
Method Article
EFAデコンボリューションとスキャッタによるSEC-SAXSデータの分析
* これらの著者は同等に貢献しました
要約
SEC-BioSAXSの生体高分子の測定は、高分子とその複合体の溶液構造を決定するための標準的なアプローチです。ここでは、一般的に発生する2種類のSECトレース(完全に解決されたピークと部分的に解決されたピークを持つクロマトグラム)からのSEC-BioSAXSデータを分析します。散布図とBioXTAS RAWによる解析とデコンボリューションの実演を行う。
要約
BioSAXSは、分子構造と構造生物学で使用される一般的な技術であり、溶液構造、粒子径と形状、表面対体積比、高分子および高分子複合体の立体構造変化を決定します。構造モデリング用の高品質の SAXS データセットは、単分散型の均一なサンプルからのものでなければならず、多くの場合、インラインクロマトグラフィーと即時 SAXS 測定の組み合わせによってのみ到達します。最も一般的には、サイズ排除クロマトグラフィーは、サンプルを分離し、単一のタンパク質種の十分に解決されたクロマトグラフィーピークからSAXS測定を行うことを可能にする目的の粒子から汚染物質および凝集を除外するために使用されます。それでも、場合によっては、インライン精製でさえ、複数の成分が互いに近すぎるか、または結合によって誘起される形状の変化が溶出時間を知覚する形の変化のために、単分散サンプルの保証ではない。これらの場合、混合物のSAXSデータをデコンボルテージして、個々の成分の理想化されたSAXS曲線を得ることができる可能性があります。ここでは、これを実現する方法を示し、SEC-SAXSデータの実用的な分析が理想的で困難なサンプルで行われます。具体的には、バクシニアE9DNAポリメラーゼエキソヌクレアーゼマイナス変異体のSEC-SAXS分析を示す。
概要
生体高分子は、最もよい光学顕微鏡でも見るには小さすぎます。それらの構造を決定する現在の方法は、一般的にタンパク質を結晶化するか、同時に同一分子の膨大な数の測定を含みます。結晶学は原子レベルに関する情報を提供するが、ほとんどの高分子が細胞内の結晶形で提示されないことを考えると、人工的なサンプル環境を表す。ここ数年、クライオ電子顕微鏡は、大きな高分子/高分子錯体の同様の高解像度構造を提供したが、サンプルは生理学的状態に近いが、依然として凍結し、したがって不動で静的である。バイオ・小角X線散乱(BioSAXS)は、生物学に関連する条件で、高分子の構造測定を提供します。この状態は、ナノメートルスケールで決定された低解像度の3次元形状として視覚化でき、高分子の立体構造空間全体を溶液中に捉えることができます。BioSAXS実験は、オリゴマー状態、ドメインおよび複雑な配置、ならびにドメイン1、2、3間の柔軟性を効率的に評価する。この方法は正確で、ほとんどが非破壊的であり、通常は最小限のサンプル調製と時間しか必要としない。しかし、データを最もよく解釈するには、サンプルを単分散にする必要があります。これは難しいことです。生体分子は、汚染の影響を受けやすい、精製不良および凝集性、例えば凍結解凍4.インラインクロマトグラフィーの開発と SAXS 測定の即時測定により、これらの効果を軽減できます。サイズ排除クロマトグラフィーはサンプルをサイズによって分離するため、ほとんどの汚染物質と凝集体を除外します5,6,7,8,9,10.しかし、場合によっては、SEC-SAXSでさえ、単分散サンプルを生成するのに十分でない場合があり、混合物はサイズが近すぎる成分またはその物理的性質またはそれらの高速ダイナミクスがSEC UVトレースのピークを重ねることにつながる可能性があるためです。これらの場合、取得したSAXSデータのソフトウェアベースのデコンボリューションステップは、個々のコンポーネント5、11、12の理想化されたSAXS曲線につながる可能性があります。一例として、プロトコルセクション2において、DNAとの複合体におけるワクシニアE9DNAポリメラーゼエキソヌクレアーゼ-変異体(E9exoマイナス)の標準的なSEC-SAXS分析を示す。ワクシニアは、ポクスウイルス科のモデル生物を表し、いくつかの病原体、例えばヒト天然痘ウイルスを含むファミリーである。ポリメラーゼは生化学的アプローチでDNAと緊密に結合することが示され、この複合体の構造は最近X線結晶学13によって解された。
ほとんどのシンクロトロン施設は、データの正規化と統合を行い、一連の減算されていないフレームを生成する自動化されたデータ処理パイプラインを提供します。しかし、この原稿に記載されているアプローチは、SEC-SAXSが実行されていれば、ラボソースでも使用できます。さらに、放射線損傷フレームを拒絶し、バッファ減算14を実行する追加の自動化が利用可能である可能性がある。前処理データに対してプライマリデータ分析を実行し、利用可能なデータを最大限に活用する方法をセクション2で説明します。
セクション3では、SEC-SAXSデータをデコンボルドし、曲線を効率的に解析する方法を示します。ガウスピークデコンボリューションのようないくつかのデコンボリューション方法があるが、US-SOMO15およびギニエ最適化された最大限の尤度法で実施され、DELAソフトウェア16で実施され、これらは一般的にピーク形状12のモデルを必要とする。我々が調査している個々のピークの有限サイズは、ピーク形状または散乱プロファイル5、11に依存することなく、重なり合うピークをデコンボリューションする特異値分解(SVD)の強化された形態として、進化因子分析(EFA)を使用することを可能にする。SAXS 固有の実装は BioXTAS RAW17にあります。EFAは、2Dダイオードアレイデータが保持時間および波長データ18に対する吸光度からマトリックスを形成することを可能にした場合にクロマトグラフィーデータに最初に使用された。EFAが優れているのは、単数形値の進化する性格、新しいコンポーネントの出現に伴ってどのように変化するかを、取得10に固有の順序があるという警告を付ける点です。幸いにも、SEC-SAXS データは、組織化された 2D データ配列に必要なすべての順序付き取得データを提供し、EFA 技術に適しています。
セクション 4 では、SAXS 曲線を引いたバッファバックグラウンドからモデル非依存 SAXS 解析の基本を説明します。モデルに依存しない解析では、パーティクルのジャイロ半径(Rg)、相関体積(Vc)、多孔質体積(Vp)、およびポロドデバイ指数(PE)が決定されます。この解析では、無次元のクラッキープロット2、4、19を介して、粒子の熱力学的状態を、コンパクトさまたは柔軟性の観点から半定量的に評価することができます。
最後に、SAXSデータは相互空間単位で測定され、SAXSデータを実空間に変換してペア距離P(r)分布関数を回復する方法を示します。P(r)分布は、パーティクル内で見つかったすべての距離のセットで、パーティクルの最大次元 dmaxが含まれます。これは熱力学的測定であるため、P(r)分布は、パーティクルの立体空間が占める物理的空間を表します。SAXS データセットを適切に分析することで、結晶学やクライオ EM の高解像度情報を補完するソリューション状態の洞察を提供できます。
プロトコル
1. タンパク質発現、精製およびSEC-SAXS測定は、公表されたプロトコル13に基づいています
- インライン SEC-SAXS データ収集プロトコル (Brennich ら6)に簡単に従ってください。
- SEC 実行バッファーの少なくとも 2 つの列ボリューム (20 mM Tris-HCl、pH 7.5、100 mM NaCl) を使用して SEC 列を平衡化します。
- E9エキソマイナス のサンプル50 μLを8\u201210 mg/mLで調製し、部分的なdsDNAのモル過剰が20%(TCAGGAAGAACAGCGGTTTAGCCとGGCTAAACCACCGCTTATCTTTTCTT)E9 exoマイナスは 、12 ± 6 nM の KD と結合します ( 補足データを参照)。
- このミックスの50 μLをSECカラム(S200増加)にインラインで注入し、SAXS測定用のフローセルを0.3 mL/minで測定します。
- 1 s 露出で 1000 フレームを収集します。
注意: BioSAXSビームラインBM29では、欧州シンクロトロン施設(ESRF)で個々のフレームがEDNAフレームワーク14内で自動的かつ独立して処理されます。データ収集後、ISPyBデータベース20 を開き、 データ取得 タブの下で [Go] ボタンを押して、データセットと自動分析21の結果にアクセスします。
- データをダウンロードします。
2. 一次データ分析
- Java ベースのプログラムを開く スキャッタIV (を参照してください。 資料表)をクリックし、サイズ除外クロマトグラフィー(SEC)データのバックグラウンド減算を実行します。
- 実験の詳細を編集し、[ 詳細を編集] ボタンを使用して、できるだけ多くのフィールドに記入し、これらは、データを収集するために使用されたソース/ビームライン、収集パラメータとサンプルの詳細にセクションが含まれています。これらはデータと共に保存され、今後のパブリケーションで「データ収集パラメータ」セクションに簡単にデータを入力できるようになります。
- [ 名前を付けて保存 ] ボックスにサンプル名を入力します。 [トレース] をクリックします。
注: これには 2 つの効果があります。まず、データの *.sec ファイルを作成します。これは、個別の *.dat ファイルからすべての実験観測を照合する単一のテキスト ファイルです。さらに 、*.sec ファイルには、バッファバックグラウンドであるフレームの平均セット、平均化で使用されるすべてのフレーム、およびSEC-SAXS実験全体でのバッファバックグラウンド減算フレームが含まれます。次に、フレーム数対積分比を背景にプロットするシグナルプロットが作成されます。バッファ減算で平均化された選択したフレーム(灰色)が表示されます。平均バッファのポイントは、データの全範囲から決定されます。しかし、不十分に平衡化またはダーティカラムまたは毛細血管汚れ22のために、不十分に定義された背景として平均化するためのバッファフレームを手動で選択することをお勧めします。 - バッファー フレームを手動で選択します。[ バッファのクリア ]をクリックし、トレースカーブ上で左クリック/ドラッグでバッファ領域を再選択します。理想的には、これは約100フレームのSECカラムのボイド体積の前のフラットな領域でなければなりません。[ バッファの設定 ] をクリックし、[ 更新] をクリックして、数分かかる可能性がある *.sec ファイルを再計算します。
- 関心のある地域 (ROI) を特定します。シグナルプロットで、左クリック&ドラッグで、対象のピークの領域を選択します。
注: 右側のパネルに 3 つのプロットが表示されます。上の 2 つのプロットは、それらの間を移動する十字線とリンクされ、2 番目の信号プロット (右上) は選択した ROI のみを示し、各フレームの強度は青、対応する Rg は赤、対応するヒート マップはダービン-Watson 自動相関分析に従って色付けされた各フレームの残差を示します。類似性の高い領域はシアン(ダービン・ワトソン、d = 2)を着色し、異なるフレームは暗い青色からピンク色に続き、最後は異なる類似性の重症度に応じて赤に続きます(d> 2)。下のプロットは、中央で選択されたフレームの(垂直線でも示される)の、引いた I 対 q 曲線です。矢印キーを使用して、減算されたフレーム間を移動できます。 I 対 q プロットは、SEC 実験から減算されたフレームの品質を示します。 - マージするフレームを選択します。ヒート マップ プロットのクロスヘアをクリックして、マージに使用するフレームのサブセットを選択します。十字線は、主に十字の右下に落ちる主にシアンの三角形の領域を識別します。これらのフレームを選択済みに設定し、上の信号プロットの対応する領域でフレームをハイライトするには、マウスクリックを使用します。これらのフレームは、理想的には安定したRgを有する領域を強調する必要があります。
注: 必要に応じて、左クリックでヒート マップを拡大し、右に左クリックでズーム アウトします。 - 選択したフレームに問題がなければ 、[MERGE]をクリックします。これにより、減算されたフレームがマージされ、[ 分析 ]タブに表示されます。
3. データのデコンボリューション
- デコンボリューションプログラムを開きます(例えば、BioXTAS Raw 2.0.0)。
- デコンボリューション プログラムで、データセットを読み込み、[ ファイル ] タブの [コントロール パネル]で、データを見つけるか、場所をコピーしてアドレス バーに貼り付けます。
注: フォルダーに含まれているのは、生の *.dat ファイルのみで、処理済みまたは平均データ ファイルがないことを確認します。 - すべての *.dat ファイルをハイライトし、 プロットシリーズ ボタンを押すと、統合強度対フレーム番号のプロットが「シリーズプロット」に描画されます。
- コントロール パネル で[ 系列 ]タブを選択し、クリックして曲線をハイライト表示します。コントロールパネルの下部にあるボタンを使用して 、LC分析 ポップアップウィンドウを開きます。このウィンドウでは、さまざまな種類の分子 (タンパク質または RNA) を選択するなど、いくつかのオプションにアクセスできます。また、ユーザーはプロットのバッファー領域を選択することもできます。最初のインスタンスで [自動 ]をクリックし、適切なバッファ領域を選択します。
注: これが失敗した場合、おそらくベースラインが不安定なために、バッファ領域を最適化するために「領域を追加」。これにより、[バッファー] ボックスに小さなボックスが表示され、バッファに使用するフレーム番号を手動で追加できます。または、「ピック」をクリックして、プロット上の領域を選択するオプションを指定します。領域を見つけ、開始位置を左クリックし、カーソルを次の位置に移動し、もう一度左クリックします。複数のバッファ位置を追加する必要がある場合があります。[ バッファを設定 ]をクリックすると、カーブが減算され、SECピークを越えてRgが計算されます。ポップアップ ボックスが表示されたら 、[OK]をクリックします。 - 進化因子分析(EFA)を開始するには、下部にあるハイライト表示されたファイルを右クリックします。 コントロール パネル を選択し、 Efa メニューから。
- データ・セットの単一値分解 (SVD) を示すポップアップ・ウィンドウが開くことを確認します。コントロール ボックスで、[フレームを 使用 ]ボックスをオンにして、デコンボルテージするピーク領域全体が強度プロットで覆われるようにします。右上の「単数値」プロットは、ベースライン上の単数形値(別々のピーク/種)の強度を示します。
注: ベースラインの上に存在するポイントの数は、存在する散乱種の数を表します。重要なのは、平坦な領域/ベースラインに対する単数形値の相対的な大きさであるという注意点です。 - 単一値の数を検証するには、最下部の 自己相関 プロットを使用します。これは、右と左の単一相関ベクトルを示しています。[ 次へ] をクリックします。
注: これらは、本質的に、解析内のベクトルの散乱または濃度プロファイルを表します。絶対サイズはベクトルの有意性を表します。重要なコンポーネントは、1 の近くに自己相関があります (経験のカットオフの法則は >0.6\u20120.7)。RAW はこれを有益に計算し、必要に応じて変更できますが、左下の#Significant SVボックスに表示されます。単一値が複数ある場合(例えば4+など)、使用されるデータの範囲を変更して、コンポーネントの2つまたは3つだけを見る必要があるかもしれません。コンポーネントの数が少ないほど、EFA分析は簡単になりますが、使用するデータが少なくなります。複雑な状況では、左右の単一ベクトル(類似)が一致しない場合、有意なSV数を減らし、左右の単一ベクトルが類似するまで使用されるフレーム数を減らします。 - EFA が各ベクトルの前方方向と後方方向のプロットを生成して計算されることを確認します。これらのプロットは、選択したSEC-SAXSデータの解析プロファイルを、コンポーネントが開始(前方プロット)および終了(逆方向プロット)したときに表示されます。RAW は、これらの範囲を識別しようとします。カウンタの横にある矢印を使用してこれらを変更し、各円がベースラインから上昇またはベースラインに落ちる変曲点の始点になるようにします。[ 次へ] をクリックします。
注: EFA の最後の段階では、SVD ベクトルがスキャッタリング カーブに戻ります。ウィンドウの左側には、前に定義した範囲が上部にプロットされます。これらの範囲は、単数形ベクトルを回転させて散乱カーブに戻す場所を定義する拘束です。右手パネルは、それぞれの分離されたピークに対応する散乱曲線プロファイルを示しています。各ピークの濃度のプロット、それは溶出プロファイルの代表であり、平均誤差加重chi2のプロットである必要があります。chi2 プロットは、デコンボリューション・データ・セットを元のデータ・セットに測定しています。理想的には、これは平坦ですが、スパイクがしばしば見られます。 - コンポーネント範囲コントロールを変更してスパイクを減らすか除去しようとし、まずどのフレームがスパイクに対応するか(chi2プロットから)、次に、どのコンポーネントがこのフレームを含む(複数の可能性がある)、矢印を使用して対応する範囲を上下に移動します。
注: これは、スパイクを増減する応答を生成する必要があります。スパイクフレームが複数のコンポーネントに存在する場合は、各コンポーネント間の試行錯誤が必要になる場合があります。 - 最小の chi2 が達成されたら、 戻るをクリックして検証チェックを実行し、前のウィンドウが表示され、変更が元の EFA プロットを大幅に変更したかどうかを確認できます。それでも有効な場合は、[ 次へ] をクリックします。 [EFA データの保存 ] をクリックしてプロットを保存し、[ 完了] をクリックします。をクリックして EFA ウィンドウを閉じます。
注: 2 つ目の検証では、各コンポーネント範囲の横にあるチェックボックスをオフにします。これらの値は、各成分に正の濃度制約を与え、オフにすると、これらの値がデータ セットに大きく影響するかどうかがチェックされます。濃度プロットに変化が見られない場合、データは有効です。
- データ・セットの単一値分解 (SVD) を示すポップアップ・ウィンドウが開くことを確認します。コントロール ボックスで、[フレームを 使用 ]ボックスをオンにして、デコンボルテージするピーク領域全体が強度プロットで覆われるようにします。右上の「単数値」プロットは、ベースライン上の単数形値(別々のピーク/種)の強度を示します。
- RAW ウィンドウに戻り、コントロール パネルの [プロファイル] タブをクリックして曲線を表示し、コントロール パネルの [操作] タブで、ファイルを右クリックしてメニュー ポップアップから選択したファイルを保存するを選択して、さらにカーブを操作するか、*.datファイルとして保存します。ファイルを保存します。さらに分析するために散布図 IV を使用します。
注: デコンボリューションと EFA BioXTAS RAW に関する詳細と説明は、https://bioxtas-raw.readthedocs.io/en/latest/
4. SAXS プロパティの決定
注: SAXS の決定に関する詳細なチュートリアルは、Bioisis.netにあります。ここでは、スキャッタで最も便利なボタンを強調する基本的なステップバイステップアプローチを示します。
- [散布図 分析 ]タブで、各サンプルファイルの右側にある手動Guinier解析ツールの G ボタンを押します。開いたプロットは、上部ボックスにln[I(q)] 対 q2、 下部のボックスに対応する残差を示します。残差に「笑顔」または「眉」機能がないようにポイントを追加または削除します。ギニエフィットで選択したデータは、最大 q x Rg 制限の 1.3 を超えないようにしてください。
- 正規化されたクラッキーボタンを押します。ポップアップするプロットは、質量と濃度のために正規化された高分子の構造状態の半定量的評価を提供します。
注意:十字線は(√3、1.1)19でギニエ・クラツキー点を指定します。コンパクトな球状のタンパク質は、ギニエ・クラツキー点で最大値を持つ単一のピークを示します。本質的に乱れたまたは円筒形の生体高分子は、十字線よりも最大大きく、減少しないであろう。折り畳まれたドメインと長い細長い非構造化領域の両方を持つタンパク質は、十字線を通して最大が増加する可能性がありますが、q x Rgの高い場合にも明らかな減少傾向を示します。 - Vcボタン (相関の体積) をクリックすると、2 つのプロット、総散乱強度、および総散乱強度の積分領域が q の関数として表示されます。プロットは、散乱曲線の品質を検証するためのクイック リファレンスとして使用されます。
注: 総散乱強度は I(0) に敏感であり、これが正しく測定されていない場合、プロットは連続した線を表示しません。積分面積プロットは、理想的には、各SAXS曲線に対して拡張された高原を有するシグモイド線を示すべきである。バッファーの不一致/減算がある場合、サンプルの凝集または粒子間干渉は、より高い q 値で鋭い傾きを観察します。 - 柔軟性の分析を開始するには、[ 柔軟性 ] ボタンを押します。ウィンドウが開き、ウィンドウの下部に 4 つのパネルとスライダーが表示されます。開かれた各パネルは、コンパクトと細長い/柔軟なバイオポリマー23の間に存在するパワー・ロー関係を利用するプロットを示しています。使用するには、ボックスの下部にあるスライダーを右から左へ動かし、マウスの左ボタンを押します。プロットの1つに高原が到達するまで、ゆっくりと左に移動し続けます。
注: プラトーが Porod-Debye プロットに見られる場合、サンプルは本質的にコンパクトであり、正規化されたクラツキープロットのギニエ・クラツキー点の単一のピークと一致するはずです。プラトーがクラッキー・デバイプロットで最初に到達した場合、サンプルは細長または柔軟である可能性が最も高いです。SIBYLSプロットが最初に高原に対するものである場合、サンプルにはコンパクトさと柔軟性の両方の領域、混合状態の粒子が含まれている可能性が最も高い。この柔軟性とPorod-Debye法との関係の理論は、ランボーで絶妙に取り上げ,他 - [ボリューム] をクリックします。ボリュームの決定は、上記からの柔軟性分析の直後に行う必要があります。柔軟性分析後に開くと、さらに3つのグラフを含むポップアップが生成されます。下の左隅のPorod-Debye プロットは、柔軟性プロットからスライダーを離れ、高原領域を示す場所を覚えています。
- パーティクルのボリュームを計算するには、矢印ボタンを使用して開始と終点を移動するか、ボックスに入力して、プロット上の青い線が高原領域に合うようにします。公平な結果の場合、右上のポロート・デバイ指数の指数力則適合の残差は、パターンを示さないはずです。
- P(r)タブを押します。実際の空間分布は、左側のパネルと右側のパネルのサンプルの散乱曲線にあります。目的は、逆空間 SAXS 曲線からサンプルの実空間表現を作成することです。理想的には、分布曲線は波が存在しない滑らかで、x軸に優しくキスする必要があります。
注:測定されたQ範囲は、緩衝性の一致、凝集、放射損傷、最適でない露光時間、粒子濃度の低さにより、完全には使用できない場合があります。P(r)決定ステップは、SAXS データセットの使用できる qmin および qmax 範囲を基本的に決定し、後続のモデリングまたは適合に使用されるこの範囲のデータである必要があります。- サンプル名を右クリックし 、[DMAX の検索 ] をクリックして新しいウィンドウを開きます。dmax の制限は、推奨される qmax (最大データポイントが使用される)、下限と上限 dの上限 、およびアルファスコアの下限と上限があらかじめ設定されています。3つのモデル(L1ノルム、レジェンドレとムーア)と背景の使用が含まれて選択することができます。最初のインスタンスでは、これらの値は変更しません。
- [スタート]ボタンを押します。左パネルに複合分布が作成され、その下に推奨される dmaxおよび alpha レベルが書き込まれます。これが許容できると思える場合は、ウィンドウを閉じて P(r) タブに戻ります。相互空間プロットは、提案されたqmaxに合わせてトリミングされます。
- モデルムーアを選択し、[背景]をクリックし、アルファレベルとdmaxをポップアップボックスから提案された値に設定します。絞り込みボタンを押します。クロス検証プロットがポップアップ表示され、ポイントが拒否される必要があるかどうかを赤色で示します。少数の点だけが拒否され、分布が良好に見える場合、モデルは良好です。
注: クロス検証プロットは、決定されたP(r)分布と矛盾するデータの領域をハイライト表示します。拒否された領域が主に低q領域、つまりy軸付近の領域である場合、これはdmax が短すぎる、凝集または高次オリゴマーの存在を示唆している可能性があります。これは、高い解像度と低解像度の情報の間の不整合を強調しています。ここでは、d max および qmin (開始値の増加) は、手動の試行錯誤アプローチを使用して調整する必要があります。同様に、拒否された領域が主に高q領域にある場合、これはバックグラウンド減算の問題や、決定されたP(r)分布によって意味を持って説明できない信号が弱すぎることを示している可能性があります。この場合、追加のデータが拒否されなくなるまで、qmax は切り捨てられる (終了を減らす) 必要があります。理想的には、拒否されたポイントはランダムに分布し、使用できるデータの5%未満を構成する必要があります。適切に定義された q分、qmax、 および "dmax"は、dmax が x 軸にキスする滑らかな分布を生成します。ただし、この値を大きくして、ギニエリージョンを完全に削除しないでください。この点は 、q x l(q) ボックス(表の上のパネルの左側)をチェックすることで簡単に見つけることができます。このカーブ上の最大変曲がギニエ領域の一部になる前に、この曲線上の散乱曲線は「総散乱強度プロット」に置き換えられます。ポイントを削除した後、もう一度 "dmax"を増減し、もう一度調整してみてください。問題が解決しない場合、特に検証曲線の先頭から多くのポイントが拒否されている場合、データが構造モデリングに適していないことを強く示唆しています。
- レポートを印刷するには、[ 分析 ] タブに戻り、サンプルを左クリックしてハイライト表示し、サンプル名を右クリックして、メニューの [単一データ セットからレポートを作成 ] に移動します。テキスト ボックスが開き、コメントを追加できます。生成されたすべての図と値を示す PDF ドキュメントが生成されます。
結果
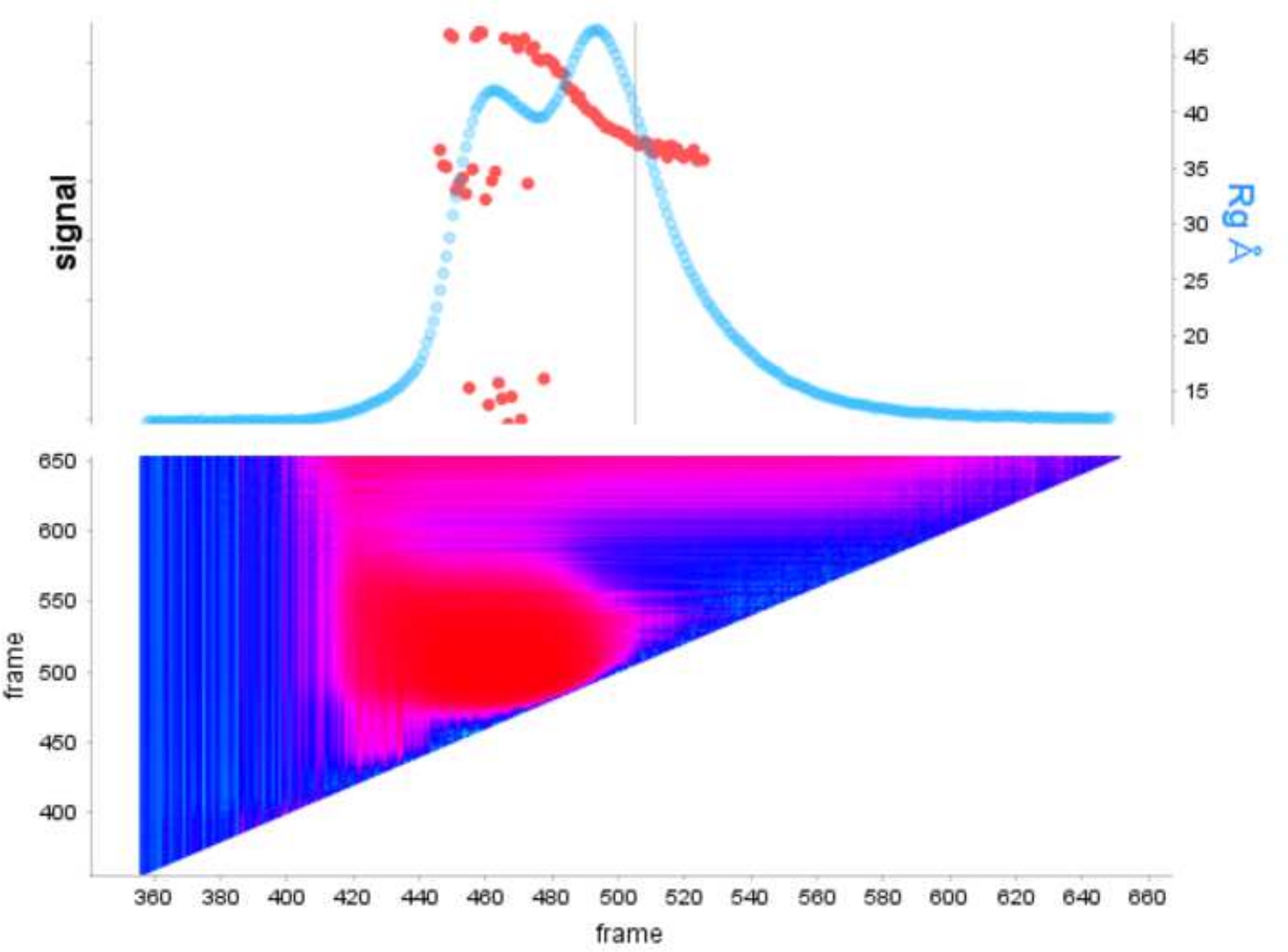
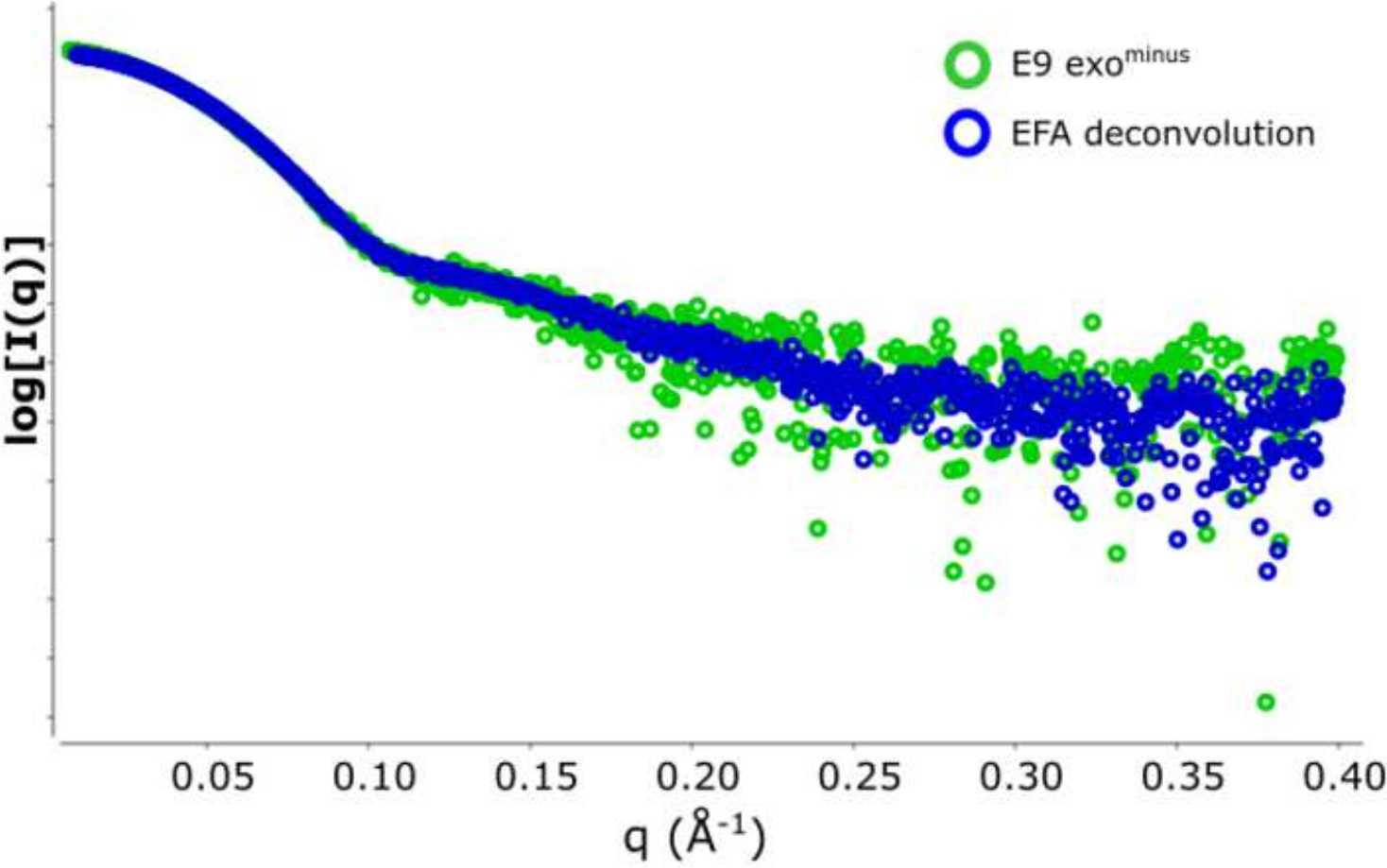
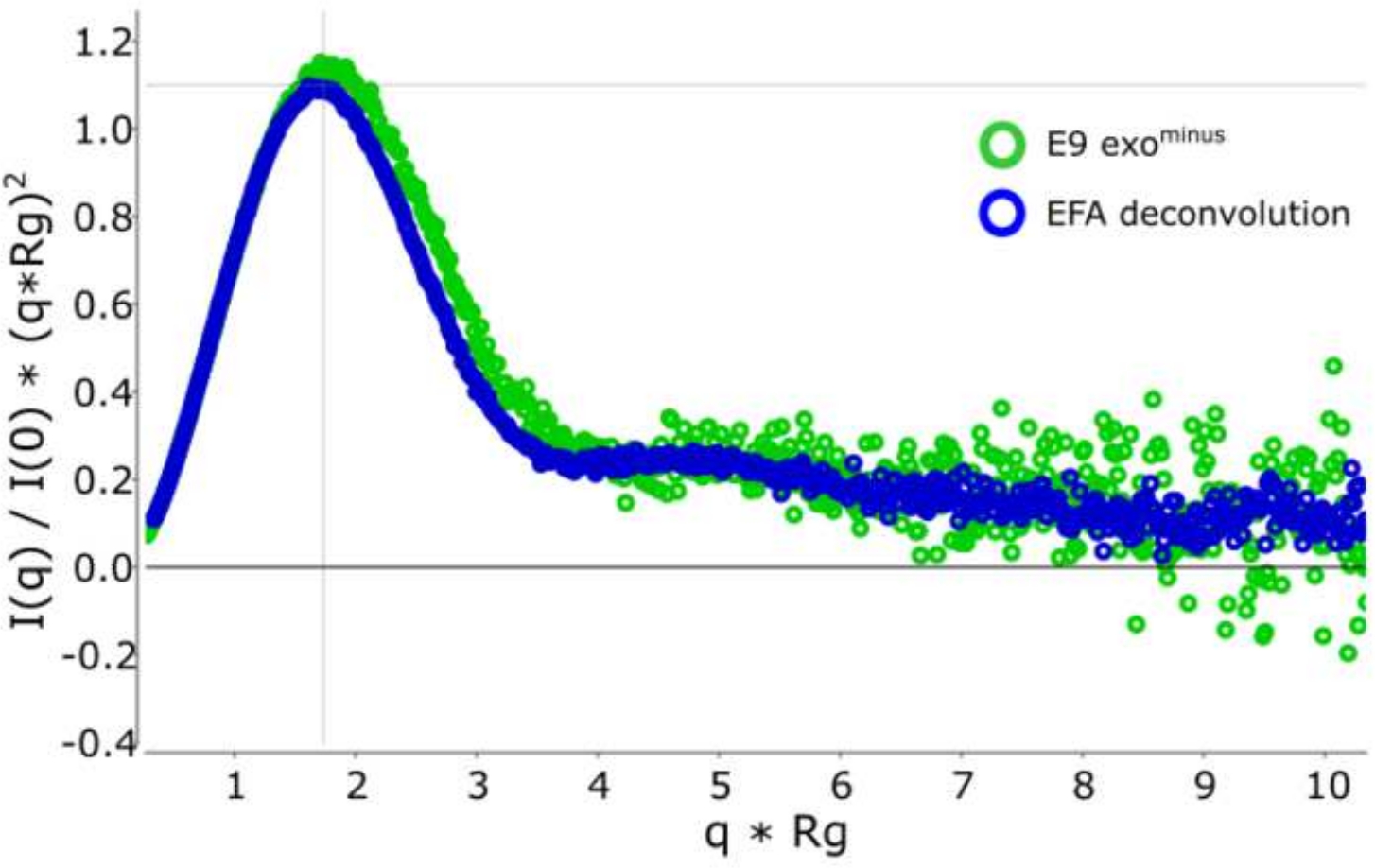
古典的なフレーム選択13に対するデコンボリューションを用いる利点は、種が互いに及ぼす影響を取り除き、単分散散乱信号を生成する。これは、多くの場合、より良い信号対ノイズ比が続きます。E9 exoマイナスがDNAに結合し、SEC-SAXSを使用して実行されると、2つのピークが観察されます(図1)。最初の大きなピーク(フレーム 420\u2012475)は、E9 exoマイナス-DNA 複合体(フレーム 475\u2012540 およそフレーム)、非バインド状態です (補足データ: 図 2を参照)。フレームを選択する古典的なアプローチは、第1のピークで複合体の安定したRgを提供しますが(補足データ:図3を参照)、第2のピークは明確にマージされ、プロット全体のRgは、対象の第2のピークがクロスピーク汚染のために安定したRgを持っていないことを示しています。半安定したRgを示す5フレームのみ使用することができ、減算するとRg = 36.3 Å(図2、緑)を与えた。第2のピークに対応する曲線をEFAを用いてデコンボルチングした場合(図2、青)を元のピークに重ね合わし、ノイズへの信号の明らかな減少を示し、低Rg、34.1Åを記録した。クラッキープロット(図3)は、デコンボルトピーク(青)がより球状である複合体を示しています。これは、デコンボルト曲線(青)に対してd max 108.5 Åを与えるP(r)曲線(図4)によって確認され、非デコンボルートはdmax 120 Å(緑)でより細長く、これは非結合E9 exoマイナスから生じる不均質性に起因する可能性が最も高い。

図1:E9 exoマイナス 単独で、DNAを複合体で示すシグナルプロット
上部パネルには、SEC-SAXS の各フレームの背景に対する積分比のプロットが表示されます (水色)。赤い点は、ピーク上の各フレームのRgを示しています。下のパネルには、ダービン・ワトソンの自己相関分析に従って色付けされた各フレームの残差を示す対応するヒートマップが表示され、類似度の高い領域はシアン色で着色され、異なるフレームは暗い青色からピンクに続き、最後は異なる類似性の重症度に応じて赤に続く。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:強度と散乱ベクトルのプロット
減算された SAXS データのオーバーレイは、E9 exoマイナス を形成します。緑色の 5 フレーム (フレーム 517\u2012522) 平均と半安定 Rg の領域から差し引くと青 で、SEC-SAXS ピークの EFA デコンボリューションに由来する代表的な散乱曲線。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:無次元のクラッキー曲線
デコンボルト(青)と非デコンボルート(緑色)クラッキー曲線のオーバーレイは、E9 exoマイナス を示し、球状である。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:P(r)曲線
E9 exoマイナスのデコンボルト(青)および非デコンボルト(緑)曲線のオーバーレイ。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
SAXS実験を開始する前に単分散サンプルを使用することが望まれますが、実際には多くのデータ収集はこれを満たしておらず、測定とインラインクロマトグラフィー(ほとんどの場合はSEC)を組み合わせることで改善する必要があります。しかしながら、サンプルの精製とデータ取得の一分散との間の時間の不足さえも保証されない。最も一般的には、コンポーネントのサイズが近すぎるか、物理的な特性が分離できないか、または高速ダイナミクスが発生しやすい実験に当てはまります。ここでは、単一値分解と進化因子分析を組み合わせたプロトコルを提供し、非連結E9 exoマイナス の影響を非連結形状から取り除き、SAXSパッケージScatter IVで分析することができた単分散散乱プロファイルを作成しました。
SEC-SAXSデータのEFAを搭載したSVDは、SAXSデータをデコンボルテし、分析を改善するために開発された非常に強力な方法ですが、制限があります。SEC-SAXSのバッファベースラインのノイズまたはドリフトを最小限に抑える必要があります。これは、サンプルの読み込み前に余分な列の平衡化を伴う(バッファーに応じて、3 列のボリュームを使用する方が良い) 場合があります。しかし、最も重要なステップは、単数の値の数と使用されるデータの範囲の選択であり、これは畳み込みの精度に大きな影響を与えます。そのため、結果は単独で取るのではなく、生物学的解釈のために分析的超遠心(AUC)や多角レーザー光散乱(MALLS)などの技術を用いてさらに分析すべきである。
Scatter IVは、研究や産業用に無料で、専門家以外でもデータを分析できる直感的なユーザーインターフェイスを備えた新しいソフトウェアパッケージです。スキャッタIVには、信号プロットにリンクされたヒートマップなど、SEC-SAXSデータの分析を改善するのに役立ついくつかの新機能があり、フレーム選択の選択により精度が向上します。一次データ分析では、P(r) 解析に関連する Guinier Peak 分析とクロス検証プロットにより、ソフトウェアに統合されたトラブルシューティング機能が提供されます。
他の多くのプログラムをプライマリデータ分析に使用できることは言及する必要があります。これらは、同じ基本的な機能を含み、また、いくつかの名前を挙げ、BioXTAS RAW17 ATSASパッケージ24 とUS-SOMO15 などの定期的に更新されます。
しかし、どの SAXS パッケージを分析に使用するかに関係なく、主な制限事項は一般的です: サンプルの準備、収集および分析の前に。図示したE9 exoマイナス 例では、信号対雑音比の改善と、単分散試料に関連するdmax のRgの減少を見ることが明らかである。これにより、既知の高解像度構造を使用して、データのフィッティングやモデリングなどのデータの処理をさらに促進できます。
開示事項
著者らは開示するものは何もない。
謝辞
我々は、フランスの助成金REPLIPOX ANR-13-BSV8-0014からのプロジェクトに対する財政的支援と、サンテ・デ・アルメサービスとデレギュレーション・ジェネラルからの研究助成金による資金援助を認める。SAXSビームタイムに対してESRFに感謝しています。この作品はグルノーブル・コンストラクテリックセンター(ISBG;)UMS 3518 CNRS-CEA-UGA-EMBL)は、FRISBI(ANR-10-INBS-05-02)とGRALが支援する構造生物学のためのグルノーブルパートナーシップ(PSB)内で、グルノーブル・アルプ大学大学院(エコールズ大学デ・レシェルシュ)CBH-EUR-GS(ANR-17-EU-000)内で資金を調達しました。IBSは、グルノーブル学際研究所(CEA)への統合を認めています。Wim P. Burmeister と Frééric Iseni の財政的および科学的なサポートに感謝し、また、APS の BioCAT のジェシー・ホプキンス博士の助けと BioXTAS RAW の開発に感謝します。
資料
| Name | Company | Catalog Number | Comments |
| Beamline control software BsXCuBE | ESRF | Pernot et al. (2013), J. Synchrotron Rad. 20, 660-664 | local development |
| BioXTAS Raw 1.2.3. | MacCHESS | http://bioxtas-raw.readthedocs.io/en/latest/index.html | First developed in 2008 by Soren Skou as part of the biological x-ray total analysis system (BioXTAS) project. Since then it has been extensively developed, with recent work being done by Jesse B. Hopkins |
| HPLC program LabSolutions | Shimadzu | n.a. | |
| ISPyB | ESRF | De Maria Antolinos et al. (2015). Acta Cryst. D71, 76-85. | local development |
| NaCl | VWR Chemicals (BDH Prolabo) | 27808.297 | |
| Scatter | Diamond Light Source Ltd | http://www.bioisis.net/tutorial/9 | Supported by SIBYLS beamline (ALS berkeley, Ca) and Bruker Cororation (Karlsruhe, Germany) |
| Superdex 200 Increase 5/150 GL column | GE Healthcare | 28990945 | SEC-SAXS column used |
| Tris base | Euromedex | 26-128-3094-B |
参考文献
- Pelikan, M., Hura, G., Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. General Physiology and Biophysics. 28 (2), 174-189 (2009).
- Brosey, C. A., Tainer, J. A. Evolving SAXS versatility: solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Current Opinion in Structural Biology. 58, 197-213 (2019).
- Gräwert, M., Svergun, D. A beginner's guide to solution small-angle X-ray scattering (SAXS). The Biochemist. 42 (1), 36-42 (2020).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly reviews of biophysics. 40 (03), 191-285 (2007).
- Meisburger, S. P., et al. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. Journal of the American Chemical Society. 138 (20), 6506-6516 (2016).
- Brennich, M. E., Round, A. R., Hutin, S. Online Size-exclusion and Ion-exchange Chromatography on a SAXS Beamline. Journal of Visualized Experiments. (119), e54861 (2017).
- Watanabe, Y., Inoko, Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. Journal of Chromatography A. 1216 (44), 7461-7465 (2009).
- Graewert, M. A., et al. Automated Pipeline for Purification, Biophysical and X-Ray Analysis of Biomacromolecular Solutions. Scientific reports. 5, (2015).
- David, G., Pérez, J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. Journal of applied crystallography. 42 (5), 892-900 (2009).
- Ryan, T. M., et al. An optimized SEC-SAXS system enabling high X-ray dose for rapid SAXS assessment with correlated UV measurements for biomolecular structure analysis. Journal of Applied Crystallography. 51 (1), 97-111 (2018).
- Gampp, H., Maeder, M., Meyer, C. J., Zuberbühler, A. D. Calculation of equilibrium constants from multiwavelength spectroscopic data-III: Model-free analysis of spectrophotometric and ESR titrations. Talanta. 32 (12), 1133-1139 (1985).
- Maeder, M., Neuhold, Y. M. . Practical Data Analysis in Chemistry. , (2007).
- Tarbouriech, N., et al. The vaccinia virus DNA polymerase structure provides insights into the mode of processivity factor binding. Nature Communications. 8 (1), (2017).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), (2016).
- Brookes, E., Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. European Biophysics Journal. 47 (7), 855-864 (2018).
- Malaby, A. W., et al. Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. Journal of Applied Crystallography. 48 (4), 1102-1113 (2015).
- Hopkins, J. B., Gillilan, R. E., Skou, S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. Journal of Applied Crystallography. 50 (5), 1545-1553 (2017).
- Maeder, M. Evolving factor analysis for the resolution of overlapping chromatographic peaks. Analytical Chemistry. 59 (3), 527-530 (1987).
- Durand, D., et al. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. Journal of Structural Biology. 169 (1), 45-53 (2010).
- De Maria Antolinos, A., et al. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta Crystallographica Section D. 71 (1), 76-85 (2015).
- Brennich, M. E., et al. Online data analysis at the ESRF bioSAXS beamline, BM29. Journal of Applied Crystallography. 49 (1), 203-212 (2016).
- Kirby, N., et al. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallographica Section D Structural Biology. 72 (12), 1254-1266 (2016).
- Rambo, R. P., Tainer, J. A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 95 (8), 559-571 (2011).
- Franke, D., et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography. 50 (4), 1212-1225 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved