbate, введя get_npt_lz.py log.myadsorbate в интерфейс командной строки. Этот сценарий выводит среднее суперячеек высота от «производство» запустить части ДНЯО моделирования в файле avg_lz.txt.
Примечание: Get_npt_lz.py сценарий предполагает, что будущее пишет длина ячейки z измерения (lz) в log.myadsorbate файл каждые 1000 fs (настраиваемый в строке 20 get_npt_lz.py скрипта), который установлен по умолчанию во входном файле предоставленный input.equil будущее. Get_npt_lz.py сценарий обнаруживает и удаляет первые 2 ns (настраиваемый в строке 19 get_npt_lz.py скрипта) на сумму lz значения в файле log.myadsorbate, поскольку они составляют часть уравновешивания моделирования, в то время как оставшиеся 3 составляют ns " часть производства» и таким образом используются get_npt_lz.py сценарий для вычисления среднего z измерение длины. В дополнение к файлу avg_lz.txt get_npt_lz.py скрипт выводит файл с названием npt_data.txt, который предоставляет значения lz как функция понадобится, а также файл с названием npt_plot.png, который участки те же данные. Участок может использоваться для проверки уравновешивания моделирования ДНЯО. - Реконструировать суперячеек, используя средняя высота определяется в ДНЯО.
- Скопируйте файл data.myadsorbate_npt в новый каталог, упоминаемые здесь как $WD2PATH и переименуйте его в data.myadsorbate.
- Измените новый файл data.myadsorbate, чтобы lz высота равна к выходу среднее значение от get_npt_lz.py сценарий, изменив zlo и zhi аргументы в файле data.myadsorbate, что zlo 0.0 и zhi lz значение из файла avg_lz.txt, произведенных в s ТЭП 3.1.
4. Создание конфигураций H2O молекул
- Скопируйте файл ввода input.prod будущее в $WD2PATH. Измените переменную группы на линии 32 для обозначения атома типа индексов для воды кислородом и атомы водорода воды и переменной группы на линии 33 для обозначения атома типа индексов для Pt и адсорбата атомов.
- Выполнение программного обеспечения будущее, набрав mpiexec - np XX lmp_mpi < input.prod в интерфейс командной строки, где XX — это число ядер процессоров для использования, и lmp_mpi — это имя исполняемого файла будущее. Это будет работать постоянно NVT моделирования на H2O молекул. Ключевые выходной файл из этого моделирования является файл dump.myadsorbate.lammpstrj.
Примечание: Продолжительность NVT моделирование должно быть достаточно, чтобы включать уравновешивания перспективе, где энергия системы приходит в устойчивое состояние, и производство, запуска, из которого ансамбля усредняет (здесь, пространственной позиции молекул воды) пробы. Во время выполнения уравновешивания энергии системы, когда заговор против времени должны выравниваться в значение устойчивого состояния. Как только это происходит, NVT моделирования можно сказать в его производства.
5. Определите Скрепление водопода жизни для надлежащего времени выборки
- Отредактируйте скрипт hb_lifetime_dist.py для указания: скорость первого кадра dump.myadsorbate.lammpstrj файла, изменив переменную actualStart на линии 22, как часто кадры записываются в файл траектории будущее, изменив переменную скорость на линия 23, первый и последний timesteps сценарий должен рассмотреть (то есть, часть производства траектории) путем изменения переменных N_first и N_last на линиях 24 и 25, ли последовательных кадров считаются или рамы пропускаются, изменив nevery переменная строки 26 и количество строк на кадр часть траектории файла, изменив переменную frameLine на линии 27. Кроме того измените линии 31, 35, указание которой атом типов в файле data.myadsorbate принадлежат к адсорбата и которой атом типы принадлежат к H2O молекул.
Примечание: Скрипт hb_lifetime_dist.py анализирует конфигурацию H2O в серийного производства и определяет, являются ли любой H2O молекулы водорода, приклеенная к адсорбата. Затем он рассчитывает время моделирования, каждый водорода облигаций остается неизменным и сообщает эту информацию как распределение существования Скрепление водопода в единицах л.с. Конкретная версия сценария, который поставляется с этот протокол предполагает, что будущее запись конфигурации H2O молекул в dump.myadsorbate.lammpstrj файл каждые 1000 fs, который установлен по умолчанию во входном файле предоставленный input.prod будущее. Он обнаруживает и удаляет первые 2 ns стоит конфигураций в файл dump.myadsorbate.lammpstrj, как они составляют часть уравновешивания моделирования, и использует оставшиеся 3 ns для вычисления Скрепление водопода продолжительностей жизни. - Выполните сценарий hb_lifetime_dist.py на файл dump.myadsorbate.lammpstrj, введя hb_lifetime_dist.py в интерфейсе командной строки. Делать это будет производить файл с названием distribution_HB_lifetime.dat.
- Печать данных в файле distribution_HB_lifetime.dat для просмотра распределения Скрепление водопода воплощений, которые произошли во время симуляции NVT.
- Определите время приращение для интервала времени выборки на основе рассчитанных Скрепление водопода продолжительностей жизни. Лучший выбор-это максимальная Скрепление водопода жизни; в качестве альтернативы можно использовать значение, которое будет захватывать 95% доверительный интервал.
6. Примеры конфигураций жидким H2O молекул
- Определите количество конфигураций с запуска производства NVT FFMD траектории для дальнейших вычислений. Количество конфигураций должны выбираться таким образом, что минимальное время между конфигурациями является равным или больше, чем интервал выборки, указанных в разделе 5.
- Измените значение по умолчанию для переменной num_frames на линии 21 lammps_frames.py сценарий для указания числа конфигураций для извлечения.
- Выполните сценарий lammps_frames.py файл dump.myadsorbate.lammpstrj, введя lammps_frames.py в интерфейсе командной строки. Это будет выводить список моделирования раз соответствует конфигурации, которые должны быть извлечены из файла dump.myadsorbate.lammpstrj. Эти конфигурации могут использоваться как начиная структуры в AIMD или QM моделирования.
Примечание: 1) lammps_frames.py сценарий автоматически определяет будущее журнал и файлы дампа, а также производство часть траектории в файле дампа и делит число конфигураций в файл дампа на 10 групп. Кроме того пользователь может указать файл журнала, файл дампа памяти и количество конфигураций из интерфейса командной строки, используя -l, -d и - n вариантов, соответственно. Сделать это, пользователь должен ввести lammps_frames.py - n XX -l $logfilename -d $dumpfilename в интерфейс командной строки, где XX это желаемое количество конфигураций, $logfilename имя logfile будущее, и $dumpfilename — это имя БУДУЩЕЕ траектории (дамп) файла. Время моделирования, которые выводятся относятся к средний раз в каждой группе. 2) Если конфигурации будет рассчитываться в VASP с флагом LDIPOLE, включен, небольшой слой вакуумного пространства следует добавить к верхней части суперячеек выше слоя воды. Это будет способствовать конвергенции электронной структуры в расчете VASP. Добавление дополнительных 3 Å вакуумного пространства выше H2O молекулы была успешной в симуляции, обсуждаются ниже.
Результаты
Этот протокол применяется для расчета энергий взаимодействия между жидкой воды и каталитического видов, т.е., ΔЕint35:
∆Eint=Eкаталитического видов + H2O+Eповерхности чистой катализатора-каталитического видовE-Eчистой катализатора поверхности + H2O
где Eкаталитического видов + H2O– это энергия конфигурацию H2O молекул вокруг каталитического видов на поверхности металла, Eповерхности чистой катализатора – это энергия поверхности чистой катализатора в вакууме, E Каталитические виды Это энергия каталитического видов на поверхности металла в вакууме, и Eочистить катализатора поверхности + H2O – это энергия конфигурации H2O над поверхностью катализатора каталитического видов удалены. Позиции H2O молекул, используемых для вычисления Eкаталитического видов + H2O и Eочистить катализатора поверхности + H2O должны быть идентичны. Все значения E рассчитываются с помощью кода VASP. Количество ΔEint включает все физические и химические взаимодействия между всеми молекул в структуре жидкой воды и каталитического видов и дает разумную оценку энтальпии сольватации из катализатора видов, который необходим для расчета его свободная энергия сольватации и общей свободной энергии. В таблице 1 приведены значения для ΔEint рассчитаны для видов на катализатор Pt(111) поверхности с химическими формулами равен CxHyOz в единицах eV (1 eV = 96.485 кДж/моль). Были рассчитаны значения покрытия ≤1/9 мл.35,46 сообщили значения являются средними, захватили 10 конфигурации жидкого H2O, и неопределенности помечаются как стандартных отклонений. Все значения являются негативными, указывающее благоприятное взаимодействие с водой.
Для создания начальной структуры для AIMD еще одно применение настоящего Протокола. Фильм 1 — фильм AIMD траектории, которая была запущена из конфигурации, создаваемые настоящим Протоколом. В начале этого фильма ког адсорбата показан на Pt(111) поверхность под структуру жидких H2O. 1 H2O молекулы подчеркивается, который сформировал водородную связь с COH. В течение фильма эта молекула H2O абстрагирует протона от адсорбата COH и депозиты второй атом водорода на поверхности Pt(111). Таким образом, молекулы H2O помогает катализируют реакции ког * + * → CO * + H *, где * s указывают каталитического сайтов. Это моделирование подчеркивается главная сила и основная цель метода мультимасштабный выборки, описанные здесь. Множество конфигураций H2O молекул создаются с FFMD, из-за его прочности в вычислительной уступчивость. Однако ограничение FFMD является, что он не может захватить Бонд взлома и формирования Если реализован реактивной силы поля. AIMD использует квантовой механики для расчета энергии и таким образом может захватить Бонд разрыва и формирования. Однако AIMD слишком требовательных для создания всех конфигураций H2O молекул необходимо убедиться, что был достигнут достаточно выборки. Таким образом этот протокол сочетает в себе два метода.
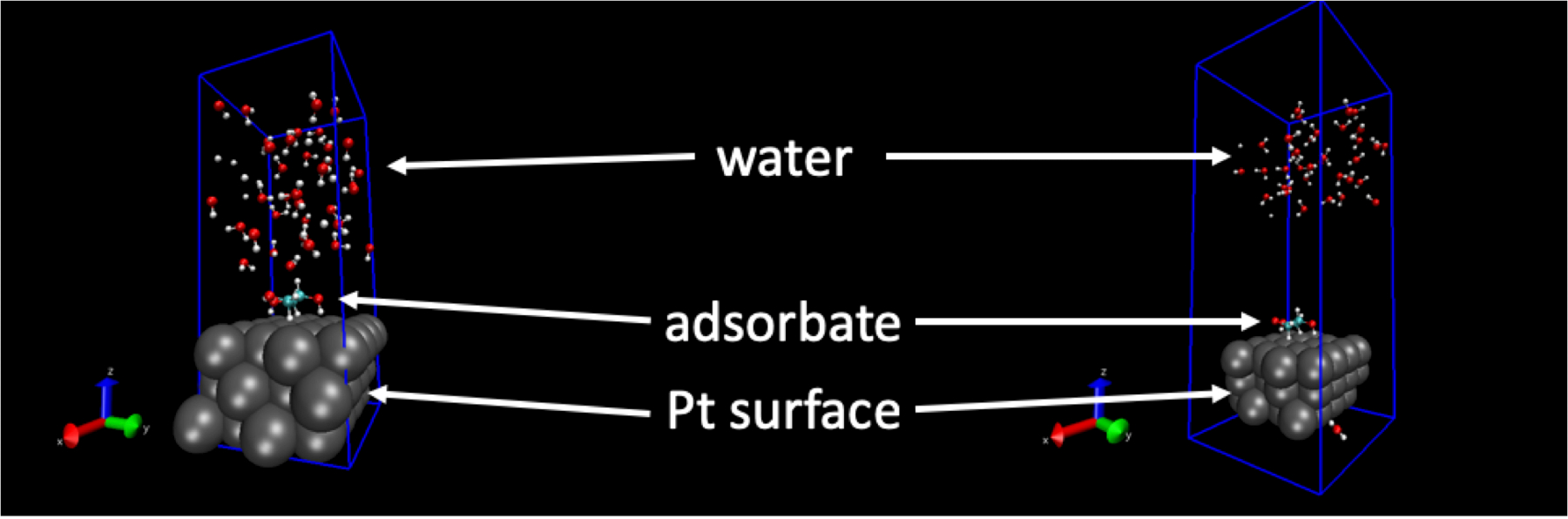
Структуры молекул жидким H2O порожденных этой процедуры зависит от входных параметров. Установка это неправильно может иметь непреднамеренные влияет на структуру воды. Например когда межмолекулярных расстояний становятся слишком мал или другие параметры во входных файлах молекулярной динамики установлены неправильно или взять на нефизическим ценности, структура воды может стать необоснованным. В этих обстоятельствах структура воды будет «взорвать» нанес во время FFMD траектории. Рисунок 1 показывает пример этого. Снимок на левой стороне Начальная структура для запуска FFMD, и снимок с правой стороны снимка в 1 ps начала моделирования. Как видно, H2O молекулы продвинулись далеко от поверхности. Это вызвано неправильной настройки, сделанные во входных файлах моделирования и не является структурой, которая может произойти в реальности.

Рисунок 1: Пример отрицательный результат. Моделирования молекулярной динамики силовое поле «взрывается» в результате нефизическим параметр или значение. Левая рука изображение: начальной геометрии поверхности Pt(111), адсорбата и структуры жидкой воды. Правая рука изображение: геометрия Pt(111) поверхности, адсорбата и жидкой воды структура менее 1 ПС позже. На правом изображении H2O молекулы отделили от поверхности из-за unphysically больших сил. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Фильм 1: Ab initio молекулярной динамики (AIMD) моделирования инициированы из конфигурации, созданные в мультимасштабный выборки. H2O молекулы, что первоначально водорода, приклеенная к адсорбата КОГ на поверхности Pt(111) рефераты протона от COH и депозиты второй водорода на поверхности Pt(111). Эта связь разрыва и формирования событий может быть захвачен, AIMD, но не с силовым полем молекулярной динамики (FFMD) Если используется поле реактивной силы. Начальную конфигурацию H2O молекул, используемых в этом моделирование AIMD был создан с помощью FFMD, как описано в этой рукописи. Пожалуйста нажмите здесь, чтобы посмотреть это видео. (Правой кнопкой мыши для загрузки.)
| Каталитические виды | ∆Eint (eV) |
| КОГ | -0.70 ± 0,07 |
| CO | -0.03 ± 0,03 |
| CH2OH | -0.64 ± 0,12 |
| Чо ЧОХ CH2OH | -0.93 ± 0,22 |
| КОГ ког CH2OH | -0.87 ± 0,23 |
| КОГ ЧОХ КОГ | -1.72 ± 0,26 |
| ЧОХ КОГ CO | -1.57 ± 0,25 |
| ЧО CO-CO | -0,31 ± 0,19 |
Таблица 1: Воды каталитические виды взаимодействия энергии результаты. Энергий взаимодействия в eV для восьми CxHyOz адсорбатов рассчитаны на Pt(111). Значения сообщили берутся средние за несколько конфигураций жидкости H2O. Неопределенности являются стандартные отклонения средних. 1 eV = 96.485 кДж/моль.
Обсуждение
Был выбран метод, представленные для своей легкости реализации, но могут быть несколько настроек. С одной стороны силовые поля, используемые в FFMD моделирования могут быть изменены. Изменение параметров силового поля и/или потенциалов может быть сделано путем редактирования файлов ввода и данных будущее. Аналогично можно было бы использовать растворители помимо H2O. Чтобы сделать это изменение, желаемого молекулы растворителя необходимо включить начиная с шаг 2.1.1, и будущее входных файлов необходимо быть отредактированы для включения надлежащих потенциалов и параметры. Вставка новой молекулы растворителя также потребует поставки внутренние координаты молекул растворителя в файле .txt, аналогично файлу water.txt.
Еще одно изменение, которое можно было бы — изменить области поверхности плиты. Результаты обсуждались в этой рукописи работало 3 Pt x 3 Pt или 4 Pt x 4 Pt поверхности плиты, которые имеют площадь менее 120 Е2. С увеличением площади поверхности плиты, также увеличивает вычислительные затраты. Вычислительных затрат имеет наибольшее влияние на раздел 5 настоящего Протокола. Если шаги обработки данных в разделе 5 становится вычислительно непомерно, большой данных пост обработки стратегии такие, как те, которые обсуждаются в Li et al. 201845 могут быть использованы.
Возможные источники неопределенности для этой процедуры включают в себя силу поле занято, метод выборки и частоту дискретизации. Структура воды определяется силовое поле, которое используется, означает, что выбор силового поля может влиять на определенные конфигурации H2O молекул. Наша Группа оценивала как выбор силового поля H2O молекулы и атомы Pt влияние энергий взаимодействия рассчитывается в FFMD и обнаружил, что выбор силового поля способствует менее чем 0.1 eV для этой энергии взаимодействия. Еще один источник неопределенности является метод выборки, который влияет на определенные конфигурации, которые используются для вычисления количества интереса. Наша группа имеет по сравнению производительности метода «время выборки» представлены в этом протоколе с методом «энергия выборки», который является предвзятым ниже конфигураций энергии H2O молекул, на взаимодействии энергий рассчитывается в DFT и нашел оба из этих методов выборки дают статистически равные значения35,46. Частота дискретизации могут также повлиять на результаты. Мы оценили, как увеличение числа конфигураций от 10 до 30 000 влияет энергии среднем взаимодействия, рассчитанных в FFMD за 40 различных C3HxO3 адсорбатов и обнаружили, что частота дискретизации способствует менее чем 0.1 eV энергии среднем взаимодействия44.
Главное ограничение этого метода является, что адсорбатов аппроксимированы структур под вакуумом, во время FFMD моделирования. В действительности адсорбатов будет экспонат конформационные изменения (Бонд тянется, угол искривления, скручивающих движений и т.д.) из-за нормальный тепловой движения, в том числе взаимодействия с молекулами растворителя. Попытки включить конформационные изменения адсорбатов в FFMD моделирования потребует детальной разработки силовых полей для каталитической поверхности адсорбатов, т.е., который включает термины, которые описывают Бонд тянется, угол искривления и крутильных термины, среди других. Как будущее направление настоящего Протокола, мы разрабатываем такие силовые поля для адсорбатов на твердых поверхностях, которые мы будем использовать, чтобы определить степень, в которой с помощью жесткой адсорбатов влияет на результаты.
Раскрытие информации
Авторы раскрывают отсутствие конфликта интересов.
Благодарности
Это исследование финансировалось национального научного фонда через награду номер CBET-1438325. Стипендии для КМД через НАСА обучения Грант NX14AN43H с благодарностью. Моделирования были исполнены в кластере Palmetto суперкомпьютера, который поддерживается группой технологии Cyberinfrastructure в Университете Клемсона. Мы благодарим д -р Пол J. Меза-Моралес для тестирования протокола.
Материалы
| Name | Company | Catalog Number | Comments |
| VASP software | Computational Materials Physics, Dept. of Physics, University of Vienna | vasp.5.4.4 | Standard parallel VASP executable in the newest version. |
| LAMMPS software | Sandia National Laboratory | 31Mar17-dp | Double-precision, parallel LAMMPS executable from 31 March 2017. |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | 1.9.3 | Standard VMD executable in the newest version. |
| MCPliQ software | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Executable and input files for the MCPliQ software availabe from the Getman Research Group GitHub page. | |
| JoVE article scripts | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Python scripts for this JoVE manuscript available from the Getman Research Group GitHub page. | |
| H2O PDB file | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University or RCSB Protein Data Bank | PDB file for a water molecule, available from the Getman Research Group GitHub page or at http://www.rcsb.org/ligand/HOH. |
Ссылки
- Liu, J. L., Cao, X. M., Hu, P. Density functional theory study on the activation of molecular oxygen on a stepped gold surface in an aqueous environment: A new approach for simulating reactions in solution. Physical Chemistry Chemical Physics. 16 (9), 4176-4185 (2014).
- Okamoto, Y., Sugino, O., Mochizuki, Y., Ikeshoji, T., Morikawa, Y. Comparative study of dehydrogenation at Pt(111)/water and Pt(111)/vacuum of methanol interfaces. Chemical Physics Letters. 377 (1-2), 236-242 (2003).
- Santana, J. A., Cabrera, C. R., Ishikawa, Y. A density-functional theory study of electrochemical adsorption of sulfuric acid anions on Pt(111). Physical Chemistry Chemical Physics. 12 (32), 9526-9534 (2010).
- Artrith, N., Kolpak, A. M. Understanding the composition and activity of electrocatalytic nanoalloys in aqueous solvents: A combination of DFT and accurate neural network potentials. Nano Letters. 14 (5), 2670-2676 (2014).
- Jinnouchi, R., Kodama, K., Morimoto, Y. DFT calculations on H, OH and O adsorbate formations on Pt(111) and Pt(332) electrodes. Journal of Electroanalytical Chemistry. 716, 31-44 (2014).
- Yoon, Y., Rousseau, R., Weber, R. S., Mei, D. H., Lercher, J. A. First-principles study of phenol hydrogenation on Pt and Ni catalysts in aqueous phase. Journal of the American Chemical Society. 136 (29), 10287-10298 (2014).
- Desai, S. K., Pallassana, V., Neurock, M. A periodic density functional theory analysis of the effect of water molecules on deprotonation of acetic acid over Pd(III). Journal of Physical Chemistry B. 105 (38), 9171-9182 (2001).
- Huang, Z. Q., Long, B., Chang, C. R. A theoretical study on the catalytic role of water in methanol steam reforming on PdZn(111). Catalysis Science & Technology. 5 (5), 2935-2944 (2015).
- Chang, C. R., Huang, Z. Q., Li, J. Hydrogenation of molecular oxygen to hydroperoxyl: An alternative pathway for O2 activation on nanogold catalysts. Nano Research. 8 (11), 3737-3748 (2015).
- Faheem, M., Heyden, A. hybrid quantum mechanics/molecular mechanics solvation scheme for computing free energies of reactions at metal-water interfaces. Journal of Chemical Theory and Computation. 10 (8), 3354-3368 (2014).
- Behtash, S., et al. Solvation effects in the hydrodeoxygenation of propanoic acid over a model Pd(211) catalyst. Journal of Physical Chemistry C. 120 (5), 2724-2736 (2016).
- Behtash, S., Lu, J. M., Walker, E., Mamun, O., Heyden, A. Solvent effects in the liquid phase hydrodeoxygenation of methyl propionate over a Pd(111) catalyst model. Journal of Catalysis. 333, 171-183 (2016).
- Norskov, J. K., et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. Journal of Physical Chemistry B. 108 (46), 17886-17892 (2004).
- Skachkov, D., Rao, C. V., Ishikawa, Y. Combined first-principles molecular dynamics/density functional theory study of ammonia electrooxidation on Pt(100) electrode. Journal of Physical Chemistry C. 117 (48), 25451-25466 (2013).
- Hibbitts, D. D., Loveless, B. T., Neurock, M., Iglesia, E. Mechanistic role of water on the rate and selectivity of Fischer-Tropsch synthesis on ruthenium catalysts. Angewandte Chemie International Edition. 52 (47), 12273-12278 (2013).
- Abdelrahman, O. A., Heyden, A., Bond, J. Q. Analysis of kinetics and reaction pathways in the aqueous-phase hydrogenation of levulinic acid to form γ-valerolactone over Ru/C. ACS Catalysis. 4 (4), 1171-1181 (2014).
- Wang, H. F., Liu, Z. P. Formic acid xxidation at Pt/H2O interface from periodic DFT calculations integratd with a continuum solvation model. Journal of Physical Chemistry C. 113, 17502-17508 (2009).
- Behtash, S., Lu, J., Faheem, M., Heyden, A. Solvent effects on the hydrodeoxygenation of propanoic acid over Pd(111) model surfaces. Green Chemistry. 16, 605-616 (2014).
- Montemore, M. M., Andreussi, O., Medlin, J. W. Hydrocarbon adsorption in an aqueous environment: A computational study of alkyls on Cu(111). The Journal of Chemical Physics. 145, 074702(2016).
- Hartnig, C., Grimminger, J., Spohr, E. Adsorption of formic acid on Pt(111) in the presence of water. Journal of Electroanalytical Chemistry. 607, 133-139 (2007).
- Hartnig, C., Grimminger, J., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (211). Electrochimica Acta. 52 (6), 2236-2243 (2007).
- Hartnig, C., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (111). Chemical Physics. 319, 185-191 (2005).
- Michel, C., et al. Role of water in metal catalyst performance for ketone hydrogenation: A joint experimental and theoretical study on levulinic acid conversion into gamma-valerolactone. Chemical Communications. 50 (83), 12450-12453 (2014).
- Zope, B. N., Hibbitts, D. D., Neurock, M., Davis, R. J. Reactivity of the gold/water interface during selective oxidation catalysis. Science. 330 (6000), 74-78 (2010).
- Pavlova, A., Meijer, E. J. Understanding the role of water in aqueous ruthenium-catalyzed transfer hydrogenation of ketones. ChemPhysChem. 13 (15), 3492-3496 (2012).
- Saavedra, J., Doan, H. A., Pursell, C. J., Grabow, L. C., Chandler, B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science. 345 (6204), 1599-1602 (2014).
- Desai, S., Neurock, M. A first principles analysis of CO oxidation over Pt and Pt66.7%Ru33.3%(111) surfaces. Electrochimica Acta. 48 (25-26), 3759-3773 (2003).
- Gohda, Y., Schnur, S., Gross, A. Influence of water on elementary reaction steps in electrocatalysis. Faraday Discussions. 140, 233-244 (2008).
- Nie, X. W., Luo, W. J., Janik, M. J., Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. Journal of Catalysis. 312, 108-122 (2014).
- Michel, C., Auneau, F., Delbecq, F., Sautet, P. C-H versus O-H bond dissociation for alcohols on a Rh(111) surface: A strong assistance from hydrogen bonded neighbors. ACS Catalysis. 1 (10), 1430-1440 (2011).
- Neurock, M., Wasileski, S. A., Mei, D. From first principles to catalytic performance: tracking molecular transformations. Chemical Engineering Science. 59 (22-23), 4703-4714 (2004).
- Camellone, M. F., Marx, D. On the impact of solvation on a Au/TiO2 nanocatalyst in contact with water. Journal of Physical Chemistry Letters. 4 (3), 514-518 (2013).
- Santana, J. A., Mateo, J. J., Ishikawa, Y. Electrochemical hydrogen oxidation on Pt(110): A combined direct molecular dynamics/density functional theory study. Journal of Physical Chemistry C. 114 (11), 4995-5002 (2010).
- Santana, J. A., Saavedra-Arias, J. J., Ishikawa, Y. Electrochemical hydrogen xxidation on Pt(100): A combined direct molecular dynamics/density functional theory study. Electrocatalysis-US. 6 (6), 534-543 (2015).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 119 (24), 13642-13651 (2015).
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics. 117, 1-19 (1995).
- MacKerell, A. D. Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 102, 3586-3616 (1998).
- Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A. III, Skiff, W. M. UFF, A full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society. 114, 10024-10035 (1992).
- Kahn, K., Bruice, T. C. Parameterization of OPLS-AA force field for the conformational analysis of macrocyclic polyketides. Journal of Computational Chemistry. 23, 977-996 (2002).
- Kresse, G., Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science. 6, 15-50 (1996).
- Kresse, G., Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B. 54, 11169-11186 (1996).
- Kresse, G., Hafner, J. Ab initio molecular dynamics for liquid metals. Physical Review B. 47, 558-561 (1993).
- Humphrey, W., Dalke, A., Schulten, K. VMD - visual molecular dynamics. Journal of Molecular Graphics and Modelling. 14, 33-38 (1996).
- Xie, T., Sarupria, S., Getman, R. B. A DFT and MD study of aqueous phase dehydrogenation of glycerol on Pt(111): Comparing chemical accuracy versus computational expense in different methods for calculating aqueous phase system energies. Molecular Simulation. 43, 370-378 (2017).
- Li, Y., Zhang, X., Srinath, A., Getman, R. B., Ngo, L. B. Combining HPC and big data infrastructures in large-scale post-processing of simulation data: A case3 study in PEARC ’18. Proceedings of the Practice and Experience on Advanced Research Computing. , Article No. 41 (2018).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Correction to "Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 120, 801(2016).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены