
Method Article
Multiscale Sampling of a Heterogeneous Water/Metal Catalyst Interface using Density Functional Theory and Force-Field Molecular Dynamics
In This Article
Summary
The goal of the protocol presented here is to generate and sample trajectories of configurations of liquid water molecules around catalytic species on a flat transition metal surface. The sampled configurations can be used as starting structures in quantum mechanics-based methods.
Abstract
A significant number of heterogeneously-catalyzed chemical processes occur under liquid conditions, but simulating catalyst function under such conditions is challenging when it is necessary to include the solvent molecules. The bond breaking and forming processes modeled in these systems necessitate the use of quantum chemical methods. Since molecules in the liquid phase are under constant thermal motion, simulations must also include configurational sampling. This means that multiple configurations of liquid molecules must be simulated for each catalytic species of interest. The goal of the protocol presented here is to generate and sample trajectories of configurations of liquid water molecules around catalytic species on flat transition metal surfaces in a way that balances chemical accuracy with computational expense. Specifically, force field molecular dynamics (FFMD) simulations are used to generate configurations of liquid molecules that can subsequently be used in quantum mechanics-based methods such as density functional theory or ab initio molecular dynamics. To illustrate this, in this manuscript, the protocol is used for catalytic intermediates that could be involved in the pathway for the decomposition of glycerol (C3H8O3). The structures that are generated using FFMD are modeled in DFT in order to estimate the enthalpies of solvation of the catalytic species and to identify how H2O molecules participate in catalytic decompositions.
Introduction
Modeling molecular phenomena involved in heterogeneous catalysis under liquid conditions is necessary for understanding catalytic function; however, this remains challenging because it requires a fine balance between chemical accuracy and computational expense. In general, since catalysis involves the breaking and forming of chemical bonds, quantum mechanics must be used to at least some degree; however, long simulations are challenging in quantum mechanics, as they require significant computer resources. Since molecules in the liquid phase are under constant thermal motion, simulations must also include configurational sampling, i.e., they must incorporate multiple spatial arrangements of the liquid molecules, as each different spatial arrangement (i.e., each configuration) has a different energy. This means that multiple configurations of liquid molecules must be simulated for each catalytic species of interest. These needs – to use quantum mechanics and to perform multiple calculations per catalytic species – can render modeling in heterogeneous catalysis under liquid phase computationally intractable. The purpose of the method described herein is to enable computationally tractable simulations of phenomena in heterogeneous catalysis under liquid phase.
We are particularly interested in heterogeneously catalyzed reactions that are carried out under liquid water. Water molecules have significant influence on catalytic phenomena, such as interacting with catalytic species (e.g., via dispersion forces and hydrogen bonding)1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23, participating in catalytic reactions1,7,8,9,15,21,22,24,25,26,27, and influencing reaction pathways and/or catalytic rates1,11,12,15,18,23,25,27,28,29,30,31. Modeling of these phenomena has been performed using QM and/or ab initio molecular dynamics (AIMD)1,2,6,7,14,22,25,27,28,32,33,34, force field molecular dynamics (FFMD)35, and quantum mechanics/molecular mechanics (QM/MM)10. In AIMD and FFMD, the atoms in the system are moved pursuant to Newton’s equations of motion according to the forces acting upon them. In AIMD, the system energy and forces are calculated with quantum mechanics, whereas in FFMD, the system energy and forces are calculated using force fields, which are algebraic expressions that are parameterized based on experimental or QM data. In QM/MM, the portion of the system where the bond breaking and forming occurs is calculated with QM, and the remainder of the system is calculated with MM, which employs force fields. Because they directly employ QM, AIMD and QM/MM are better suited for capturing the bond breaking and forming that occurs in aqueous phase heterogeneous catalysis; however, FFMD is significantly more computationally tractable and thus better suited for generating the configurations of liquid H2O molecules. The method presented in this protocol balances chemical accuracy and computational expense by employing a combination of QM and FFMD.
Specifically, this method uses FFMD simulations for generating configurations of liquid H2O and QM to calculate system energies. FFMD is carried out using LAMMPS.36 The force fields used in FFMD in this work employ Lennard-Jones + Coulomb (LJ+C) potentials, where the LJ parameters have been taken from the TIP3P/CHARMM model37 for H2O, the universal force field38 (UFF) for Pt, and the OPLS-AA force field39 for catalytic species, and the Coulomb parameters have been taken from the TIP3P/CHARMM37 model for H2O and the OPLS-AA force field39 for catalytic species. The Coulomb parameters for Pt atoms have been set to 0. QM calculations are performed using the VASP code40,41,42, which is a density functional theory (DFT) code. Water molecule insertions are performed with a code developed in-house called Monte Carlo Plug-in for Quantum Methods (MCPliQ). File conversions from VASP to LAMMPS in this protocol are performed with the Visual Molecular Dynamics (VMD) software43.
The protocol is intended to generate configurations of liquid water molecules around catalytic species on flat transition metal surfaces at low coverage. Coverage is denoted θ and defined as the number of adsorbates per surface metal atom (i.e., the number of surface adsorbates normalized by the number of metal atoms in the topmost layer of the metal slab in the catalyst model). In this manuscript, low coverage is defined as θ ≤ 1/9 monolayer (ML), where 1 ML means one catalytic species per surface metal atom. The catalyst models should be placed in periodic simulation boxes. The simulation boxes do not have to be cubes. This manuscript demonstrates the use of the protocol for generating configurations of liquid H2O that can be used to calculate quantities of interest in aqueous phase heterogeneous catalysis.
This protocol requires that the user has access to installed and working versions of the VASP, MCPliQ, LAMMPS, and VMD software. More information about VASP (https://www.vasp.at/), LAMMPS (https://Lammps.sandia.gov/), and VMD (https://www.ks.uiuc.edu/Research/vmd/) are available on their respective websites. The MCPliQ software is documented at https://github.com/getman-research-group/JoVE_article, along with all input files and Python scripts mentioned in this protocol. This protocol assumes that the executables and scripts mentioned within will be run on a high-performance research computer and are installed in a directory that is in the user’s $PATH variable. If an executable or script is placed in a location that is not in the user’s $PATH, then the path to the executable must be included to execute it. Executables and scripts are executed in steps 2.1.2, 2.2.1, 2.2.8, 3.1, 4.2, 5.2, and 6.1.2. For example, to execute the MCPliQ code in step 2.1.2 from a directory that is not in the user’s $PATH, the user would type $PATHTOMCPLIQ/mcpliq at the command-line interface instead of mcpliq, where $PATHTOMCPLIQ is the location where the mcpliq executable has been stored (e.g., $PATHTOMCPLIQ might be ~/bin). Before starting this protocol, all executables and scripts should be given executable permissions (e.g., in Linux, this could be done by typing chmod +x mcpliq at the command-line interface from the directory where the mcpliq executable is stored). Further, any modules required by any of the software or scripts should be loaded (these dependencies will be specific to individual installations of the various software and the computer where the simulations will be run).
Protocol
1. Generate the adsorbate structure
- Create a VASP POSCAR file comprising a supercell with periodic boundary conditions, as you would if you were performing simulations of adsorbates on metal surfaces under vacuum. The supercell should include an initial guess of the adsorbate structure and the metal surface as well as vacuum space above the adsorbate for adding H2O molecules. Details on how to do this are provided in previous work35,44.
NOTE: It is important that the height of the vacuum space be at least 12 Å above the top of the adsorbate. - Relax the structure and minimize its energy using the VASP code. Details on how to do this are provided in previous work35,44. This will produce a file called CONTCAR, which will be used in the next section.
2. Add explicit H2O molecules
- Add N water molecules to the vacuum space in the CONTCAR created in step 1.2 using the MCPliQ code, where N = ρV, ρ is the density of water, and V is the volume of the vacuum space above the adsorbate.
NOTE: ρ should be taken as the density of water as determined by the TIP3P/CHARMM water model at the simulation temperature. V will be refined in the next step.- Specify the following information in the MCPliQ master_input.txt file: The number of H2O molecules to add (N) by changing the first argument in line 28, the path to the water.txt file by changing the second argument in line 28, and the minimum and maximum height of the supercell that can be occupied by water molecules by changing the Minimum z-coordinate in line 11 and the Maximum z-coordinate in line 12.
- Execute the MCPliQ code by typing mcpliq from the command-line interface to insert water molecules into the CONTCAR file. The code will output one or more files with the file extension of .POSCAR.
NOTE: If more than one POSCAR files are produced, they will be named POSCAR_n.POSCAR. Select the file where n is largest.
- Generate the LAMMPS input files for an NPT simulation and equilibrate the cell volume using FFMD in the NPT ensemble in LAMMPS.
- Execute the script lmps_bond_angle.py on the .POSCAR file generated in step 2.1.2 by typing lmps_bond_angle.py $filename.POSCAR at the command-line interface, where $filename is the name of the .POSCAR file generated in step 2.1.2. This script creates a file called $filename.POSCAR.bond_angle_info.txt which lists bonds and angles that will be used in the LAMMPS data file.
- Open VMD and select File > New Molecule in the main window to open the Molecule File Browser window. Select VASP_POSCAR from the Determine File Type dropdown menu. Click Browse and navigate to the $filename.POSCAR file. Click Load to open the $filename.POSCAR file.
- Open the Tk Console within VMD by selecting Extensions > Tk Console from the VMD Main window.
- Execute the following command in the Tk console: topo writelammpsdata $WDPATH/data.myadsorbate full, where $WDPATH is the directory on the computer where VMD will write the LAMMPS data file and data.myadsorbate is the name of the LAMMPS data file.
- Delete the Bonds and Angles section at the bottom of the data.myadsorbate file. Then, append the bond and angle lists in the file $filename.POSCAR.bond_angle_info.text into data.myadsorbate.
NOTE: The indexes for the O-H bond type and H-O-H angle type for the water molecules in the $filename.POSCAR.bond_angle_info.txt file are both set to 1. Thus, bond and angle types for adsorbates should start counting at 2. - Edit the data.myadsorbate file by adding the Lennard-Jones parameters to the Pair Coeffs section and the Coulomb parameters to the Atoms section. Lennard-Jones and Coulomb parameters for the H2O molecules, adsorbate atoms, and metal surface atoms need to be added.
NOTE: The Lennard-Jones parameters for Pt atoms, water molecules, and adsorbate atoms in this protocol are obtained from the UFF38, TIP3P/CHARMM37, and OPLS-AA39 force fields, respectively. Coulomb parameters for water molecules and adsorbates atoms are obtained from the TIP3P/CHARMM37 and OPLS-AA39 force fields, respectively. Coulomb parameters for Pt atoms are set to 0 in this protocol. Alternatively, calculated partial charges could be used for the Coulomb parameters for adsorbate atoms and Pt atoms. - Copy the LAMMPS input file input.equil into the directory $WDPATH. Edit the group variable on line 34 to indicate the atom type indexes for the water oxygen and water hydrogen atoms and the group variable on line 35 to indicate the atom type indexes for the Pt and adsorbate atoms.
- Execute the LAMMPS software by typing mpiexec -np XX lmp_mpi < input.equil at the command-line interface, where XX is the number of CPU cores to use, and lmp_mpi is the name of the LAMMPS executable. Doing this will run an energy minimization to refine the H2O configuration, followed by a FFMD simulation performed at constant number of H2O molecules (N), volume (V), and temperature (T) to bring the water to the simulation temperature, followed by a FFMD simulation run at constant N, pressure (P), and temperature (T) to determine the physically correct height of the simulation box. Output files that will be used in section 3 are called data.myadsorbate_npt and log.myadsorbate.
NOTE: The duration of the NPT simulation should be long enough to comprise an “equilibration” run, where the volume of the supercell comes to steady state, and a “production” run, which is used to sample the ensemble averages (here, the height of the supercell). During the equilibration run, the volume of the supercell when plotted against time should level off to a steady state value. Once this occurs, the NPT simulation can be said to be in its production run. Verify equilibration of the NPT simulation by ensuring that the fluctuations in the height of the supercell (lz) are minimal or have converged to a steady value. If large fluctuations occur, then re-generate a H2O configuration by decreasing the timestep on line 92 in the input.equil file and repeating step 2.2.8 or starting again from step 2.1.1 .
3. Extract the proper height of the supercell
- Execute the script get_npt_lz.py on the log.myadsorbate file by typing get_npt_lz.py log.myadsorbate at the command-line interface. This script outputs the average supercell height from the “production” run portion of the NPT simulation in the avg_lz.txt file.
NOTE: The get_npt_lz.py script assumes that LAMMPS writes the length of the cell z-dimension (lz) to the log.myadsorbate file every 1000 fs (customizable at line 20 of the get_npt_lz.py script), which is the default in the provided input.equil LAMMPS input file. The get_npt_lz.py script detects and discards the first 2 ns (customizable at line 19 of the get_npt_lz.py script) worth of lz values in the log.myadsorbate file, as they comprise the equilibration portion of the simulation, while the remaining 3 ns comprise the “production” portion and are thus used by the get_npt_lz.py script to compute the average z-dimension length. In addition to the avg_lz.txt file, the get_npt_lz.py script outputs a file called npt_data.txt, which provides values of lz as a function of the timestep, as well as a file called npt_plot.png, which plots the same data. The plot can be used to verify equilibration of the NPT simulation. - Reconstruct the supercell using the average height determined in NPT.
- Copy the data.myadsorbate_npt file into a new directory, referred to here as $WD2PATH, and rename it data.myadsorbate.
- Edit the new data.myadsorbate file so that the lz height is equal to the average value output from the get_npt_lz.py script by changing the zlo and zhi arguments in the data.myadsorbate file such that zlo is 0.0 and zhi is the lz value from the avg_lz.txt file produced in step 3.1.
4. Generate configurations of H2O molecules
- Copy the LAMMPS input file input.prod into $WD2PATH. Edit the group variable on line 32 to indicate the atom type indexes for the water oxygen and water hydrogen atoms and the group variable on line 33 to indicate the atom type indexes for the Pt and adsorbate atoms.
- Execute the LAMMPS software by typing mpiexec -np XX lmp_mpi < input.prod into the command-line interface, where XX is the number of CPU cores to use, and lmp_mpi is the name of the LAMMPS executable. Doing this will run a constant NVT simulation on the H2O molecules. The key output file from this simulation is the dump.myadsorbate.lammpstrj file.
NOTE: The duration of the NVT simulation should be long enough to comprise an equilibration run, where the energy of the system comes to steady state, and a production run, from which the ensemble averages (here, the spatial positions of the water molecules) are sampled. During the equilibration run, the energy of the system when plotted against time should level off to a steady state value. Once this occurs, the NVT simulation can be said to be in its production run.
5. Determine the hydrogen bond lifetime for proper time sampling
- Edit the hb_lifetime_dist.py script to specify: the timestep of the first frame of the dump.myadsorbate.lammpstrj file by changing the actualStart variable on line 22, how often frames are written to the LAMMPS trajectory file by changing the timestep variable on line 23, the first and last timesteps the script should consider (i.e., the production portion of the trajectory) by changing the N_first and N_last variables on lines 24 and 25, whether consecutive frames are considered or frames are skipped by changing the nevery variable on line 26, and the number of lines per frame section of the trajectory file by changing the frameLine variable on line 27. Additionally, edit lines 31 through 35 to specify which atom types within the data.myadsorbate file belong to the adsorbate and which atom types belong to the H2O molecules.
NOTE: The hb_lifetime_dist.py script analyzes the H2O configurations in the production run and determines if any H2O molecules are hydrogen bonded to the adsorbate. It then counts the simulation time that each hydrogen bond remains intact and reports this information as a distribution of hydrogen bond lifetimes in units of ps. The specific version of the script that is provided with this protocol assumes that LAMMPS writes the configuration of H2O molecules to the dump.myadsorbate.lammpstrj file every 1000 fs, which is the default in the provided input.prod LAMMPS input file. It detects and discards the first 2 ns worth of configurations in the the dump.myadsorbate.lammpstrj file, as they comprise the equilibration portion of the simulation, and uses the remaining 3 ns to calculate hydrogen bond lifetimes. - Execute the script hb_lifetime_dist.py on the dump.myadsorbate.lammpstrj file by typing hb_lifetime_dist.py at the command-line interface. Doing this will produce a file called distribution_HB_lifetime.dat.
- Plot the data in the distribution_HB_lifetime.dat file to view the distribution of hydrogen bond lifetimes that occurred during the NVT simulation.
- Determine the time increment to use for the time sampling interval based on the calculated hydrogen bond lifetimes. The best choice is the maximum hydrogen bond lifetime; alternatively, a value that would capture the 95% confidence interval can be used.
6. Sample configurations of liquid H2O molecules
- Determine the number of configurations from the production run of the NVT FFMD trajectory for further calculations. The number of configurations should be selected so that the minimum time between configurations is equal to or greater than the time sampling interval identified in section 5.
- Edit the default value for the num_frames variable on line 21 of the lammps_frames.py script to specify the number of configurations to extract.
- Execute the script lammps_frames.py on the file dump.myadsorbate.lammpstrj by typing lammps_frames.py at the command-line interface. Doing this will output a list of simulation times corresponding to the configurations that should be extracted from the dump.myadsorbate.lammpstrj file. These configurations can be used as starting structures in AIMD or QM simulations.
NOTE: 1) The lammps_frames.py script automatically detects the LAMMPS log and dump files as well as the production portion of the trajectory within the dump file and divides the number of configurations within the dump file into 10 groups. Alternatively, the user can specify the log file, the dump file, and the number of configurations from the command-line interface using the -l, -d, and -n options, respectively. To do so, the user should type lammps_frames.py -n XX -l $logfilename -d $dumpfilename at the command-line interface, where XX is the desired number of configurations, $logfilename is the name of the LAMMPS logfile, and $dumpfilename is the name of the LAMMPS trajectory (dump) file. The simulation times that are output refer to the median times in each group. 2) If the configurations will be calculated in VASP with the LDIPOLE flag turned on, a small layer of vacuum space should be added to the top of the supercell above the water layer. This will facilitate convergence of the electronic structure in the VASP calculation. Adding an additional 3 Å of vacuum space above the H2O molecules has been successful in the simulations discussed below.
Results
One use of this protocol is to calculate the energies of interaction between the liquid water and catalytic species, i.e., ΔEint35:
∆Eint=ECatalytic species+H2O+EClean catalyst surface-ECatalytic species-EClean catalyst surface+H2O
where ECatalytic species+H2O is the energy of a configuration of H2O molecules around a catalytic species on a metal surface, EClean catalyst surface is the energy of the clean catalyst surface in vacuum, ECatalytic species is the energy of the catalytic species on a metal surface in vacuum, and EClean catalyst surface+H2O is the energy of the configuration of H2O over the catalyst surface with the catalytic species removed. The positions of the H2O molecules used to calculate ECatalytic species+H2O and EClean catalyst surface+H2O should be identical. All values of E are calculated using the VASP code. The quantity ΔEint includes all of the physical and chemical interactions between all of the molecules in the liquid water structure and the catalytic species and gives a reasonable estimate of the enthalpy of solvation of the catalytic species, which is needed to calculate its free energy of solvation and total free energy. Table 1 provides values for ΔEint calculated for species on a Pt(111) catalyst surface with chemical formulas equal to CxHyOz in units of eV (1 eV = 96.485 kJ/mol). The values were calculated at coverages ≤1/9 ML.35,46 The values reported are the averages taken over 10 configurations of liquid H2O, and the uncertainties are reported as standard deviations. All the values are negative, indicating favorable interactions with water.
Another application of this protocol is to generate starting structures for AIMD. Movie 1 is a movie of an AIMD trajectory that was started from a configuration generated by this protocol. At the start of this movie, a COH adsorbate is shown on a Pt(111) surface under a structure of liquid H2O. One H2O molecule is emphasized, which formed a hydrogen bond with COH. Over the course of the movie, this H2O molecule abstracts the proton from the COH adsorbate and deposits a second hydrogen atom on the Pt(111) surface. The H2O molecule thus helps to catalyze the reaction COH* + * → CO* + H*, where the *s indicate catalytic sites. This simulation highlights the main strength and the main purpose of the multiscale sampling method described herein. Numerous configurations of H2O molecules are generated with FFMD, due to its strength in computational tractability. However, a limitation of FFMD is that it cannot capture bond breaking and forming unless a reactive force field is implemented. AIMD uses quantum mechanics to calculate energies and thus can capture bond breaking and forming. However, AIMD is too computationally demanding to generate all of the configurations of H2O molecules necessary to ensure sufficient sampling has been achieved. Thus, this protocol combines the two methods.
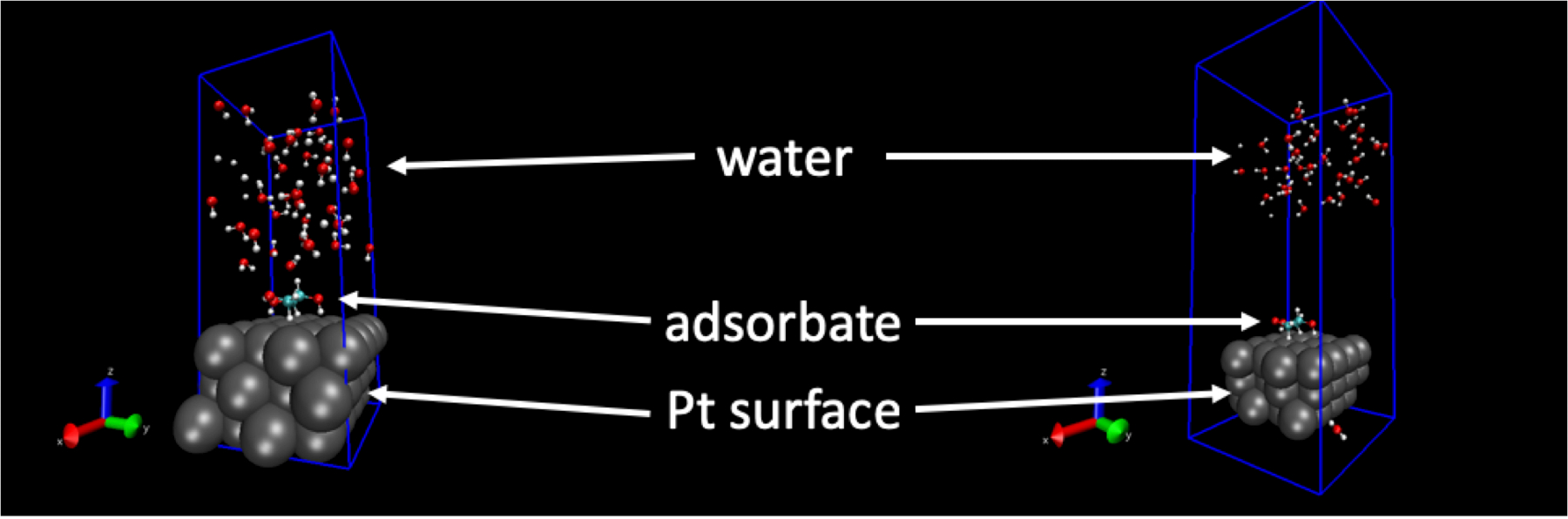
The structures of liquid H2O molecules generated by this procedure are dependent on input settings. Setting these improperly can have unintended influences on the water structures. For example, when the intermolecular distances become too small or when other parameters in the molecular dynamics input files are set improperly or take on unphysical values, the water structure can become unreasonable. Under these circumstances, the structure of water will “blow up” unintendedly during the FFMD trajectory. Figure 1 shows an example of this. The snapshot on the left-hand side is the starting structure for a FFMD run, and the snapshot on the right-hand side is a snapshot taken within 1 ps of starting the simulation. As can be seen, the H2O molecules have moved far away from the surface. This is caused by improper settings made in the simulation input files and is not a structure that is likely to occur in reality.

Figure 1: Example of a negative result. The force field molecular dynamics simulation “blows up” due to an unphysical setting or value. Left hand image: The starting geometry of the Pt(111) surface, adsorbate, and liquid water structure. Right hand image: The geometry of the Pt(111) surface, adsorbate, and liquid water structure less than 1 ps later. In the right-hand image, the H2O molecules have separated from the surface due to unphysically large forces. Please click here to view a larger version of this figure.
{kind=link}

Movie 1: Ab initio molecular dynamics (AIMD) simulation initiated from a configuration generated in multiscale sampling. A H2O molecule that is originally hydrogen bonded to a COH adsorbate on a Pt(111) surface abstracts the proton from COH and deposits a second hydrogen on the Pt(111) surface. This bond breaking and forming event can be captured by AIMD but not with force field molecular dynamics (FFMD) unless a reactive force field is used. The initial configuration of H2O molecules used in this AIMD simulation was generated using FFMD as described in this manuscript. Please click here to view this video. (Right-click to download.)
| Catalytic Species | ∆Eint (eV) |
| COH | -0.70 ± 0.07 |
| CO | -0.03 ± 0.03 |
| CH2OH | -0.64 ± 0.12 |
| CHO-CHOH-CH2OH | -0.93 ± 0.22 |
| COH-COH-CH2OH | -0.87 ± 0.23 |
| COH-CHOH-COH | -1.72 ± 0.26 |
| CHOH-COH-CO | -1.57 ± 0.25 |
| CHO-CO-CO | -0.31 ± 0.19 |
Table 1: Water-catalytic species interaction energy results. Interaction energies in eV calculated for eight CxHyOz adsorbates on Pt(111). Values reported are the averages taken over multiple configurations of liquid H2O. The uncertainties are the standard deviations of the averages. 1 eV = 96.485 kJ/mol.
Discussion
The method as presented was selected for its ease of implementation, but multiple customizations could be made. For one, the force fields used in the FFMD simulations can be modified. Changing the force field parameters and/or potentials can be done by editing the LAMMPS input and data files. Similarly, solvents other than H2O could be employed. To make this modification, the desired solvent molecule would need to be inserted starting from step 2.1.1, and the LAMMPS input files would need to be edited to incorporate the appropriate potentials and parameters. Inserting the new solvent molecule would also require supplying the internal coordinates of the solvent molecule in a .txt file analogous to the water.txt file.
Another modification that could be made is to modify the area of the surface slab. The results discussed in this manuscript employed 3 Pt x 3 Pt or 4 Pt x 4 Pt surface slabs, which have surface areas less than 120 Å2. As the slab surface area increases, the computational expense also increases. Computational expense has the largest impact on section 5 of this protocol. If the data processing steps in section 5 become computationally prohibitive, big data post processing strategies such as those discussed in Li et al. 201845 can be employed.
Possible sources of uncertainty for this procedure include the force field employed, the sampling method, and the sampling frequency. The water structure is determined by the force field that is used, meaning that the choice of force field could influence the specific configurations of H2O molecules. Our group has assessed how the choices of force field for H2O molecules and Pt atoms influence the interaction energies calculated in FFMD and found that the choice of force field contributes less than 0.1 eV to this interaction energy. Another source of uncertainty is the sampling method, which influences the specific configurations that are used to calculate a quantity of interest. Our group has compared the performance of the “time sampling” method presented in this protocol with an “energy sampling” method, which is biased to lower energy configurations of H2O molecules, on the interaction energies calculated in DFT and found both of these sampling methods give statistically equal values35,46. The sampling frequency can also influence the results. We have assessed how increasing the number of configurations from 10 to 30,000 influences the average interaction energies calculated in FFMD for 40 different C3HxO3 adsorbates and found that the sampling frequency contributes less than 0.1 eV to the average interaction energy44.
The main limitation to this method is that the adsorbates are approximated by the structures under vacuum during the FFMD simulations. In reality, the adsorbates would exhibit conformational changes (bond stretches, angle bends, torsional motions, etc.) due to normal thermal movements, including interactions with solvent molecules. Attempts to include conformational changes of adsorbates into the FFMD simulations would require detailed development of force fields for catalytic surface adsorbates, i.e., which comprise terms that describe bond stretches, angle bends, and torsional terms, amongst others. As a future direction of this protocol, we are developing such force fields for adsorbates at solid surfaces, which we will use to determine the extent to which using rigid adsorbates influences the results.
Disclosures
The authors disclose no conflicts of interest.
Acknowledgements
This research was funded by the National Science Foundation through award number CBET-1438325. Fellowship support for CJB through NASA Training Grant NX14AN43H is gratefully acknowledged. Simulations were performed on the Palmetto Supercomputer Cluster, which is maintained by the Cyberinfrastructure Technology Group at Clemson University. We thank Dr. Paul J. Meza-Morales for testing the protocol.
Materials
| Name | Company | Catalog Number | Comments |
| VASP software | Computational Materials Physics, Dept. of Physics, University of Vienna | vasp.5.4.4 | Standard parallel VASP executable in the newest version. |
| LAMMPS software | Sandia National Laboratory | 31Mar17-dp | Double-precision, parallel LAMMPS executable from 31 March 2017. |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | 1.9.3 | Standard VMD executable in the newest version. |
| MCPliQ software | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Executable and input files for the MCPliQ software availabe from the Getman Research Group GitHub page. | |
| JoVE article scripts | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University | Python scripts for this JoVE manuscript available from the Getman Research Group GitHub page. | |
| H2O PDB file | Getman Research Group, Dept. of Chemical and Biomolecular Engineering, Clemson University or RCSB Protein Data Bank | PDB file for a water molecule, available from the Getman Research Group GitHub page or at http://www.rcsb.org/ligand/HOH. |
References
- Liu, J. L., Cao, X. M., Hu, P. Density functional theory study on the activation of molecular oxygen on a stepped gold surface in an aqueous environment: A new approach for simulating reactions in solution. Physical Chemistry Chemical Physics. 16 (9), 4176-4185 (2014).
- Okamoto, Y., Sugino, O., Mochizuki, Y., Ikeshoji, T., Morikawa, Y. Comparative study of dehydrogenation at Pt(111)/water and Pt(111)/vacuum of methanol interfaces. Chemical Physics Letters. 377 (1-2), 236-242 (2003).
- Santana, J. A., Cabrera, C. R., Ishikawa, Y. A density-functional theory study of electrochemical adsorption of sulfuric acid anions on Pt(111). Physical Chemistry Chemical Physics. 12 (32), 9526-9534 (2010).
- Artrith, N., Kolpak, A. M. Understanding the composition and activity of electrocatalytic nanoalloys in aqueous solvents: A combination of DFT and accurate neural network potentials. Nano Letters. 14 (5), 2670-2676 (2014).
- Jinnouchi, R., Kodama, K., Morimoto, Y. DFT calculations on H, OH and O adsorbate formations on Pt(111) and Pt(332) electrodes. Journal of Electroanalytical Chemistry. 716, 31-44 (2014).
- Yoon, Y., Rousseau, R., Weber, R. S., Mei, D. H., Lercher, J. A. First-principles study of phenol hydrogenation on Pt and Ni catalysts in aqueous phase. Journal of the American Chemical Society. 136 (29), 10287-10298 (2014).
- Desai, S. K., Pallassana, V., Neurock, M. A periodic density functional theory analysis of the effect of water molecules on deprotonation of acetic acid over Pd(III). Journal of Physical Chemistry B. 105 (38), 9171-9182 (2001).
- Huang, Z. Q., Long, B., Chang, C. R. A theoretical study on the catalytic role of water in methanol steam reforming on PdZn(111). Catalysis Science & Technology. 5 (5), 2935-2944 (2015).
- Chang, C. R., Huang, Z. Q., Li, J. Hydrogenation of molecular oxygen to hydroperoxyl: An alternative pathway for O2 activation on nanogold catalysts. Nano Research. 8 (11), 3737-3748 (2015).
- Faheem, M., Heyden, A. hybrid quantum mechanics/molecular mechanics solvation scheme for computing free energies of reactions at metal-water interfaces. Journal of Chemical Theory and Computation. 10 (8), 3354-3368 (2014).
- Behtash, S., et al. Solvation effects in the hydrodeoxygenation of propanoic acid over a model Pd(211) catalyst. Journal of Physical Chemistry C. 120 (5), 2724-2736 (2016).
- Behtash, S., Lu, J. M., Walker, E., Mamun, O., Heyden, A. Solvent effects in the liquid phase hydrodeoxygenation of methyl propionate over a Pd(111) catalyst model. Journal of Catalysis. 333, 171-183 (2016).
- Norskov, J. K., et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. Journal of Physical Chemistry B. 108 (46), 17886-17892 (2004).
- Skachkov, D., Rao, C. V., Ishikawa, Y. Combined first-principles molecular dynamics/density functional theory study of ammonia electrooxidation on Pt(100) electrode. Journal of Physical Chemistry C. 117 (48), 25451-25466 (2013).
- Hibbitts, D. D., Loveless, B. T., Neurock, M., Iglesia, E. Mechanistic role of water on the rate and selectivity of Fischer-Tropsch synthesis on ruthenium catalysts. Angewandte Chemie International Edition. 52 (47), 12273-12278 (2013).
- Abdelrahman, O. A., Heyden, A., Bond, J. Q. Analysis of kinetics and reaction pathways in the aqueous-phase hydrogenation of levulinic acid to form γ-valerolactone over Ru/C. ACS Catalysis. 4 (4), 1171-1181 (2014).
- Wang, H. F., Liu, Z. P. Formic acid xxidation at Pt/H2O interface from periodic DFT calculations integratd with a continuum solvation model. Journal of Physical Chemistry C. 113, 17502-17508 (2009).
- Behtash, S., Lu, J., Faheem, M., Heyden, A. Solvent effects on the hydrodeoxygenation of propanoic acid over Pd(111) model surfaces. Green Chemistry. 16, 605-616 (2014).
- Montemore, M. M., Andreussi, O., Medlin, J. W. Hydrocarbon adsorption in an aqueous environment: A computational study of alkyls on Cu(111). The Journal of Chemical Physics. 145, 074702 (2016).
- Hartnig, C., Grimminger, J., Spohr, E. Adsorption of formic acid on Pt(111) in the presence of water. Journal of Electroanalytical Chemistry. 607, 133-139 (2007).
- Hartnig, C., Grimminger, J., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (211). Electrochimica Acta. 52 (6), 2236-2243 (2007).
- Hartnig, C., Spohr, E. The role of water in the initial steps of methanol xxidation on Pt (111). Chemical Physics. 319, 185-191 (2005).
- Michel, C., et al. Role of water in metal catalyst performance for ketone hydrogenation: A joint experimental and theoretical study on levulinic acid conversion into gamma-valerolactone. Chemical Communications. 50 (83), 12450-12453 (2014).
- Zope, B. N., Hibbitts, D. D., Neurock, M., Davis, R. J. Reactivity of the gold/water interface during selective oxidation catalysis. Science. 330 (6000), 74-78 (2010).
- Pavlova, A., Meijer, E. J. Understanding the role of water in aqueous ruthenium-catalyzed transfer hydrogenation of ketones. ChemPhysChem. 13 (15), 3492-3496 (2012).
- Saavedra, J., Doan, H. A., Pursell, C. J., Grabow, L. C., Chandler, B. D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science. 345 (6204), 1599-1602 (2014).
- Desai, S., Neurock, M. A first principles analysis of CO oxidation over Pt and Pt66.7%Ru33.3%(111) surfaces. Electrochimica Acta. 48 (25-26), 3759-3773 (2003).
- Gohda, Y., Schnur, S., Gross, A. Influence of water on elementary reaction steps in electrocatalysis. Faraday Discussions. 140, 233-244 (2008).
- Nie, X. W., Luo, W. J., Janik, M. J., Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. Journal of Catalysis. 312, 108-122 (2014).
- Michel, C., Auneau, F., Delbecq, F., Sautet, P. C-H versus O-H bond dissociation for alcohols on a Rh(111) surface: A strong assistance from hydrogen bonded neighbors. ACS Catalysis. 1 (10), 1430-1440 (2011).
- Neurock, M., Wasileski, S. A., Mei, D. From first principles to catalytic performance: tracking molecular transformations. Chemical Engineering Science. 59 (22-23), 4703-4714 (2004).
- Camellone, M. F., Marx, D. On the impact of solvation on a Au/TiO2 nanocatalyst in contact with water. Journal of Physical Chemistry Letters. 4 (3), 514-518 (2013).
- Santana, J. A., Mateo, J. J., Ishikawa, Y. Electrochemical hydrogen oxidation on Pt(110): A combined direct molecular dynamics/density functional theory study. Journal of Physical Chemistry C. 114 (11), 4995-5002 (2010).
- Santana, J. A., Saavedra-Arias, J. J., Ishikawa, Y. Electrochemical hydrogen xxidation on Pt(100): A combined direct molecular dynamics/density functional theory study. Electrocatalysis-US. 6 (6), 534-543 (2015).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 119 (24), 13642-13651 (2015).
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics. 117, 1-19 (1995).
- MacKerell, A. D., et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 102, 3586-3616 (1998).
- Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A., Skiff, W. M. UFF, A full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society. 114, 10024-10035 (1992).
- Kahn, K., Bruice, T. C. Parameterization of OPLS-AA force field for the conformational analysis of macrocyclic polyketides. Journal of Computational Chemistry. 23, 977-996 (2002).
- Kresse, G., Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science. 6, 15-50 (1996).
- Kresse, G., Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B. 54, 11169-11186 (1996).
- Kresse, G., Hafner, J. Ab initio molecular dynamics for liquid metals. Physical Review B. 47, 558-561 (1993).
- Humphrey, W., Dalke, A., Schulten, K. VMD - visual molecular dynamics. Journal of Molecular Graphics and Modelling. 14, 33-38 (1996).
- Xie, T., Sarupria, S., Getman, R. B. A DFT and MD study of aqueous phase dehydrogenation of glycerol on Pt(111): Comparing chemical accuracy versus computational expense in different methods for calculating aqueous phase system energies. Molecular Simulation. 43, 370-378 (2017).
- Li, Y., Zhang, X., Srinath, A., Getman, R. B., Ngo, L. B. Combining HPC and big data infrastructures in large-scale post-processing of simulation data: A case3 study in PEARC ’18. Proceedings of the Practice and Experience on Advanced Research Computing. , (2018).
- Bodenschatz, C. J., Sarupria, S., Getman, R. B. Correction to "Molecular-level details about liquid H2O interactions with CO and sugar alcohol adsorbates on Pt(111) calculated using density functional theory and molecular dynamics. Journal of Physical Chemistry C. 120, 801 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved