
Method Article
Analyse des données SEC-SAXS via la déconvolution de l’EPT et Scatter
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Les mesures SEC-BioSAXS des macromolécules biologiques sont une approche standard pour déterminer la structure des solutions des macromolécules et de leurs complexes. Ici, nous analysons les données SEC-BioSAXS de deux types de traces de SEC couramment rencontrées : les chromatogrammes avec des pics entièrement résolus et partiellement résolus. Nous démontrons l’analyse et la déconvolution à l’aide de scatter et BioXTAS RAW.
Résumé
BioSAXS est une technique populaire utilisée en biologie moléculaire et structurelle pour déterminer la structure de la solution, la taille et la forme des particules, le rapport surface/volume et les changements conformationnels des macromolécules et des complexes macromoléculaires. Un ensemble de données SAXS de haute qualité pour la modélisation structurale doit être à partir d’échantillons monodisperse et homogènes et cela n’est souvent atteint que par une combinaison de chromatographie inline et de mesure immédiate de SAXS. Le plus souvent, la chromatographie d’exclusion de taille est utilisée pour séparer les échantillons et exclure les contaminants et les agrégations de la particule d’intérêt permettant de faire des mesures SAXS à partir d’un pic chromatographique bien résolu d’une seule espèce protéique. Pourtant, dans certains cas, même la purification en ligne n’est pas une garantie d’échantillons de monodisperse, soit parce que plusieurs composants sont trop proches les uns des autres dans la taille ou les changements de forme induits par la liaison modifier le temps d’élitution perçue. Dans ces cas,, il peut être possible de déconvoluter les données SAXS d’un mélange pour obtenir les courbes SAXS idéalisées de composants individuels. Ici, nous montrons comment cela est réalisé et l’analyse pratique des données SEC-SAXS est effectuée sur des échantillons idéaux et difficiles. Plus précisément, nous montrons l’analyse SEC-SAXS de la vaccinia E9 ADN polymerase exonuclease moins mutant.
Introduction
Les macromolécules biologiques sont trop petites pour être vues même avec les meilleurs microscopes légers. Les méthodes actuelles pour déterminer leurs structures impliquent généralement la cristallisation de la protéine ou des mesures sur un grand nombre de molécules identiques en même temps. Bien que la cristallographie fournit des informations sur le niveau atomique, elle représente un environnement d’échantillon artificiel, étant donné que la plupart des macromolécules ne sont pas présentées sous une forme cristalline dans la cellule. Au cours des deux dernières années, la microscopie cryo-électronique a livré des structures similaires à haute résolution de grands complexes macromolécules/macromoléculaires, mais bien que les échantillons soient plus proches de l’état physiologique, ils sont encore gelés, donc immobiles et statiques. La diffusion des rayons X à petit angle (BioSAXS) fournit une mesure structurelle de la macromolécule, dans des conditions pertinentes pour la biologie. Cet état peut être visualisé comme une forme 3D à basse résolution déterminée sur l’échelle nanométrique et capture tout l’espace conformationnel de la macromolécule en solution. Les expériences BioSAXS évaluent efficacement l’état oligomérique, le domaine et les arrangements complexes ainsi que la flexibilité entre lesdomaines 1,2,3. La méthode est précise, la plupart du temps non destructive et ne nécessite généralement qu’un minimum de préparation de l’échantillon et de temps. Toutefois, pour la meilleure interprétation des données, les échantillons doivent être monodisperse. C’est un défi; les molécules biologiques sont souvent sensibles aux contaminations, à une mauvaise purification et à une agrégation, par exemple à la dégelationdu gel 4. Le développement de la chromatographie inline suivie d’une mesure immédiate du SAXS permet d’atténuer ces effets. La chromatographie d’exclusion de taille sépare les échantillons par taille excluant ainsi la plupart des contaminants et agrégations5,6,7,8,9,10. Toutefois, dans certains cas, même sec-SAXS n’est pas suffisant pour produire un échantillon monodisperse, parce que le mélange peut se composer de composants qui sont trop proches en taille ou leurs propriétés physiques ou leur dynamique rapide conduire à des pics qui se chevauchent dans la trace UV SEC. Dans ces cas, une étape de déconvolution logicielle des données SAXS obtenues pourrait conduire à une courbe SAXS idéalisée du composantindividuel 5,11,12. À titre d’exemple, dans la section protocolaire 2, nous montrons l’analyse standard SEC-SAXS de la vaccinia E9 ADN polymése exonuclease moins mutant (E9 exomoins) en complexe avec de l’ADN. La vaccinia représente l’organisme modèle des Poxviridae, une famille contenant plusieurs agents pathogènes, par exemple le virus de la variole humaine. Il a été démontré que la polymése se lie étroitement à l’ADN dans les approches biochimiques, la structure du complexe ayant été récemment résolue par cristallographie aux rayons X13.
La plupart des installations de synchrotron fourniront un pipeline automatisé de traitement des données qui effectuera la normalisation et l’intégration des données produisant un ensemble d’images non sous-traitées. Mais l’approche décrite dans ce manuscrit pourrait également être utilisé avec une source de laboratoire à condition sec-SAXS est effectuée. En outre, une automatisation supplémentaire peut être disponible qui rejettera les cadres endommagés par les radiations et effectuera la soustraction tampon14. Nous montrerons comment effectuer l’analyse des données primaires sur les données pré-traitées et tirer le meilleur parti des données disponibles dans la section 2.
Dans la section 3, nous montrons comment déconvoluter les données SEC-SAXS et analyser efficacement les courbes. Bien qu’il existe plusieurs méthodes de déconvolution telles que la déconvolution du pic gaussien, mise en œuvre dans US-SOMO15 et la méthode de probabilité maximale optimisée Guinier, implémentée dans le logiciel DELA16, celles-ci nécessitent généralement un modèle pour la formede pointe 12. La taille limitée des pics individuels que nous étudions permet l’utilisation de l’analyse évolutive des facteurs (EPT), comme forme améliorée de décomposition de la valeur singulière (SVD) aux pics de chevauchement décontvolutes, sans s’appuyer sur la forme de pointe ou le profil de diffusion5,11. Une implémentation spécifique à SAXS se trouve dans BioXTAS RAW17. L’EPT a d’abord été utilisé sur les données de chromatographie lorsque les données du réseau de diodes 2D ont permis de former des matrices à partir de l’absorption par rapport au temps de rétention et aux données de longueurd’onde 18. Là où l’EPT excelle, c’est qu’il met l’accent sur le caractère évolutif des valeurs singulières, comment elles changent avec l’apparition de nouveaux composants, avec la mise en garde qu’il y a un ordre inhérent dans l’acquisition10. Heureusement, les données SEC-SAXS fournissent toutes les données d’acquisition ordonnées nécessaires dans les tableaux de données 2D organisés, se prêtant bien à la technique de l’EPT.
Dans la section 4, nous démontrerons les bases de l’analyse SAXS indépendante du modèle à partir de la courbe SAXS soustraite en arrière-plan tampon. L’analyse indépendante du modèle détermine le rayon de gyration (Rg) de la particule, le volume de corrélation (Vc), le volume de Porod (Vp) et l’exposant porod-debye (PE). L’analyse fournit une évaluation semi-quantitative de l’état thermodynamique de la particule en termes de compacité ou de flexibilité via la parcelle kratky sans dimension2,4,19.
Enfin, les données SAXS sont mesurées en unités spatiales réciproques et nous montrerons comment transformer les données SAXS en espace réel pour récupérer la fonction de distribution à distance paire, P(r). La distribution P(r) est l’ensemble de toutes les distances trouvées dans la particule et comprend la dimension maximale de la particule, dmax. Puisqu’il s’agit d’une mesure thermodynamique, la distribution P(r) représente l’espace physique occupé par l’espace conformationnel des particules. Une analyse appropriée d’un ensemble de données SAXS peut fournir des informations d’état de solution qui complètent les informations haute résolution de la cristallographie et du cryo-EM.
Protocole
1. L’expression des protéines, la purification et la mesure SEC-SAXS sont basées sur le protocolepublié 13
- Suivez le protocole de collecte de données SEC-SAXS en ligne (Brennich et coll.6)en bref.
- Equilibrer la colonne SEC avec au moins 2 volumes de colonnes de tampon de fonctionnement SEC (20 mM Tris-HCl, pH 7,5, 100 mM NaCl).
- Préparer 50 μL d’échantillon d’E9 exomoins à 8\u201210 mg/mL avec 20% d’excès molaire d’un dsDNA partiel (TCAGGAAGATAACAGCGGTTTAGCC et GGCTAAACCGCTGTTATCTT). E9 exomoins se lie avec un KD de 12 ± 6 nM (voir Données supplémentaires).
- Injectez 50 μL de ce mélange sur une colonne SEC (Augmentation S200) en ligne avec la cellule d’écoulement pour les mesures SAXS à 0,3 mL/min.
- Collecter 1000 cadres à 1 s d’exposition chacun.
REMARQUE : Sur la ligne de faisceau BM29 de BioSAXS, à l’Installation européenne de synchrotron (ESRF), les cadres individuels sont traités automatiquement et indépendamment dans le cadre EDNA14. Après la collecte de données, ouvrez la base de données ISPyB20 et sous l’onglet Acquisition de données appuyez sur le bouton Go pour accéder à l’ensemble de données et aux résultats de l’analyseautomatique 21.
- Téléchargez les données.
2. Analyse primaire des données
- Ouvrez le programme basé sur Java Scatter IV (voir le Tableau des matériaux) et effectuer une soustraction d’arrière-plan pour les données de chromatographie d’exclusion de taille (SEC).
- Ouvrez l’onglet SEC. Faites glisser et déposer les fichiers de donnéesréduits ( *.dat) dans la fenêtre " Déposer les données ci-dessous « . Définissez l’annuaire de sortie « Out Dir :: » en cliquant sur le bouton Dir de sortie marqué bleu.
REMARQUE : Si vos données ont été collectées en nm-1,une boîte de conversion devra être cochée (en bas à gauche du panneau) lorsque vous déposez des fichiers dans la fenêtre ou dans l’onglet soustraction. - Modifiez les détails expérimentaux, utilisez le bouton Modifier les détails et remplissez autant de champs que possible, y compris les sections sur lesquelles la source/faisceau a été utilisée pour recueillir des données, les paramètres de collecte et les détails de l’échantillon. Ceux-ci seront enregistrés avec les données et permettront de remplir plus facilement la section « Paramètres de collecte de données » dans les publications futures.
- Entrez le nom de l’échantillon dans l’enregistrer sous forme de boîte. Cliquez sur TRACE.
REMARQUE: Cela a deux effets. Tout d’abord, il va créer un fichier *.sec pour les données. Il s’agit d’un fichier texte unique qui rassemblera toutes les observations expérimentales à partir des fichiers distincts *.dat. En outre, le fichier *.sec contient l’ensemble moyen d’images qui est l’arrière-plan tampon, tous les cadres utilisés dans la moyenne ainsi que les cadres soustraits d’arrière-plan tampon sur l’ensemble de l’expérience SEC-SAXS. Deuxièmement, une parcelle de signal est créée qui trace le nombre d’images par rapport au rapport intégral à l’arrière-plan. Cela montre les images sélectionnées (gris) qui ont été moyennes pour la soustraction tampon. Les points du tampon moyen sont déterminés à partir de l’ensemble de la gamme des données. Toutefois, il est conseillé de choisir manuellement les cadres tampons pour une moyenne car un arrière-plan mal défini peut se produire en raison de colonnes mal calibrées ou sales ou d’encrassementcapillaire 22. - Sélectionnez manuellement les cadres tampons. Cliquez sur Tampons clairs puis resélectionner une zone tampon avec une traînée de clic gauche, sur la courbe de traçabilité. Idéalement, il devrait s’agir d’une région plate avant le volume nul de la colonne SEC d’environ 100 images. Cliquez sur DÉFINIR BUFFER puis Mettre à jour pour recalculer le fichier *.sec qui peut prendre quelques minutes.
- Identifier une région d’intérêt (ROI). Sur la parcelle de signal, sélectionnez la région du pic d’intérêt, avec un clic-traîné gauche.
REMARQUE : Cela peuple trois parcelles dans le panneau droit. Les deux premières parcelles sont reliées à des réticules se déplaçant entre elles, une deuxième parcelle de signal (en haut à droite) ne montre que le roi sélectionné, avec l’intensité de chaque image en bleu et le Rg correspondant de chaque image en rouge et une carte thermique correspondante ci-dessous, montrant les résidus pour chaque image colorée selon l’analyse de corrélation automatique Durbin-Watson. Les régions de haute similitude sont de couleur cyan (Durbin-Watson, d = 2) tandis que les cadres dissemblables suivront blues plus foncé aux roses et enfin aux rouges en fonction de la gravité de la dissemblable (d > 2). La parcelle inférieure est une courbe I versus q soustrayée pour le cadre central sélectionné (également indiqué par une ligne verticale). Les touches fléchées peuvent être utilisées pour naviguer à travers les cadres soustraits. L’intrigue I versus q démontrera la qualité des cadres soustraits de l’expérience SEC. - Sélectionnez les cadres à fusionner. Cliquez sur le réticule dans l’intrigue de la carte thermique pour sélectionner le sous-ensemble de cadres qui seront utilisés pour la fusion. Le réticule identifiera une zone triangulaire de cyan prédominant qui tombe vers le bas à droite du réticule. Utilisez un clic de souris pour définir ces images telles que sélectionnées et mettre en surbrillance les images dans la zone correspondante de l’intrigue signal ci-dessus. Ces cadres devraient idéalement mettre en valeur une région avec un Rg stable.
REMARQUE : Au besoin, zoomez sur la carte thermique avec une traînée de clic gauche et effectuez un zoom arrière avec un balayage du clic gauche vers la droite. - Lorsqu’ils sont satisfaits des images sélectionnées cliquez sur MERGE. Cela fusionnera les cadres soustraits et les présentera dans l’onglet ANALYSE.
- Ouvrez l’onglet SEC. Faites glisser et déposer les fichiers de donnéesréduits ( *.dat) dans la fenêtre " Déposer les données ci-dessous « . Définissez l’annuaire de sortie « Out Dir :: » en cliquant sur le bouton Dir de sortie marqué bleu.
3. Déconvolution des données
- Ouvrez le programme de déconvolution (p. ex., BioXTAS Raw 2.0.0).
- Dans le programme de déconvolution, chargez l’ensemble de données, sous l’onglet Fichiers, dans le panneau de contrôle,utilisez le symbole foldether pour localiser les données ou copier et coller l’emplacement dans la barre d’adresse.
REMARQUE : Assurez-vous que le dossier ne contient que les fichiers bruts *.dat et aucun fichier de données traité ou moyen. - Mettez en surbrillance tous les fichiers *.dat, appuyez sur le bouton Plot Series, un tracé d’intensité intégrée par rapport au numéro d’image sera dessiné dans le « Plot série ».
- Dans le panneau de contrôle sélectionnez l’onglet Série, puis cliquez pour mettre en surbrillance la courbe. Ouvrez la fenêtre pop-up LC Analysis à l’aide du bouton à la base du panneau de commande. Cette fenêtre donne accès à plusieurs options, telles que la sélection de différents types de molécules (protéines ou ARN). Il permet également à l’utilisateur de sélectionner la zone tampon pour le tracé. Dans un premier temps cliquez sur Auto; cela devrait sélectionner une zone tampon appropriée.
REMARQUE : Si cela échoue, peut-être en raison d’une ligne de base instable, alors « Ajouter de la région » pour optimiser la région tampon. Cela peuple la boîte tampon avec une boîte plus petite dans laquelle on peut ajouter manuellement les numéros d’image à utiliser pour le tampon. Sinon, cliquez sur « Choisir » pour donner la possibilité de sélectionner une zone sur l’intrigue. Localisez la zone, cliquez une fois sur la position de départ, déplacez le curseur vers la position suivante et cliquez à nouveau à gauche. Il peut être nécessaire d’ajouter plus d’un emplacement tampon. Cliquez sur Définir tampon et les courbes seront soustraites et le Rg calculé à travers le pic SEC. Si une boîte pop-up apparaît, cliquez sur OK. - Pour démarrer l’analyse des facteurs évolutifs (EPT), cliquez à droite sur le fichier mis en surbrillance au bas de la Panneau puis sélectionnez Ept du menu.
- Vérifiez qu’une fenêtre contexturée s’ouvre qui affiche la décomposition de la valeur unique (SVD) de l’ensemble de données. Dans la boîte de contrôle, cochez la case Cadres d’utilisation de sorte que toute la zone de pointe à déconvolute est couverte dans la parcelle d’intensité. L’intrigue « Valeurs singulières », en haut à droite, montre l’intensité des valeurs singulières (pics/espèces distincts) au-dessus de la ligne de base.
REMARQUE : Le nombre de points présents au-dessus de la ligne de base représente le nombre d’espèces de dispersion présentes. Avec la mise en garde que c’est l’ampleur relative de la valeur singulière à la zone plate / ligne de base qui importe. - Pour aider à valider le nombre de valeurs unique, utilisez le tracé autocorrérelation inférieur. Cela montre les vecteurs de corrélation unique droite et gauche. Cliquez ensuite.
REMARQUE : Il s’agit essentiellement de profils de dispersion ou de concentration pour le vecteur de la solution. Lorsque la taille absolue représente la signification du vecteur. Un composant important aura une autocorrépendation près de 1 (une règle de coupure de base est >0.6\u20120.7). RAW calcule utilement ceci et est montré dans la boîte #Significant SVs, en bas à gauche, bien que vous puissiez changer ceci si nécessaire. S’il existe plusieurs valeurs simples (p. ex. 4+), il peut être nécessaire d’examiner seulement 2 ou 3 des composants, en modifiant la portée des données utilisées. Plus le nombre de composants est faible, plus l’analyse de l’EPT sera facile, mais au prix de l’utilisation de moins de données. Dans une situation complexe, où les vecteurs singuliers gauche et droit, qui devraient être similaires, ne correspondent pas réduire le nombre significatif de SV et diminuer le nombre d’images utilisées jusqu’à ce que les vecteurs singuliers gauche et droit soient similaires. - Vérifiez que l’EPT est calculé en générant des parcelles dans les directions avant et arrière pour chaque vecteur. Ces parcelles montrent quand les composants démarrent (tracé vers l’avant) et sortent (intrigue vers l’arrière) le profil de solution pour les données SEC-SAXS sélectionnées. RAW tente d’identifier ces plages; les modifier à l’aide des flèches à côté des compteurs de sorte que chaque cercle est au début d’un point d’inflexion s’élevant ou tombant à la ligne de base. Cliquez ensuite.
REMARQUE : La dernière étape de l’EPT transforme les vecteurs SVD en courbes de diffusion. Sur la gauche de la fenêtre, les plages précédemment définies sont tracées en haut. Ces plages sont les contraintes pour définir où faire pivoter les vecteurs singuliers dans des courbes de diffusion. Le panneau de droite affiche ces profils de courbe de diffusion correspondants, pour chaque pic séparé. Un tracé pour la concentration de chaque pic, qui devrait être représentatif des profils d’élitution et un tracé pour l’erreur moyenne pondérée chi2. L’intrigue chi2 mesure l’ensemble de données de déconvolution à l’ensemble de données d’origine. Idéalement, ce sera plat, mais des pointes peuvent souvent être vues. - Essayez de réduire ou d’éliminer les pointes en modifiant les commandes de plage des composants,d’abord identifier approximativement quel cadre correspond à la pointe (à partir de chi2 parcelle) puis, dans les contrôles de portée, quel composant contient ce cadre (il pourrait être plus d’un), en utilisant les flèches, déplacer vers le haut ou vers le bas de la plage correspondante.
REMARQUE : Cela devrait produire une réponse, augmentant ou diminuant le pic. Si le cadre de pointe était présent dans plus d’un composant, un peu d’essais et d’erreurs entre chaque composant peut être nécessaire. - Lorsqu’un chi2 minimum a été atteint, effectuez une vérification de validation en cliquant en arrière,la fenêtre précédente semble permettre de vérifier si les modifications apportées ont radicalement modifié les parcelles ept d’origine. S’ils ont toujours l’air valides, cliquez sur Suivant. Cliquez sur Enregistrer les données de l’EPT pour enregistrer les parcelles, puis cliquez sur Done; pour fermer la fenêtre de l’EPT.
REMARQUE : Une deuxième validation est de cliquer sur la case à cochée à côté de chaque plage de composants, à son tour. Ceux-ci fournissent une contrainte de concentration positive à chaque composant et l’éteignement vérifiera si ceux-ci affectent considérablement l’ensemble de données. Si aucun changement n’est observé dans le tracé de concentration, les données sont valides.
- Vérifiez qu’une fenêtre contexturée s’ouvre qui affiche la décomposition de la valeur unique (SVD) de l’ensemble de données. Dans la boîte de contrôle, cochez la case Cadres d’utilisation de sorte que toute la zone de pointe à déconvolute est couverte dans la parcelle d’intensité. L’intrigue « Valeurs singulières », en haut à droite, montre l’intensité des valeurs singulières (pics/espèces distincts) au-dessus de la ligne de base.
- De retour dans la fenêtre RAW, cliquez sur l’onglet Profils dans le panneau de contrôle pour afficher les courbes et dans l’onglet Manipulation du panneau de contrôle,manipuler les courbes plus loin ou enregistrer les courbes sous forme de fichiers *.dat en cliquant à droite sur le fichier et en sélectionnant enregistrer les fichiers sélectionnés à partir du menu pop up. Enregistrez le fichier. Utilisez Scatter IV pour une analyse plus approfondie.
REMARQUE : De plus amples informations et instructions sur la déconvolution et l’EPT BioXTAS RAW se trouvent https://bioxtas-raw.readthedocs.io/en/latest/
4. Déterminer les propriétés SAXS
REMARQUE : Un didacticiel détaillé pour la détermination SAXS se trouve à Bioisis.net. Ici, nous montrons une approche de base étape par étape, mettant en évidence les boutons les plus utiles dans Scatter.
- Dans l’onglet Analyse Scatter, appuyez sur le bouton G pour l’outil manuel d’analyse Guinier, à droite de chaque fichier d’échantillon. L’intrigue qui s’ouvre montre le ln [I (q)] contre q2 dans la boîte supérieure et les résidus correspondants dans la boîte inférieure. Ajoutez ou supprimez des points de sorte que les résidus n’aient pas de fonction « sourire » ou « froncement de sourcils ». Les données sélectionnées dans l’ajustement Guinier ne doivent pas dépasser la limite maximale q x Rg de 1,3.
- Appuyez sur le bouton Kratky normalisé; l’intrigue qui apparaît fournit une évaluation semi-quantitative de l’état structurel de la macromolécule, normalisée pour la masse et la concentration.
NOTE: Le réticule désigne le point Guinier-Kratky à (√3, 1,1)19. Une protéine compacte et sphérique affichera un pic unique avec la valeur maximale au point Guinier-Kratky. Un biopolymère intrinsèquement désordonné ou cylindrique aurait un maximum plus grand que le réticule et ne diminuerait pas. Une protéine qui avait à la fois des domaines pliés et de longues régions allongées non structurées pourrait présenter un maximum accru à travers le réticule, mais montrerait également une tendance à la baisse évidente à q x Rg plus élevé. - Cliquez sur le bouton Vc (Volume de corrélation), qui évoque deux parcelles, l’intensité totale dispersée et une zone intégrée de l’intensité totale dispersée en fonction de q. Les parcelles sont utilisées comme référence rapide pour valider la qualité de la courbe de diffusion.
REMARQUE: L’intensité totale dispersée est sensible à la I(0) et si cela n’a pas été mesuré correctement, alors l’intrigue ne montrera pas une ligne continue. La parcelle de zone intégrée, idéalement, devrait montrer une ligne sigmoïde avec un plateau étendu pour chaque courbe SAXS. S’il y a inadéquation/soustraction tampon, agrégation ou interférence interparticule dans l’échantillon, une pente forte sera observée à des valeurs q plus élevées. - Appuyez sur le bouton Flexibilité pour commencer l’analyse de flexibilité. Cela ouvrira une fenêtre avec quatre panneaux et un curseur en bas. Chaque panneau ouvert montre un complot exploitant une relation pouvoir-droit qui existe entre les biopolymères compacts et allongés/flexibles23. Pour l’utiliser, déplacez le curseur en bas de la boîte de droite à gauche avec le bouton de la souris gauche appuyé. Continuez à avancer lentement vers la gauche jusqu’à ce qu’un plateau dans l’une des parcelles soit atteint.
REMARQUE : Si le plateau est observé dans la parcelle de Porod-Debye, alors l’échantillon est de nature compacte, ce qui devrait être compatible avec un seul pic au point Guinier-Kratky dans une parcelle kratky normalisée. Si le plateau est atteint en premier dans la parcelle Kratky-Debye, l’échantillon est très probablement allongé ou flexible. Si la parcelle SIBYLS est d’abord au plateau, alors l’échantillon contient très probablement des zones de compacité et de flexibilité, une particule aux états mixtes. La théorie de cette relation de flexibilité avec la loi Porod-Debye est abordée de façon exquise dans Rambo, et coll.23 - Cliquez sur Volume. La détermination du volume doit être effectuée immédiatement après l’analyse de flexibilité d’en haut. Lorsqu’il est ouvert après l’analyse de flexibilité, un pop up avec trois graphiques de plus est généré. Dans le coin inférieur gauche, la parcelle Porod-Debye se souvient où l’on a laissé le curseur de la parcelle de flexibilité, montrant la zone plafonnée.
- Pour calculer le volume de la particule, déplacez le début et les points de terminaison à l’aide des boutons fléchés ou tapez dans les boîtes, de sorte que la ligne bleue sur la parcelle s’adapte à la région plafonnée. Pour un résultat impartial, les résidus en haut à droite Porod-Debye exponent la loi du pouvoir ajustement, ne devrait montrer aucun modèle.
- Appuyez sur l’onglet P(r). La distribution de l’espace réel se trouve dans le panneau gauche et la courbe de diffusion de l’échantillon dans le panneau de droite. L’objectif est de créer une représentation de l’espace réel de l’échantillon à partir de la courbe de l’espace réciproque SAXS. Idéalement, la courbe de distribution sera lisse sans vagues présentes et devrait juste embrasser doucement l’axe x.
REMARQUE : La plage q mesurée de l’instrument peut ne pas être entièrement utilisable en raison d’une faible correspondance tampon, d’une agrégation, de dommages causés par les radiations, de temps d’exposition sous-optimaux et de faibles concentrations de particules. L’étape de détermination P(r)déterminera fondamentalement la plage qmin et qmaxutilisable de l’ensemble de données SAXS et c’est cette gamme de données qui est utilisée pour toute modélisation ou ajustement ultérieur.- Cliquez à droite sur le nom de l’échantillon puis cliquez sur Trouver DMAX pour ouvrir une nouvelle fenêtre. Les limites pour le dmax sont pré-définies avec le qmax suggéré (points de données maximaux utilisés), leslimites maximales inférieures et supérieures et un score alpha inférieur et supérieur. Trois modèles peuvent être choisis (L1-norm, Legendre et Moore) et l’utilisation de l’arrière-plan inclus. Laissez-les inchangés en premier lieu.
- Appuyez sur le bouton Démarrer. Une distribution composite est créée dans le panneau gauche avec le niveau dmax et alpha suggéré écrit en dessous. Si cela semble acceptable, fermez la fenêtre et retournez à l’onglet P(r). La parcelle spatiale réciproque aura été recadrée pour correspondre au qmax suggéré.
- Choisissez le modèle Moore, cliquez sur Arrière-plan, puis définissez le niveau alpha et dmax aux valeurs suggérées à partir de la boîte pop-up. Appuyez sur le bouton affiner. Un tracé de validation croisée apparaîtra montrant si des points devaient être rejetés, marqués en rouge. S’il n’y a que quelques points rejetés et que la distribution semble bonne, alors le modèle est bon.
REMARQUE : Le tracé de validation croisée mettra en évidence les régions des données incompatibles avec la distribution déterminée de P(r). Si la région rejetée se trouve principalement dans la région à faible q, c’est-à-dire la région proche de l’axe Y, cela suggère probablement un dmax trop court, une présence d’agrégation ou des oligomers d’ordre supérieur. Il met en évidence une incohérence entre les informations de résolution supérieure et inférieure. Ici, dmax et qmin (augmentation de la valeur de démarrage) doivent être ajustés à l’aide d’une approche manuelle, tâtonnements. De même, si la région rejetée se trouve principalement dans la région à q élevé, cela peut indiquer un problème avec la soustraction de fond ou que le signal est trop faible pour être expliqué de façon significative par la répartition déterminée de P(r).- Dans ce cas, q max doitêtre tronqué (fin décroissante) jusqu’à ce qu’aucune donnée supplémentaire ne soit rejetée. Idéalement, les points rejetés devraient être distribués au hasard et être moins de 5 % des données utilisables. Un q min biendéfini,qmax et " dmax" produira une distribution en douceur où le dmax embrasse l’axe x. Cependant, n’augmentez pas tellement cette valeur qu’elle supprime complètement la région du Guinier. Ce point est facilement trouvé en cochant la case q x l(q) (à gauche du panneau au-dessus de la table). La courbe de diffusion est remplacée par la « parcelle d’intensité totale dispersée », sur cette courbe tous les points avant l’inflexion maximale font partie de la région de Guinier. Après avoir supprimé des points essayer à nouveau d’augmenter / diminuer le « dmax »et ensuite affiner une fois de plus. Si des problèmes persistent, surtout lorsque de nombreux points sont rejetés dès le début de la courbe de validation, cela suggère fortement que les données ne sont pas idéales pour la modélisation structurelle.
- Pour imprimer un rapport, revenez à l’onglet Analyse, cliquez à gauche pour mettre en surbrillance l’échantillon puis cliquez à droite sur le nom de l’échantillon et déplacez-vous pour créer un rapport à partir d’un seul ensemble de données dans le menu. Une boîte de texte s’ouvre pour permettre l’ajout de commentaires. Un document PDF est produit montrant tous les chiffres et valeurs générés.
Résultats
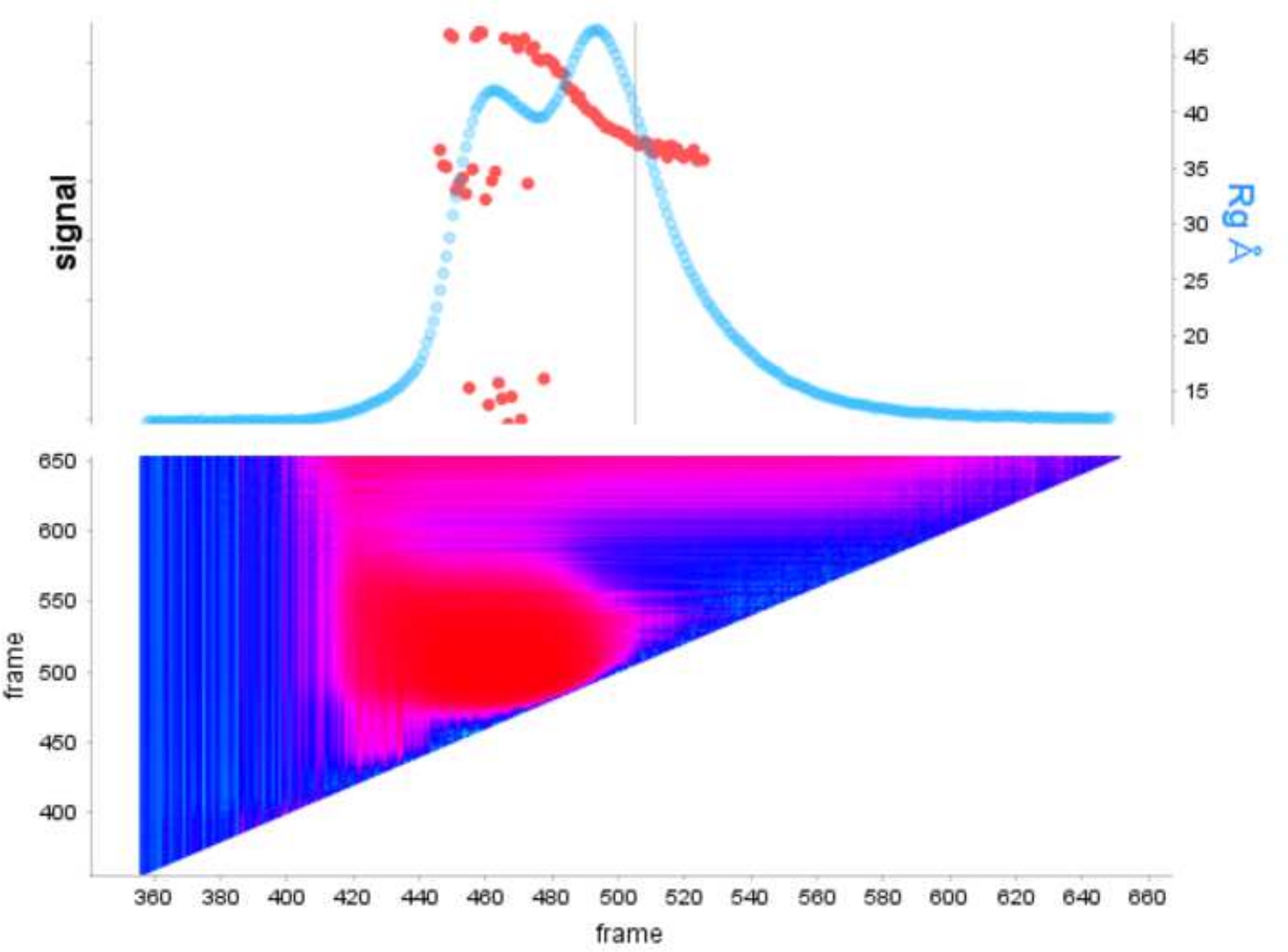
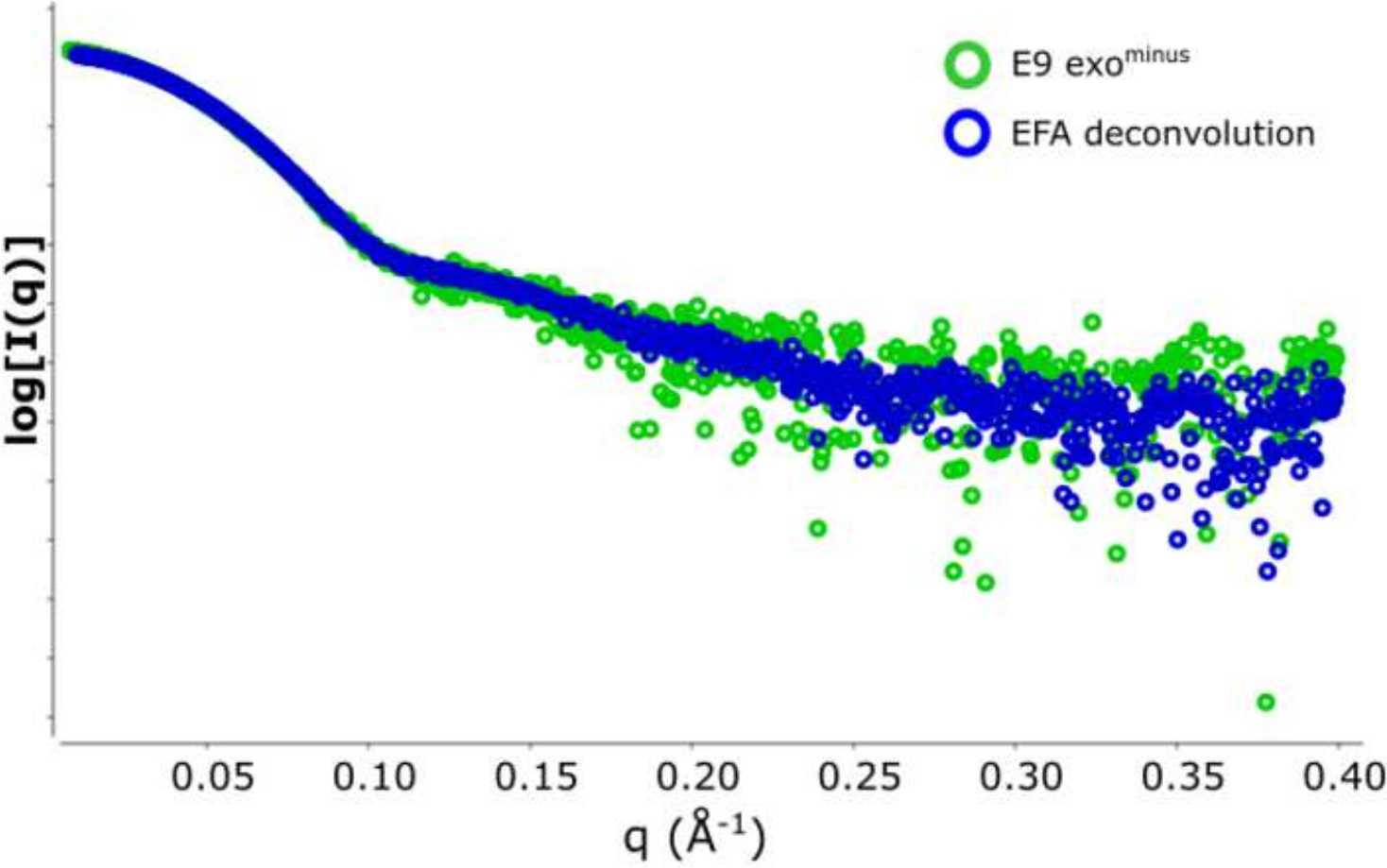
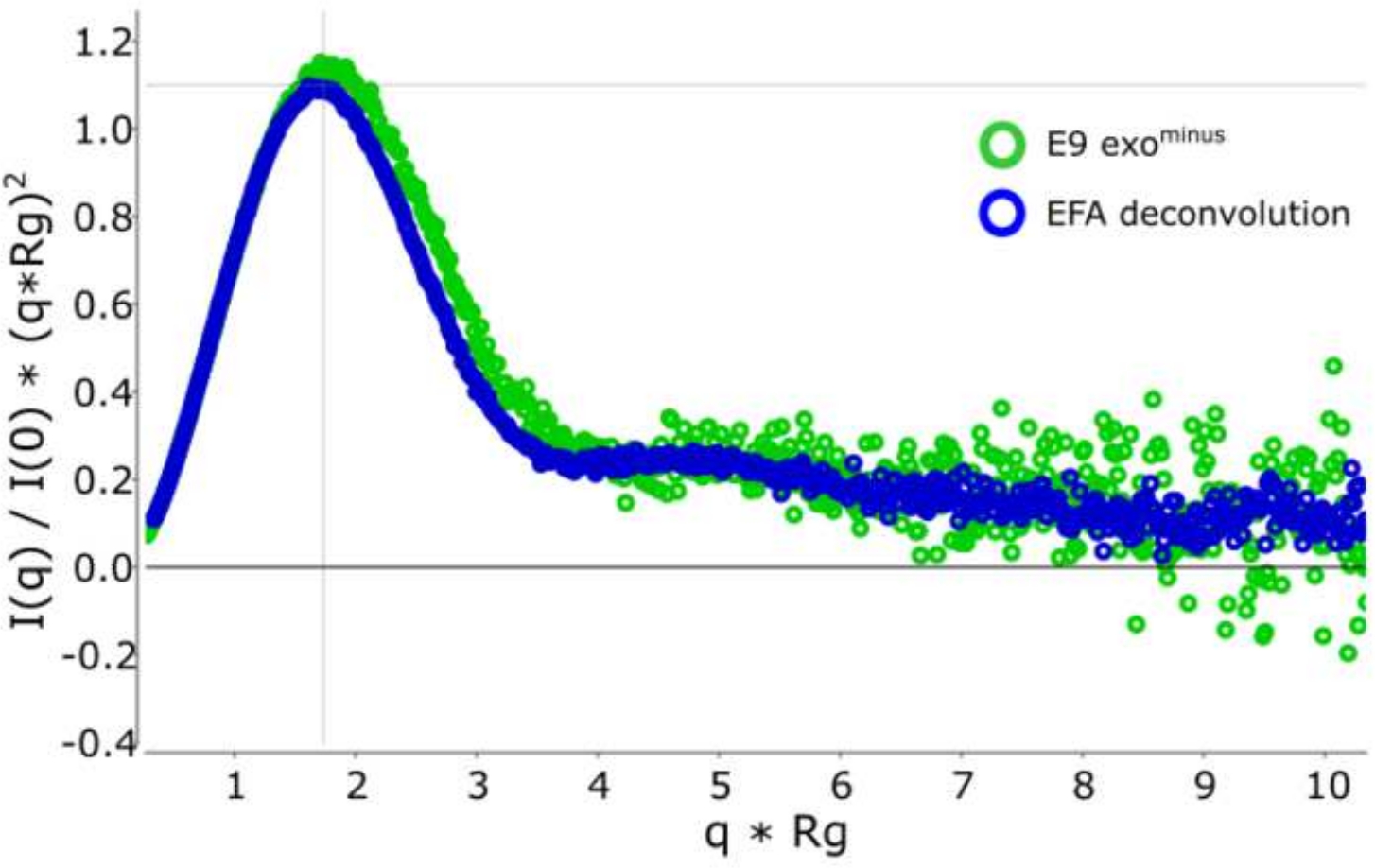
L’avantage d’utiliser la déconvolution par rapport à la sélection classiquedu cadre 13 est d’éliminer l’influence des espèces les unes sur les autres, produisant un signal de diffusion monodisperse. Ceci est également souvent suivi d’un meilleur rapport signal/bruit. Lorsque e9 exomoins est lié à l’ADN et exécuté à l’aide de SEC-SAXS, deux pics sont observés ( Figure1). Le premier, grand pic (environ cadres 420\u2012475) est le complexe E9 exomoins-ADN le second (environ cadres 475\u2012540), l’état non lié (voir Données supplémentaires: Figure 2). Alors que l’approche classique de sélection des cadres fournit un Rg stable du complexe dans le premier pic (voir données supplémentaires: Figure 3), le deuxième pic est clairement fusionné et le Rg à travers la parcelle montre que le deuxième pic d’intérêt n’a pas un Rg stable, en raison de la contamination croisée. Seulement 5 cadres pouvaient être utilisés qui montraient un Rg semi-stable, lorsqu’ils étaient soustraits, ils donnaient un Rg = 36,3 Å(figure 2, vert). Lorsque les pics ont été décontaminés à l’aide de l’EPT, la courbe correspondante du deuxième pic(figure 2, bleu) a été superposée à l’original et a montré une nette diminution du signal au bruit, et un Rg inférieur, 34,1 Å a été enregistré. L’intrigue kratky (Figure 3) montre le complexe avec le pic décontvoluté (bleu) est plus globulaire. Ceci est confirmé par la courbe P(r) (Figure 4) qui donne un dmax 108.5 Å pour la courbe décontvolutée (bleu) tandis que le non-décontrvoluté est plus allongé avec un dmax 120 Å (vert), cela est très probablement dû à l’hétérogénéité résultant de l’exo E9 non liémoins.

Figure 1 : Parcelle de signal de l’exo E9moins seul et avec de l’ADN en complexe.
Le panneau supérieur montre une parcelle du rapport intégral à l’arrière-plan pour chaque image d’une course SEC-SAXS (bleu clair). Les points rouges montrent le Rg à chaque image au-dessus du pic. Le panneau inférieur montre la carte thermique correspondante montrant les résidus pour chaque image colorée selon l’analyse de corrélation automatique Durbin-Watson, les régions de haute similitude sont de couleur cyan tandis que les cadres dissemblables suivent des bleus plus foncés aux roses et enfin au rouge en fonction de la gravité de la dissemblable. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Parcelle d’intensité par rapport au vecteur de diffusion.
Une superposition des données SAXS soustraites forme l’exo E9moins . Dans le vert 5 cadres (cadre 517\u2012522) en moyenne et soustrait d’une zone de Rg semi-stable et en bleu la courbe de diffusion représentative dérivée de la déconvolution de l’EPT du pic SEC-SAXS. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Courbe de Kratky sans dimension.
La superposition de la courbe kratky décontvolutée (bleue) et non décontvolutée (verte) montrant E9 exomoins est globulaire. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Courbe P(r).
La superposition des courbes décont alambiquées (bleues) et non décont alambiquées (vertes) pour l’exo E9moins. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
Données supplémentaires. S’il vous plaît cliquez ici pour télécharger ce fichier
Discussion
Il est souhaité d’avoir un échantillon monodisperse avant de commencer une expérience SAXS, mais en réalité, de nombreuses collections de données ne satisfont pas à cela et doivent être améliorées en combinant la mesure avec la chromatographie inline- SEC dans la plupart des cas. Cependant, même le manque de temps entre la purification et la monodispersité d’acquisition de données de l’échantillon n’est pas garanti. Le plus souvent, cela s’applique aux expériences où les composants sont trop proches en taille ou dans leurs propriétés physiques pour être séparés ou sont sujets à une dynamique rapide. Ici, nous avons fourni un protocole combinant décomposition de valeur unique avec l’analyse des facteurs en évolution pour supprimer l’influence de DNAbound E9 exomoins de son profil de diffusion monodisperse que nous avons ensuite été en mesure d’analyser avec le paquet SAXS Scatter IV.
SVD avec EFA de données SEC-SAXS sont des méthodes très puissantes développées pour déconvoluter les données SAXS et améliorer l’analyse, mais ils ont des limites. Ils exigent que le bruit ou la dérive dans la ligne de base tampon du SEC-SAXS soit maintenu au minimum. Cela peut impliquer un équilibre supplémentaire de la colonne (mieux vaut utiliser plus de 3 volumes de colonnes, selon le tampon) avant le chargement de l’échantillon. Toutefois, l’étape la plus critique est le choix du nombre de valeurs singulières et de la gamme de données utilisées, car cela affectera considérablement l’exactitude de la déconvolution. C’est pour cette raison que les résultats ne doivent pas être pris par eux-mêmes, mais analysés plus en détail à l’aide de techniques telles que l’ultracentrifugation analytique (AUC) ou la diffusion de la lumière laser à angle multiples (MALLS) pour l’interprétation biologique.
Scatter IV est un nouveau logiciel, gratuit pour la recherche et l’utilisation industrielle avec une interface utilisateur intuitive qui permet même aux non-experts d’analyser leurs données. Scatter IV a plusieurs nouvelles fonctionnalités qui aident à améliorer l’analyse des données SEC-SAXS, telles que la carte thermique liée à la parcelle de signal, permettant une plus grande précision avec le choix de la sélection du cadre. Dans l’analyse des données primaires, l’analyse guinier peak et le tracé de validation croisée associé à l’analyse P(r) offrent une capacité intégrée de dépannage dans le logiciel.
Il convient de mentionner que de nombreux autres programmes peuvent être utilisés pour l’analyse des données primaires; ceux-ci contiennent les mêmes caractéristiques de base et sont également mis à jour régulièrement tels que BioXTAS RAW17 ATSAS paquet24 et US-SOMO15 pour n’en nommer que quelques-uns.
Mais quel que soit le paquet SAXS utilisé pour l’analyse, les principales limitations sont communes : la préparation de l’échantillon, avant la collecte et l’analyse. Dans l’exemple E9 exomoins montré, il est clair de voir l’amélioration du rapport signal/bruit et avec une réduction du Rg le dmax associé à un échantillon monodisperse. Cela facilitera grandement le traitement ultérieur des données telles que le montage ou la modélisation avec des structures à haute résolution connues.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous reconnaissons le soutien financier au projet de la subvention de Français REPLIPOX ANR-13-BSV8-0014 et par les subventions de recherche du Service de Santé des Armées et de la Délégation Générale pour l’Armement. Nous remercions l’ESRF pour le temps de faisceau SAXS. Ce travail a utilisé les plates-formes du centre Grenoble Instruct-ERIC (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) dans le cadre du Partenariat grenoblois pour la biologie structurale (PSB), soutenu par frisbi (ANR-10-INBS-05-02) et GRAL, financé au sein de l’Ecole supérieure grenoble Alpes (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). IBS reconnaît son intégration à l’Institut interdisciplinaire de recherche de Grenoble (IRIG, CEA). Nous remercions Wim P. Burmeister et le Pèreédéric Iseni pour leur soutien financier et scientifique et nous remercions également le Dr Jesse Hopkins de BioCAT à l’APS pour son aide et pour le développement de BioXTAS RAW.
matériels
| Name | Company | Catalog Number | Comments |
| Beamline control software BsXCuBE | ESRF | Pernot et al. (2013), J. Synchrotron Rad. 20, 660-664 | local development |
| BioXTAS Raw 1.2.3. | MacCHESS | http://bioxtas-raw.readthedocs.io/en/latest/index.html | First developed in 2008 by Soren Skou as part of the biological x-ray total analysis system (BioXTAS) project. Since then it has been extensively developed, with recent work being done by Jesse B. Hopkins |
| HPLC program LabSolutions | Shimadzu | n.a. | |
| ISPyB | ESRF | De Maria Antolinos et al. (2015). Acta Cryst. D71, 76-85. | local development |
| NaCl | VWR Chemicals (BDH Prolabo) | 27808.297 | |
| Scatter | Diamond Light Source Ltd | http://www.bioisis.net/tutorial/9 | Supported by SIBYLS beamline (ALS berkeley, Ca) and Bruker Cororation (Karlsruhe, Germany) |
| Superdex 200 Increase 5/150 GL column | GE Healthcare | 28990945 | SEC-SAXS column used |
| Tris base | Euromedex | 26-128-3094-B |
Références
- Pelikan, M., Hura, G., Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. General Physiology and Biophysics. 28 (2), 174-189 (2009).
- Brosey, C. A., Tainer, J. A. Evolving SAXS versatility: solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Current Opinion in Structural Biology. 58, 197-213 (2019).
- Gräwert, M., Svergun, D. A beginner's guide to solution small-angle X-ray scattering (SAXS). The Biochemist. 42 (1), 36-42 (2020).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly reviews of biophysics. 40 (03), 191-285 (2007).
- Meisburger, S. P., et al. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. Journal of the American Chemical Society. 138 (20), 6506-6516 (2016).
- Brennich, M. E., Round, A. R., Hutin, S. Online Size-exclusion and Ion-exchange Chromatography on a SAXS Beamline. Journal of Visualized Experiments. (119), e54861 (2017).
- Watanabe, Y., Inoko, Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. Journal of Chromatography A. 1216 (44), 7461-7465 (2009).
- Graewert, M. A., et al. Automated Pipeline for Purification, Biophysical and X-Ray Analysis of Biomacromolecular Solutions. Scientific reports. 5, (2015).
- David, G., Pérez, J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. Journal of applied crystallography. 42 (5), 892-900 (2009).
- Ryan, T. M., et al. An optimized SEC-SAXS system enabling high X-ray dose for rapid SAXS assessment with correlated UV measurements for biomolecular structure analysis. Journal of Applied Crystallography. 51 (1), 97-111 (2018).
- Gampp, H., Maeder, M., Meyer, C. J., Zuberbühler, A. D. Calculation of equilibrium constants from multiwavelength spectroscopic data-III: Model-free analysis of spectrophotometric and ESR titrations. Talanta. 32 (12), 1133-1139 (1985).
- Maeder, M., Neuhold, Y. M. . Practical Data Analysis in Chemistry. , (2007).
- Tarbouriech, N., et al. The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding. Nature Communications. 8 (1), (2017).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), (2016).
- Brookes, E., Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. European Biophysics Journal. 47 (7), 855-864 (2018).
- Malaby, A. W., et al. Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. Journal of Applied Crystallography. 48 (4), 1102-1113 (2015).
- Hopkins, J. B., Gillilan, R. E., Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. Journal of Applied Crystallography. 50 (5), 1545-1553 (2017).
- Maeder, M. Evolving factor analysis for the resolution of overlapping chromatographic peaks. Analytical Chemistry. 59 (3), 527-530 (1987).
- Durand, D., et al. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. Journal of Structural Biology. 169 (1), 45-53 (2010).
- De Maria Antolinos, A., et al. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallographica Section D. 71 (1), 76-85 (2015).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), 203-212 (2016).
- Kirby, N., et al. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallographica Section D Structural Biology. 72 (12), 1254-1266 (2016).
- Rambo, R. P., Tainer, J. A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 95 (8), 559-571 (2011).
- Franke, D., et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 50 (4), 1212-1225 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.