
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Ablations volumiques profondes et spatialement contrôlées à l’aide d’un microscope à deux photons dans la Gastrula du poisson-zèbre
Dans cet article
Résumé
Le développement embryonnaire nécessite une coordination à grande échelle du mouvement cellulaire. L’ablation laser médiée par l’excitation à deux photons permet l’ablation en 3 dimensions contrôlée spatialement de grands groupes de cellules profondes. De plus, cette technique permet de sonder la réaction des cellules qui migrent collectivement in vivo à des perturbations dans leur environnement mécanique.
Résumé
La morphogenèse implique de nombreux mouvements cellulaires pour organiser les cellules en tissus et organes. Pour un bon développement, tous ces mouvements doivent être étroitement coordonnés, et l’accumulation de preuves suggère que cela est réalisé, au moins en partie, par des interactions mécaniques. Tester cela dans l’embryon nécessite des perturbations physiques directes. Les ablations au laser sont une option de plus en plus utilisée qui permet d’alléger les contraintes mécaniques ou d’isoler physiquement deux populations cellulaires l’une de l’autre. Cependant, de nombreuses ablations sont effectuées avec un laser ultraviolet (UV), qui offre une résolution axiale et une pénétration tissulaire limitées. Une méthode est décrite ici pour ablation de volumes profonds, significatifs et spatialement bien définis à l’aide d’un microscope à deux photons. Les ablations sont démontrées dans une lignée transgénique de poisson zèbre exprimant la protéine fluorescente verte dans le mésendoderme axial et utilisées pour couper le mésendoderme axial sans affecter l’ectoderme sus-jacent ou la cellule vitellin sous-jacente. Le comportement cellulaire est surveillé par imagerie en direct avant et après l’ablation. Le protocole d’ablation peut être utilisé à différents stades de développement, sur n’importe quel type de cellule ou de tissu, à des échelles allant de quelques microns à plus d’une centaine de microns.
Introduction
Les interactions cellule-cellule jouent un rôle essentiel dans le développement. Les cellules fournissent des signaux que leurs voisines directes, ou des cellules plus éloignées, peuvent percevoir, influençant ainsi leur destin et / ou leur comportement. Beaucoup de ces signaux sont de nature chimique. Par exemple, dans les événements d’induction bien caractérisés, un groupe cellulaire produit des molécules diffusibles affectant le devenir d’une autre population cellulaire1. D’autres signaux, cependant, sont mécaniques; les cellules exercent des forces et des contraintes sur leurs voisins, que les voisins perçoivent et auxquels ils réagissent2.
Une façon d’étudier l’importance de ces interactions cellule-cellule in vivo est d’éliminer certaines cellules et d’observer le développement ultérieur. Malheureusement, les techniques disponibles pour éliminer ou détruire les cellules sont limitées. Les cellules peuvent être enlevées chirurgicalement3,4, à l’aide d’aiguilles ou de petits fils, mais ces traitements sont invasifs, peu précis et généralement effectués sous stéréomicroscope, empêchant l’imagerie immédiate au microscope. De plus, cibler les cellules profondes implique de percer un trou dans les tissus sus-jacents, créant ainsi des perturbations indésirables. Des photosensibilisateurs génétiquement codés, tels que KillerRed, ont été utilisés pour induire la mort cellulaire via l’éclairage lumineux5. Les photosensibilisateurs sont des chromophores qui génèrent des espèces réactives de l’oxygène lors de l’irradiation lumineuse. Leur principale limite est qu’ils nécessitent de longs éclairages lumineux (environ 15 min), ce qui peut être difficile à réaliser si les cellules se déplacent, et qu’ils induisent la mort cellulaire par apoptose, qui n’est pas immédiate.
Enfin, les ablations au laser ont été développées et largement utilisées au cours des 15 dernières années6,7,8,9,10,11,12. Un faisceau laser est focalisé sur la cellule/le tissu ciblé. Il induit son ablation par chauffage, photoablation ou ablation induite par plasma; le processus impliqué dépend de la densité de puissance et du temps d’exposition13. La plupart des protocoles d’ablation utilisent des lasers UV pour leur haute énergie. Cependant, la lumière UV est à la fois absorbée et diffusée par les tissus biologiques. Ainsi, le ciblage des cellules profondes nécessite une puissance laser élevée, qui induit ensuite des dommages dans des tissus plus superficiels et hors plan. Cela limite l’utilisation des lasers UV aux structures superficielles et explique leur résolution axiale relativement faible. L’optique non linéaire (appelée microscopie à deux photons) utilise les propriétés non linéaires de la lumière pour exciter un fluorophore avec deux photons d’environ une demi-énergie dans le domaine infrarouge. Lorsqu’il est appliqué aux ablations, cela présente trois avantages principaux. Tout d’abord, la lumière infrarouge est moins diffusée et moins absorbée que la lumière UV par les tissus biologiques14, ce qui permet d’atteindre des structures plus profondes sans augmenter la puissance laser requise. Deuxièmement, l’utilisation d’un laser pulsé femtoseconde fournit des densités de puissance très élevées, créant une ablation par induction plasmatique qui, contrairement au chauffage, ne diffuse pas spatialement15. Troisièmement, la densité de puissance induisant la formation de plasma est atteinte au point focal uniquement. Grâce à ces propriétés, les ablations laser à deux photons peuvent être utilisées pour cibler avec précision les cellules profondes sans affecter l’environnement tissulaire environnant.
Les migrations collectives sont un excellent exemple de processus de développement dans lesquels les interactions cellule-cellule sont fondamentales. Les migrations collectives sont définies comme des migrations cellulaires dans lesquelles les cellules voisines influencent le comportement d’une cellule16. La nature de ces interactions (chimiques ou mécaniques) et la façon dont elles affectent la migration cellulaire peuvent varier considérablement et ne sont souvent pas entièrement comprises. La capacité d’éliminer les cellules et d’observer comment cela affecte les autres est essentielle pour démêler davantage ces processus collectifs. Il y a quelques années, nous avons établi – à l’aide d’approches chirurgicales – que la migration du polster lors de la gastrulation du poisson-zèbre est une migration collective17. Le polster est un groupe de cellules qui constitue les premières cellules internalisantes de la face dorsale de l’embryon18. Ces cellules, marquées en vert dans la lignée transgénique Tg(gsc:GFP), sont situées profondément dans l’embryon, sous plusieurs couches de cellules épiblastiques. Au cours de la gastrulation, ce groupe mène l’extension du mésoderme axial, migrant de l’organisateur embryonnaire au pôle animal19,20,21,22,23 (Figure 1A). Nous avons établi que les cellules ont besoin d’un contact avec leurs voisins pour orienter leur migration dans la direction du pôle animal. Cependant, mieux comprendre les bases cellulaires et moléculaires de cette migration collective implique d’enlever certaines cellules pour voir comment cela influence les autres. Nous avons donc développé des ablations de grands volumes profonds à l’aide d’une configuration de microscopie à deux photons. Ici, nous démontrons l’utilisation de ce protocole pour couper le polster en son milieu et observer les conséquences sur la migration cellulaire en suivant les noyaux marqués avec Histone2B-mCherry.
Protocole
Tous les travaux d’animaux ont été approuvés par le Comité d’Éthique N 59 et le Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche sous le numéro de dossier APAFIS#15859-2018051710341011v3. Certaines des étapes décrites ci-dessous sont spécifiques à nos équipements et logiciels, mais pourraient être facilement adaptées à différents équipements.
1. Préparation par injection
- Préparer 75 mL de solution d’agarose à 1 % dans un milieu embryonnaire (EM).
- Placez le moule injecteur dans une boîte de Petri de 90 mm et versez environ 50 mL d’agarose, assez pour que le moule flotte. Laissez l’agarose se solidifier et retirez le moule d’injection.
- Préparez un plat enrobé d’agarose en versant 1 mL d’agarose dans une boîte de Petri de 30 mm.
- Préparer 4 μL de solution d’ARNm Histone2B-mCherry de 30 ng/μL en diluant la solution mère dans de l’eau exempte de RNase et conserver sur la glace.
REMARQUE: Prenez soin de porter des gants lors de la manipulation de l’ARNm pour éviter la dégradation médiée par la RNase. - Tirez une aiguille d’injection d’un capillaire à l’aide de l’extracteur de micropipette.
2. Préparation de l’embryon
- Une fois que les poissons ont pondu des œufs, collectez, rincez et récoltez dans une boîte de Petri de 90 mm dans EM. Placez les embryons dans un incubateur à 28,5 °C.
- Attendez 20 min pour que la première cellule devienne visible.
- Transférer 30 embryons sur la plaque d’injection remplie d’EM. Pressez les embryons dans les rainures à l’aide de pinces légèrement émoussées et orientez-les avec le pôle de l’animal vers le haut.
- À l’aide d’une pointe de microchargeur, remplissez une aiguille d’injection avec 2 μL de solution d’ARNm. Insérez l’aiguille dans le porte-capillaire placé dans un micro-manipulateur relié à un tube en polytétrafluoroéthylène (PTFE) à un injecteur d’air.
- Sous le stéréomicroscope, cassez soigneusement le bout de l’aiguille.
- Injecter la solution d’ARNm dans les embryons au stade 1 en insérant l’aiguille dans la cellule.
REMARQUE: Le volume injecté représente environ un tiers du volume de la cellule. - Replacez les embryons injectés dans l’incubateur à 28,5 °C.
3. Préparation du microscope à deux photons
REMARQUE: Deux lasers sont utilisés dans ce protocole. L’un est utilisé pour imager la GFP (à 920 nm) et effectuer des ablations (à 820 nm). On l’appellera le laser vert/ablation. L’autre est utilisé à 1160 nm pour imager mCherry. On l’appellera le laser rouge.
- Réglez le laser vert/ablation sur 820 nm (longueur d’onde d’ablation) et le laser rouge sur 1160 nm (excitation mCherry).
- À l’aide de miroirs mobiles sur le chemin optique, alignez les faisceaux laser verts/ablation et rouges à l’entrée et à la sortie de la tête de balayage.
REMARQUE: Cela augmente la mise au point du faisceau laser et minimise le volume focal pour l’excitation et l’ablation. - Mesurez la puissance maximale du laser vert/ablation à 820 nm sous l’objectif. Pour ce faire, placez le capteur de puissance sous l’objectif, fermez la chambre noire, réglez la puissance laser verte / ablation à 100% et ouvrez les volets. Calculez le pourcentage de puissance laser nécessaire pour atteindre 300 mW.
- Réglez le laser vert/ablation à 920 nm (excitation GFP) et réglez la puissance du laser à 7 %. Réglez la puissance du laser rouge sur 15%.
- Activer les détecteurs epi-PhotoMultiplier Tubes (PMT) pour les lignes vertes et rouges; réglez la sensibilité PMT des lignes vertes et rouges sur 65.
- Réglez le champ de vision sur 400 x 400 μm, la résolution de l’image sur 512 x 512 pixels et la fréquence de numérisation sur 800 Hz.
- Sélectionnez le mode 3D Timelapse Imaging . Ensuite, créez un dossier et activez l’enregistrement automatique des données après chaque acquisition.
- Assemblez la chambre de chauffage et réglez-la à 28 °C. Attendez au moins 10 min que la chambre et l’objectif se réchauffent.
4. Montage de l’embryon
- Sous un stéréomicroscope à fluorescence, identifier les embryons à 70% d’épibole qui expriment la GFP.
REMARQUE: Sélectionnez des embryons avec un signal lumineux dans le mésoderme axial et sans fluorescence de fond pour une meilleure qualité d’imagerie. - Transférer trois à quatre embryons sélectionnés dans le plat enrobé d’agarose (étape 1.3) à l’aide d’une pipette Pasteur en plastique et les dépéchorioner soigneusement à l’aide de pinces fines.
REMARQUE: Les embryons décononnés sont très délicats et éclatent au contact de l’air ou du plastique. - Verser 1 mL d’agarose à 0,2 % dans 1x EM de pénicilline-streptomycine dans un petit flacon en verre. Placer le flacon dans un bloc chauffant préchauffé à 42 °C.
REMARQUE: Les étapes suivantes doivent être effectuées rapidement pour permettre l’orientation de l’embryon avant les ensembles d’agarose. - Transférer un embryon déséchorioné dans le flacon en verre d’agarose à 0,2 % à l’aide d’une pipette en verre poli au feu. Veillez à ne pas ajouter trop d’EM dans l’agarose pour éviter de le diluer. Jetez l’EM restant de la pipette et aspirez l’embryon avec suffisamment d’agarose pour couvrir la lame du fond en verre avant que l’embryon ne tombe de la pipette.
- Soufflez l’agarose et l’embryon sur la lame de verre du plat. Veillez à ne pas laisser l’embryon toucher l’air ou le côté plastique du plat. Ensuite, remplissez la chambre autour de la glissière de verre avec de l’agarose.
- Utilisez un cil pour orienter l’embryon de manière à ce que la région ciblée soit en haut (Figure 1B).
REMARQUE: Lors de l’orientation des embryons, veillez à ne toucher que le blastoderme, pas le jaune très fragile. L’agarose s’installera en environ 1 min, en fonction de la température ambiante. - Attendez ~ 5 min pour que l’agarose se fixe complètement, puis ajoutez quelques gouttes de pénicilline-streptomycine EM.
5. Localisation de l’embryon et imagerie pré-ablation
- Placez le plat inférieur en verre sous l’objectif dans la chambre chauffée. Immerger l’objectif dans la pénicilline-streptomycine EM et fermer la chambre chauffée.
- Déplacez le curseur pour définir le trajet de la lumière sur les oculaires. Ensuite, à l’aide d’oculaires, de lampes fluorescentes et d’un contrôle de scène, trouvez un embryon et réglez le foyer sur la surface de l’embryon.
- Éteignez la lampe à fluorescence, réglez le trajet de la lumière sur les PMT et fermez la chambre noire.
REMARQUE: Veillez à éteindre toutes les sources lumineuses dans la chambre noire car cela pourrait endommager les PMT. - Commencez l’imagerie en direct et localisez le mésoderme axial. Ajustez les puissances laser verte/ablation et rouge pour avoir un bon signal (c’est-à-dire entre 1 000 et 20 000 photons par pixel pour les zones d’expression GFP). Utilisez le canal rouge pour déplacer la scène tout en haut de l’embryon et définissez cette position sur Z = 0.
- Choisissez un pas de temps de 1 min et un pas de Z de 2 μm. Un cours Z de 110 μm est suffisant pour englober l’ensemble du polster et est acquis en moins de 1 min avec ces réglages. Placer la première tranche à 15 μm au-dessus du mésoderme axial (dans l’ectoderme plus superficiel).
REMARQUE : Le polster se déplace le long d’une ligne courbe de sorte que la tranche inférieure de la pile Z doit être placée 30 μm plus loin que la position la plus profonde du polster pour s’adapter à son mouvement pendant l’imagerie time-lapse (Figure 1E). - Enregistrez 10-15 min de film de pré-ablation.

Figure 1: Résultat positif des ablations au laser. (A) Schéma d’un embryon gastrulant à 70% d’épibolye en vue dorsale; pAM: mésoderme axial postérieur; la flèche noire marque la direction de la migration des polsters; le carré noir indique un champ de vision typique pour les ablations dans le polster. (B) Schéma de montage d’embryons pour la séparation des polsters. Vue latérale. L’embryon est monté de telle sorte que le plan du polster soit perpendiculaire à l’axe optique. (C) la survie et (D) la morphologie des embryons témoins et ablés 24 heures après la fécondation. La barre d’échelle est de 300 μm. (E) Séquence temporelle de l’ablation au laser dans le polster d’un embryon Tg(gsc:GFP) exprimant Histone2B-mCherry. Les vues avec le canal vert uniquement sont des projections maximales. Le gros plan affiche la zone ablée contenant des débris cellulaires. Les vues avec des canaux verts et rouges (affichés sous forme magenta) sont des tranches XY et XZ avant et après l’ablation (l’éclair vert représente l’ablation). Les tranches XZ montrent que les tissus sus-jacents (noyaux magenta sans expression de GFP) n’ont pas été affectés par l’ablation des structures sous-jacentes. La boîte en pointillés jaune correspond au ROI sélectionné pour le traitement d’ablation au laser. La barre d’échelle est de 50 μm dans les grandes vues et de 25 μm dans le gros plan. Veuillez cliquer ici pour voir une version agrandie de cette figure.
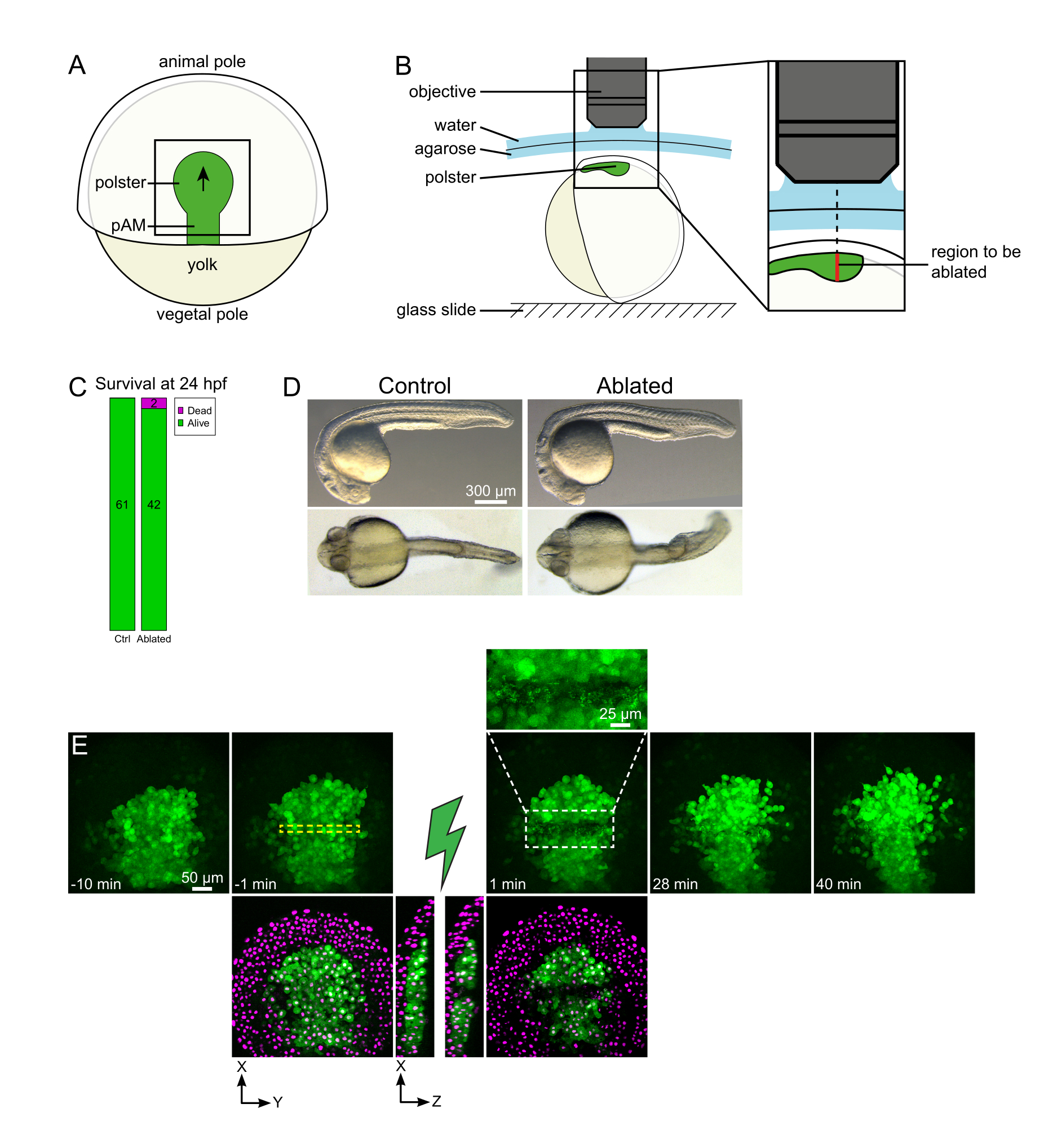{kind=link}
6. Emplacement de la cible et ablation au laser
- Localisez le contour du polster sur l’imagerie en direct et, à l’aide de l’outil EOM (Electro-Optic Modulator Region of Interest), dessinez un grand rectangle de 20 pixels (15 μm) qui couvre la largeur du polster. Placez ce rectangle au milieu du polster (Figure 1E).
- Notez la position axiale des plans les plus hauts et les plus bas contenant des cellules polster. Des ablations seront effectuées tous les 10 μm entre ces deux plans. Veillez à ce que le retour sur investissement ne chevauche pas la cellule jaune sur aucun de ces plans.
- Placez la scène à la position Z la plus basse de l’intervalle. Les ablations doivent être effectuées de bas en haut car les débris absorbent la lumière.
- Réglez la longueur d’onde du laser vert/ablation sur 820 nm et réglez le pourcentage de puissance pour obtenir une puissance de sortie de 300 mW (étape 3.3).
- Réglez la fréquence d’imagerie sur 200 Hz.
- Réglez la MOE d’imagerie laser verte/ablation sur 0 et sélectionnez le mode ROI-Treat .
- Allumez la moE et réglez le traitement pour qu’il commence immédiatement (après 0 image).
- Réglez le mode d’imagerie sur Timelapse et désactivez l’enregistrement automatique.
- Réglez le pas de temps sur le mode rapide.
- Définissez le nombre de trames de traitement et le nombre d’images sur la valeur correspondant à la profondeur ciblée (tableau 1).
| Profondeur (μm) | Cadres de traitement |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Tableau 1 : Nombre suggéré de cadres de traitement au laser en fonction de la profondeur cellulaire ciblée dans l’embryon (0 étant la surface de l’embryon).
- Commencez l’imagerie. L’acquisition est noire car l’obturateur de PMT se ferme pendant le traitement EOM.
- Montez sur la scène jusqu’à la position Z suivante de la liste (étape 6.2).
- Répétez les étapes 6.10 à 6.12 jusqu’à ce que le sommet du polster soit atteint.
7. Vérification et imagerie post-ablation
- Réglez le laser vert/ablation sur 920 nm et 5 % de puissance. Réglez la moE d’imagerie laser verte/ablation sur 100 et sélectionnez le mode Plein champ.
- Réglez la fréquence d’imagerie sur 800 Hz. Désactivez la moe.
- Parcourez toute la pile en mode live pour vérifier si chaque plan a été ablation. Si ce n’est pas le cas, revenez à l’étape 6.2.
REMARQUE: L’ablation induit parfois un déplacement vertical des tissus voisins, de sorte que la pile Z pourrait devoir être redéfinie. - Réglez le mode d’imagerie sur Timelapse 3D et réactivez l’enregistrement automatique. Enregistrez 40 à 60 minutes de film post-ablation.
- Vérifiez, dans le film post-ablation, si les cellules ciblées ont été effectivement ablées. La récupération par fluorescence, ou les cellules ciblées occupant l’espace et empêchant les cellules suiveuses de se déplacer, indiquent que les cellules ciblées n’étaient que photoblanchis et non ablées (figure 1E et figure 2A).

Figure 2 : Résultats négatifs des ablations au laser. (A) Exemples typiques de défaillances potentielles de l’ablation au laser. Les grandes vues XY sont des projections maximales, la vue XZ est une section reconstruite. La zone traitée au laser est située entre les deux pointes de flèches blanches. Trois plans focaux sont mis en évidence dans la section reconstruite et affichés à droite. Ils correspondent à trois types d’échecs différents. Le plan 1 montre que les cellules au-dessus du polster ont été ablées. Ceci peut être identifié par la présence de débris autofluorescents sur ce plan focal (voir gros plan) au-dessus du polster (voir position du plan 1 sur la section reconstruite). Cela résulte probablement d’une définition incorrecte de la région à abréger. Le plan 2 montre des cellules qui ont été blanchies mais pas ablées. Ils peuvent être identifiés car le signal de faible fluorescence révèle encore des contours cellulaires intacts (voir gros plan). Le plan 3 affiche des cellules intactes, qui ont à peine été blanchies par un traitement au laser. Cela pourrait résulter d’une définition incorrecte de la région à ablation ou d’un mauvais traitement. Dans les situations décrites dans les plans 2 et 3, il est possible de réappliquer le traitement d’ablation aux cellules ciblées non ablées. La barre d’échelle est de 50 μm en grandes vues et de 20 μm en gros plans. (B) Un exemple typique de bulles (marquées par des astérisques blancs) formées par cavitation à cause d’un traitement au laser trop intense. De telles bulles ne se limitent pas à un plan Z, parfois même s’étendant sur toute la hauteur du polster, déformant les tissus voisins. La barre d’échelle est de 50 μm. Veuillez cliquer ici pour voir une version plus grande de cette figure.
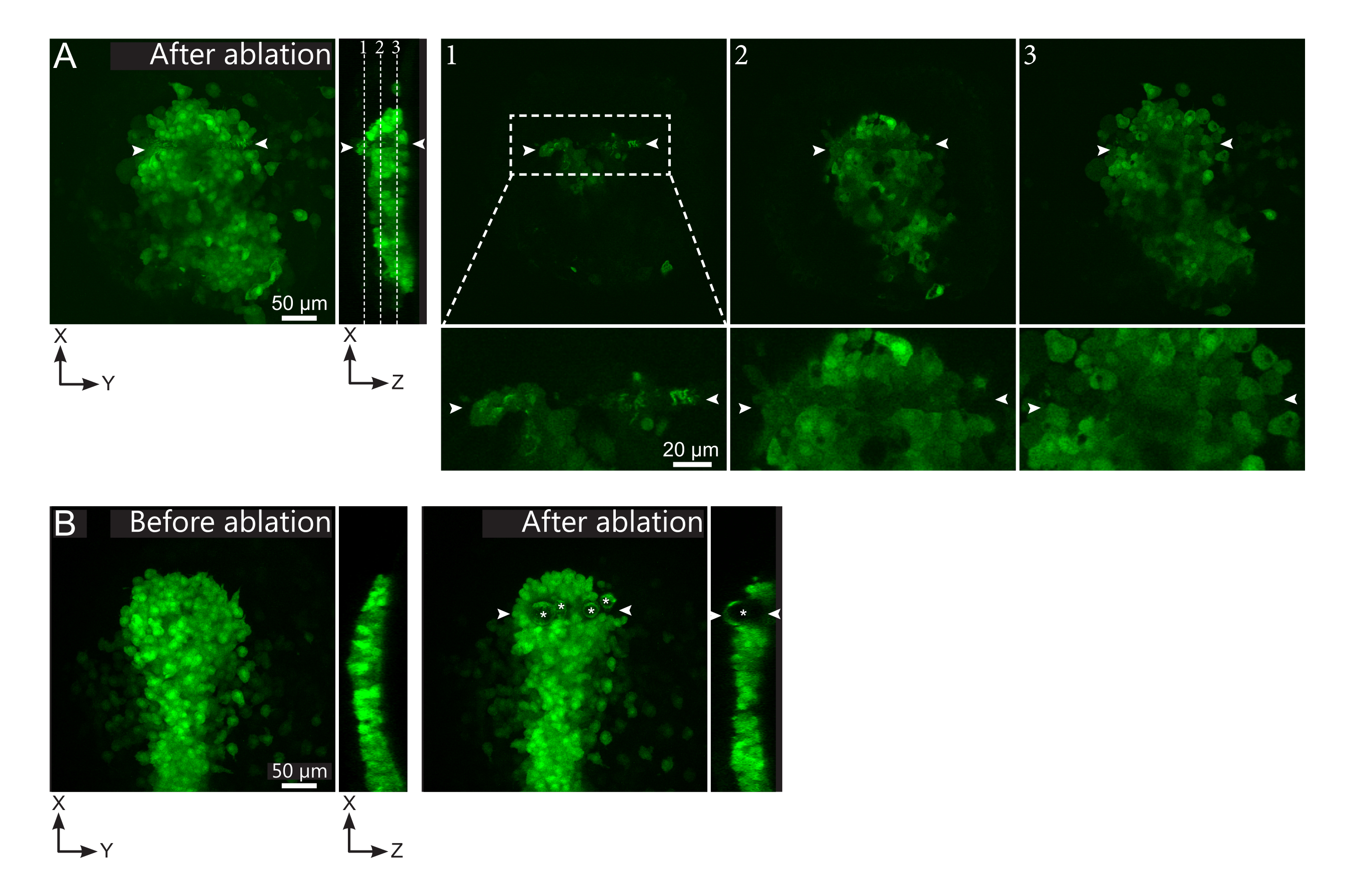{kind=link}
8. Analyse des données
- Ouvrez la série time-lapse avec le logiciel d’analyse d’image et définissez la taille de pixel correcte.
- Dans la fonction Spot , définissez la taille de l’objet sur 10 μm, car il s’agit de la taille moyenne du noyau pendant la gastrulation. Ensuite, exécutez la fonction Spot pour détecter et suivre les noyaux.
REMARQUE: La détection peut être légèrement améliorée en tenant compte de la résolution axiale inférieure, en adaptant une forme ellipsoïdale de 12 μm de long le long de l’axe Z. - Utilisez des filtres pour supprimer les faux positifs. Dans la lignée Tg(Gsc:GFP), les cellules de l’axe embryonnaire et certaines cellules endodermiques sont marquées en vert. Par conséquent, le filtrage sur l’intensité verte permet une sélection rapide de ces cellules (Figure 3A).
- Définissez la distance maximale entre les points consécutifs sur une valeur compatible avec la vitesse des cellules.
REMARQUE: Veillez à prendre en compte l’intervalle de temps entre deux images. Les cellules polster migrent à 2,8 ± 0,8 μm/min. Par conséquent, permettre 4 μm de déplacement maximum pour un pas de temps de 1 min supprime la plupart des traces artéfactuelles. - Autoriser les intervalles sur un ou deux points temporels fournit des pistes continues plus longues, mais peut introduire des erreurs de suivi. Si un noyau n’est pas détecté correctement à un point unique, envisagez de relancer la détection ponctuelle avec différents paramètres/filtres.
- Vérifiez visuellement les pistes et, si nécessaire, corrigez-les.
- Exportez les résultats sous forme de fichier .xlsx. Traitez le fichier à l’aide de routines de feuille de calcul publiées24 (Figure 3B) et de routines personnalisées sur un logiciel d’analyse de données (disponible sur demande).

Figure 3 : L’isolement de la moitié antérieure du polster affecte la directionnalité cellulaire. (A) Reconstructions 3D d’un embryon Tg(gsc:GFP) exprimant Histone2B-mCherry (affiché en magenta), avant et après une ablation au laser coupant le polster en son milieu. Les noyaux appartenant à la moitié antérieure du polster sont marqués d’un point magenta et suivis dans le temps avant et après l’ablation (voir le film S1). La barre d’échelle est de 50 μm. (B) Comme mesure de la persistance de la migration, auto-corrélation de direction des cellules appartenant à la partie antérieure du polster avant et après l’ablation. Les cellules affichent un mouvement continu avant l’ablation, qui diminue considérablement après l’ablation, indiquant une perte de migration orientée vers le collectif. L’auto-corrélation de direction a également été mesurée sur des cellules formant la moitié antérieure du polster d’un embryon non ablée, comme témoin. Les enveloppes du graphique indiquent une erreur type. Veuillez cliquer ici pour voir une version agrandie de cette figure.
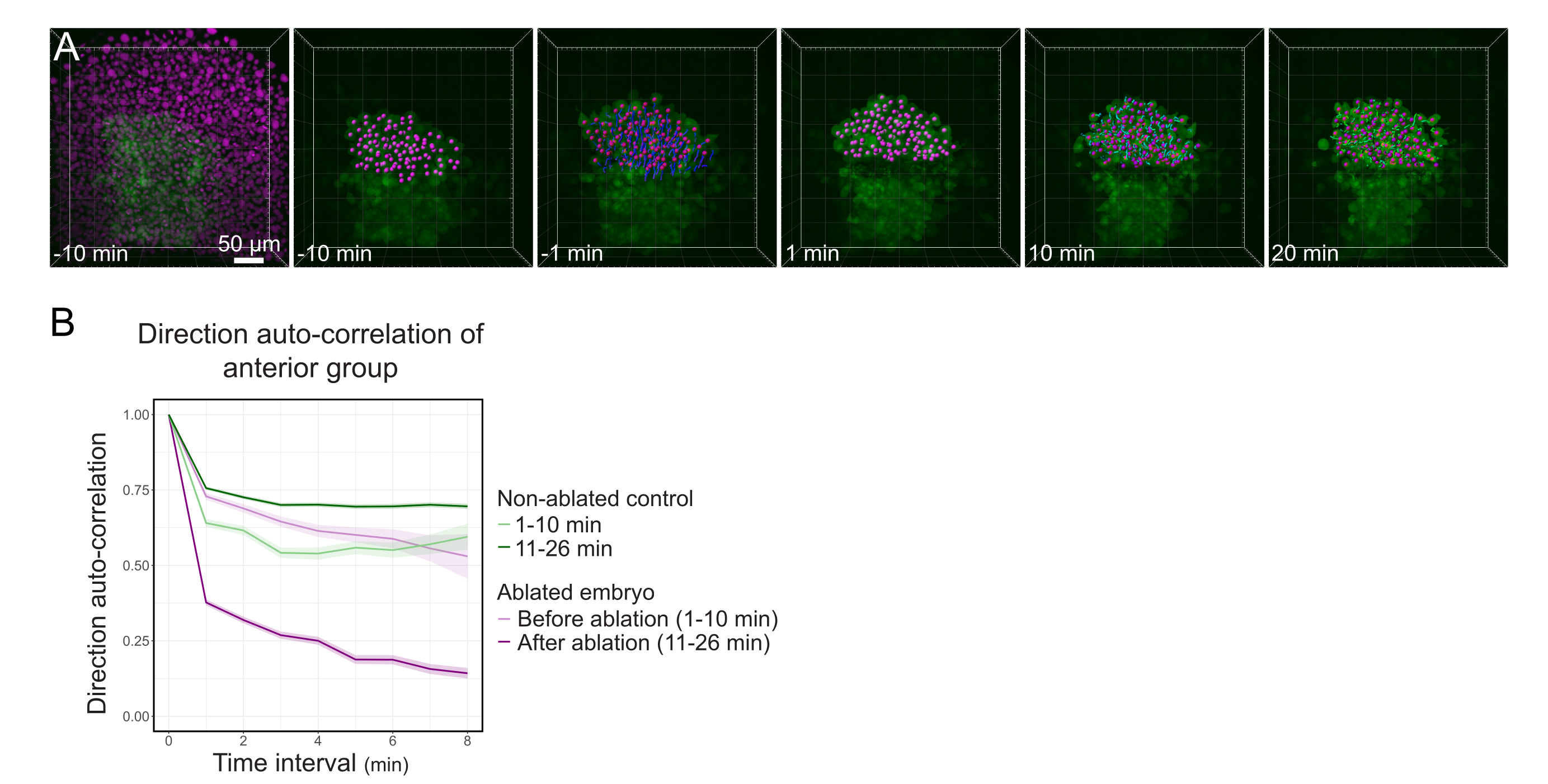{kind=link}
Résultats
Pour couper le polster en son milieu, un embryon Tg(gsc:GFP), injecté avec des ARNm Histone2B-mCherry a été monté au stade épiboly à 70%, comme décrit à l’étape 4. Le polster a été identifié par l’expression de GFP, et l’embryon a été monté de manière à ce que le plan du polster soit perpendiculaire à l’axe optique (Figure 1B). Incliner l’embryon loin de cette position compliquera la procédure. La lumière devra traverser plus de tissus pour atteindre le...
Discussion
Nous décrivons ici un protocole qui utilise une optique non linéaire pour effectuer des ablations volumiques profondes et spatialement bien définies. L’étape la plus critique du protocole est de trouver des conditions de traitement qui fournissent suffisamment d’énergie pour permettre des ablations, mais pas trop d’énergie, afin d’éviter des débris excessifs ou une cavitation. La quantité d’énergie délivrée sur le site cible dépend principalement de: (1) la puissance de sortie du laser, (2) la qual...
Déclarations de divulgation
Les auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Nous remercions Emilie Menant pour les soins aux poissons, le Centre polytechnique de bioimagerie, en particulier Pierre Mahou, pour son aide à l’imagerie en direct sur leurs équipements en partie soutenus par la Région Ile-de-France (interDIM) et l’Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging). Ces travaux ont été soutenus par les subventions ANR 15-CE13-0016-1, 18-CE13-0024, 20-CE13-0016 et le programme de recherche et d’innovation Horizon 2020 de l’Union européenne dans le cadre de la convention de subvention Marie Skłodowska-Curie n° 840201, du ministère de l’Enseignement Supérieur et de la Recherche et du Centre National de la Recherche Scientifique.
matériels
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
Références
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.