
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
転写開始点を決定するための蛍光に基づくプライマー伸長技術とのRNaseの切断サイト
要約
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
要約
蛍光ベースのプライマー伸長(FPE)は、転写開始点またはRNA分子のプロセシング部位を決定するための分子方法である。これは、特定の蛍光標識プライマーおよび変性ポリアクリルアミドゲル電気泳動によって得られたcDNA断片のその後の分析を使用して、目的のRNAを逆転写することによって達成される。同時に、従来のサンガー配列決定反応は、正確な対応するベースにcDNA断片の末端をマッピングするためにゲル上で実行される。生成物をクローン化しなければならず、複数の候補を配列決定5'-RACE(cDNA末端の迅速増幅)とは対照的に、プライマー伸長によって生成されたcDNA断片の大部分は、同時に1つのゲルの実行を検出することができる。また、(逆転写の結果の最終分析に)全体の手順は、1営業日で完了することができる。蛍光標識されたプライマーを用いることによって、危険な放射性同位体の使用は、試薬を標識回避することができ、生成物は、電気泳動手順中に検出することができるように、処理時間が低減される。
以下のプロトコールでは、確実かつ迅速にS.(例えば、毒素-抗毒素システムコンポーネントによって)開始点及びRNAプロセッシング部位、転写を推定するRNAの5 '末端を検出するためのin vivo蛍光プライマー伸長方法を記載球菌、E。 coliおよび他の細菌。
概要
プライマー伸長は、1つの基地解像度までの特定のRNA分子の5 '末端を決定するための分子方法である。そのような5'-RACE(cDNA末端の迅速増幅)のような他の方法の利点は、高速のターンアラウンドタイムと容易にRNA分子の異なる長さの混合物を分析する能力である。
この方法は、特定の長さのcDNA断片を生成する、特異的な蛍光プライマーを用いて逆転写反応するRNA分子を供することによって動作する。これらのcDNA分子は、変性ポリアクリルアミドゲル上で従来のサンガー配列決定反応2と一緒に実行される起因蛍光標識プライマーの使用に、それらの蛍光によって検出することができる。 cDNA断片の長さは、5 'RNA末端のマッピングを可能にする、シークエンシングラダーとの比較によって評価される。
伝統的に、プライマー伸長反応は、組み合わせて使用される放射性同位体でX線フィルム上のcDNA分子を検出することができる。それらの感度が若干低いとはいえ起因する健康被害、廃棄物処理の問題や取り扱いの容易さのために、より新しいプロトコルは、自動化されたシーケンサーを持つプライマー伸長を検出するための蛍光を利用している。蛍光プライマーは(我々の手で一年以上)、長い間安定しているように、蛍光標識されたプライマーを用いて、定期的な放射性標識化手順は、省略することができる。
ここで記述する方法は、自動化されたゲルシーケンサーを利用したが、若干の変更を加えて、キャピラリーシーケンサーも、cDNAの分離および検出の3のために使用することができる。ゲル分析の並列性は、RNA切断または加工の小さな量を検出することが可能となる。別の利点は、検出することができる末端切断、あるいは一つの塩基の処理として、この方法は、高解像度である。
RNA切断または処理、tは検出に関してはypicallyプライマー拡張の2つのタイプが区別される。他の場合には、処理は、生体内で行われ、得られたRNAが精製されるのに対し、あるケースでは、酵素処理は、精製されたRNAおよび精製された酵素を用いてインビトロで行われる。プライマー伸長は、in vitroで実施する両方の場合において、RNAは、RNAの供給源に依存して、しかし、供され、この方法は、in vitroまたはin vivoでのプライマー伸長と呼ばれるか。ここで提示したプロトコルでは、使いやすさのために(無精製タンパク質必要)と同時に、転写開始点と処理を決定する可能性を、in vivoでのプライマー伸長のみに焦点を当てる。しかし、in vitroでのプライマー拡張は同じように設定する原則にあり、このプロトコルは、出発点として役立つことができる。
ここに示された方法は、それらが高に適しているように、多くの細菌種に適用することができ純度および核酸の高収率製剤。
私たちの研究室での研究は、毒素-抗毒素(TA-)システム4,5、プライマー伸長法が広く使用されているフィールドの規制範囲に焦点を当てています。 TA-システムが安定しており、内因的に積極的な毒性タンパク質や毒性6,7を打ち消す主に不安定なタンパク質またはRNA抗毒素で構成され、原核生物のゲノム中に存在する小さな遺伝的要素である。毒素活性は、しばしば複製の阻害、細胞壁合成、または他の機構によって作用するが、ほとんどの場合RNase活性8,9によってれる。一般的に、RNアーゼ特異性プライマー伸長法であるそのうちの一つの異なる試験を行うことによって決定される。切断された完全長断片の混合物が同時にそれらの5 '末端を決定するために分析することができるように、プライマー伸長反応は、この用途に非常に適している。 、in vitroおよびin vivoプライマーエクステンション内のミックスを使用特定の毒素のRNase切断は、 例えば、配列特異性は、10-13を決定することができる。

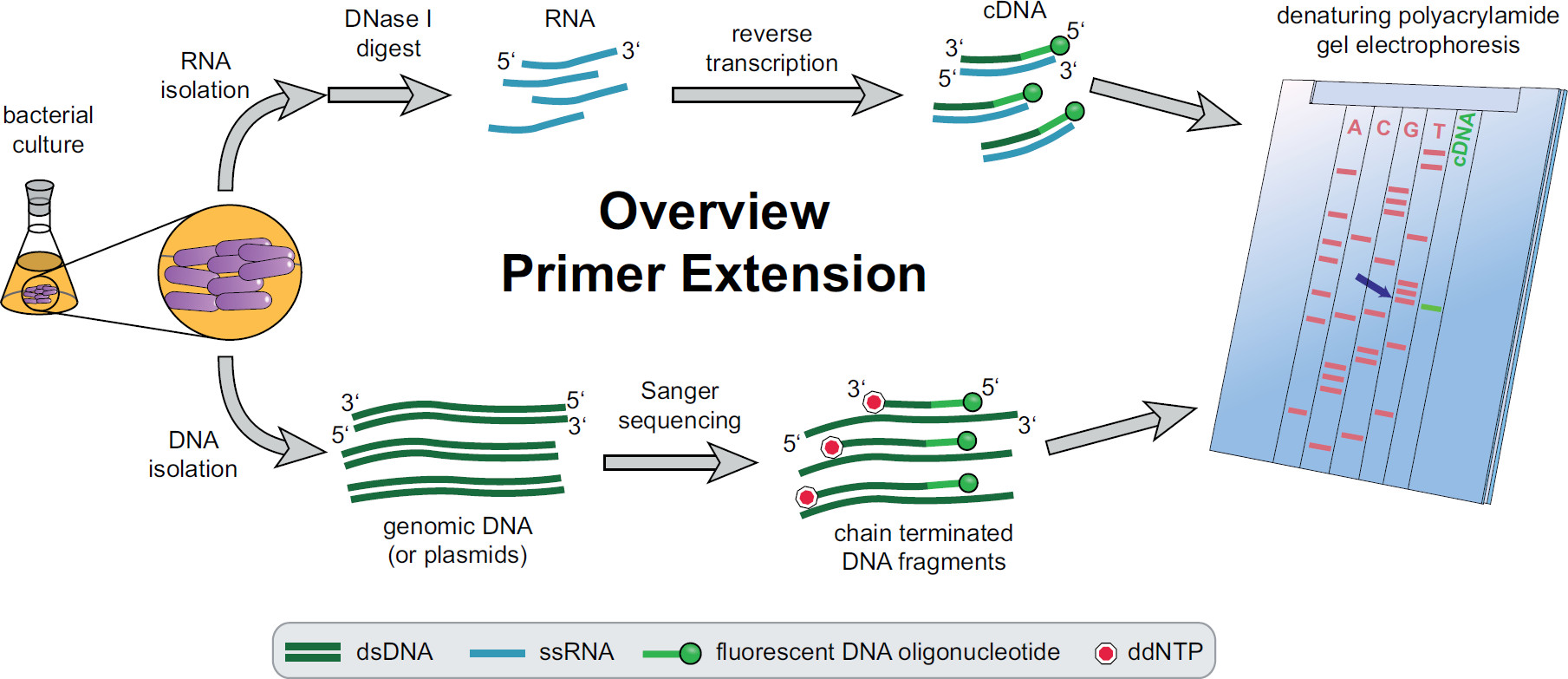
プライマー伸長手順の概要を図1。細菌培養実験のニーズに応じてインキュベートし、処理される。全RNAを、細胞から抽出したDNAの痕跡を除去するためにDNアーゼIで処理したcDNAを得た標的特異的蛍光DNAプライマーを用いて逆転写反応に供される。ゲノムDNAまたはプラスミドを抽出し、続いてcDNA断片とサイズ比較用蛍光サンガー配列決定反応のために使用される。プライマー伸長産物は、変性尿素ポリアクリルアミドゲル上サンガーシークエンシング産物と一緒に実行して、自動化されたレーザー顕微鏡で分析する。ラcDNAのバンドとラインアップしているシーケンシングベースSTの5 'cDNAの端(青い矢印)のベース。 Feketeの中でより多くの情報、 ら 3 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
全体のプライマー伸長手順の概要を図1に見ることができる。簡単に説明すると、細菌細胞を培養し、収穫し、細胞ペレットを溶解し、RNAを抽出した。精製されたRNAは、逆転写酵素のためのテンプレートとして作用し得るDNA分子の痕跡を除去するためにDNアーゼIで処理する。特異的な蛍光プライマーは、RNAに付加関心領域にハイブリダイズし、続いて一本鎖の相補的DNA(cDNA)の結果として、逆転写されている。配列決定ラダーは、蛍光プライマーを用いた従来のサンガー配列決定によって作成され、プライマー伸長cDNA断片の横に変性ポリアクリルアミドゲル上で分離する。結果として生じるゲルは、対象の5 '末端の同定を可能にする、蛍光バンドを比較することによって分析される。転写開始点およびプロセシング部位は、次いで配列比較によって個別に評価される。
プロトコル
1.高収量のRNAの準備
- RNAの単離
NOTE:トータルRNAの高濃度は、プライマー伸長反応のために必要とされる。スピンカラムキットは、通常必要とされるRNAの量を得られない(〜5から16μgの5μlの体積中)。したがって、酸グアニジニウムチオシアネート - フェノール - クロロホルム抽出法を用いて精製を以下に概説する、推奨される。
注:フェノールは、発がん性、有毒で腐食性のある。化学物質等安全データシートを読み、適切な保護とヒュームフードの下でご利用下さい!- 4600×gおよび4℃で10分間の遠心分離によって所望の収穫として、細菌細胞(この例では、 黄色ブドウ球菌や大腸菌 ) を成長させるか、治療する。注: - 70細胞ペレットを-20℃で数週間保存することができる一般的に、我々は20の合計OD 600が収穫。
- 酸グアニジニウムチオシアネート - フェノール - クロロホルム溶液1mlで細胞ペレットを再懸濁し、に転送2ミリリットルは0.1ミリメートルガラスジルコニウム/シリカビーズの0.5ミリリットルを含むカップをねじ込みます。
- 溶解細胞をそれぞれの実行後5分間氷上でサンプルを冷却する3つのラウンドのために30秒間6.5メートル/秒での高速予備校/ビーズビーターで三回、。注:ホモジナイズ試料は、数週間にわたって、-80℃で保存することができる。
- RTで5分間ライセートをインキュベートし、次に200μlのクロロホルムを追加します。
- 激しく振りまたはRNAを抽出するために30秒間のサンプルをボルテックス。
- 15,000×gおよび4℃ - 13000で15分間遠心後、3分間室温でインキュベートする。 (クリア、RNAが含まれています)溶剤が下層の有機相に分離されている(ピンク、タンパク質を含む)、相間(白、DNAを含む)と、上部水相:注意してください。
注:この手順から、使用上の唯一のRNaseフリーの試薬およびプラスチック食器! - 適切に新鮮なRNaseフリー1.5ミリリットルの反応チューブ、ラベルを用意し、ほぼ同じVOを使用する(100%RNaseを含まないイソプロパノール各約500μlを添加する以前のチューブ内の水相としてLUME)。
- 角度でチューブを握り、RNaseを含まないチップを使用して準備されたチューブに水相(約500μl)を転送する。相間を乱すことがありません。
- 数回転倒し、RTで10分間インキュベートすることによりRNAを沈殿させる。
- 遠心13000で15分間サンプル- 、15,000×gで 4℃、ピペッティングまたは吸引(ウォータージェットポンプと哺乳瓶)によって上清を除去する。一番下に白い透明なRNAペレットを乱さないようにしてください。
- 洗浄するために70-80%のRNaseフリーのエタノール(ボルテックスはしない)の1ミリリットルを追加します。注意:エタノール中のRNAを-20℃で数週間保存することができる。
- 7500×gおよび4℃で5分間遠心し、ピペッティングまたは好ましくは吸引により上清を捨てる。
- ヒュームフード下で30分 - 15のための空気の乾燥RNAペレット。過乾燥しないでください、そうでなければペレットは再溶解するのが難しいかもしれません。
- 5でペレットを再懸濁RNアーゼフリーのddH 2 OまたはRNA記憶バッファの0μlの。
- 微量紫外可視分光光度計や石英キュベット(従来光度計)を有するRNA濃度を測定し、DNアーゼI消化に進む。
- DNAの痕跡を除去するために、RNAのDNアーゼI消化
注:DNAは逆転写(プライマー伸長)反応においてスプリアスの鋳型として作用することができるので、試料から除去されるべきである。 RNA溶液からDNAを除去するための様々な方法は、通常、DNase消化に頼るが利用可能である。 DNA除去のためのシンプルだが効果的かつコスト効率的な方法を以下に概説する。- 37℃に予熱水浴。
- 1.5ミリリットルの反応管に、表1に列挙した化合物を混合する。
- 水浴中37℃で1時間混合物をインキュベートし、その後フェノール/クロロホルム抽出に直接進む。注:これは、RNAを低下させる可能性がありとしてDNアーゼIの熱不活性化は、推奨されません。
- フェノール/クロロホルムDNアーゼI消化後のRNAの抽出
注:RNAをDNアーゼI消化から遊離ヌクレオチド、DNAフラグメントおよび緩衝液成分を除去するために精製されなければならない。フェノール/クロロホルム抽出は、RNAサンプルの高い回収率および濃度を可能にし、したがって、以下に概説する。彼らはこれらの要件を満たす場合にRNA精製のための他の方法もまた、使用され得る。- 私は2ミリリットルの反応チューブに2250μlのサンプルに混ぜて消化を500μlのDNaseを分割します。
- (:24:5 - 1、pH4.5の水飽和フェノールで、クロロホルムとイソペンタノール、25の比率)酸性P / C / I溶液の1容量(250μl)を追加します。
注:P / C / I溶液は、発がん性有毒で腐食性のある。化学物質等安全データシートを読み、適切な保護とヒュームフードの下でご利用下さい! - 3分 - 1のためのボルテックスプラットフォーム内で激しく渦や場所。
- 15,000×gおよび4℃ - 13000で30分間遠心する。
- アッパー(水性)相を収集し、新しいチューブ(250μl)をへ移す。
- 3 M酢酸ナトリウムpHが5.2の1/9量(28μl)を追加します。
- 純エタノール(700μl)を2.5-3のボリュームを追加します。
- 3時間 - 30分または2のために-20℃で-80℃でまもなくと場所ボルテックスで混ぜる。必要に応じて、-20℃でのRNAのO / Nを格納します。
- 15,000×gおよび4℃ - 13000で60分間- 30遠心する。
- ピペッティングまたは吸引により上清を除去します。
- ペレットに70%エタノール1mlを添加することにより、ペレットを洗浄する。サンプルをボルテックスしないでください。
- 15,000×gおよび4℃ - 13000で5分間遠心分離サンプル。
- ピペッティングまたは吸引により上清を除去します。
- ヒュームフード下の空気の乾燥ペレット。 CO / Nの必要に応じて-20℃でペレットを保管してください。
- 2分間ボルテックスすることによって、H 2 O を処理した30μlのDEPCでペレットを溶解し、PEを溶解するために、このソリューションを使用サンプルあたりの対応する第二の管のllet(つの抽出ペアあたり30μlの溶液)。
- RNA濃度を測定し、平均的に発現したmRNAのプライマー伸長で使用するために1μg/μlのを超えていることを確認してください。
- 必要に応じて、数ヶ月までの数週間-20℃でRNAを保存する。
2.プライマー伸長反応
- プライマーデザイン
注:プライマー伸長実験のためのプライマーを設計する場合、PCRプライマーの設計の一般的なガイドラインに従う(本論文ではより多くの情報と議論のセクションのためのマニュアルに付随する自動化されたゲルシーケンサーを参照)。- 、比AT(iv)の程度のアニーリング温度を有している。具体的には、バランスのとれたGCを有し、(ii)の(iii)の3 '末端にGまたはCを有し、プライマーは、(i)塩基の実行が含まれていないことを保証する55 - 関心領域の下流に60°Cおよび(v)結合の少なくとも50塩基対、より良好な100 bpが鮮明な画像を受信する。
- プライマー伸長反応
注:プライマー伸長反応(cDNA合成)鋳型RNAを多量に必要とする。使用されるRNAの量が低いように選択された場合、信号を検出するには低すぎるかもしれません!上記のように私たちはそのためのRNAの精製をお勧めします。
注:注意:使用のRNaseフリーの試薬およびプラスチック食器!!!- 95°Cの温度にサーモサイクラーを予熱し、使用し、再現性の容易さのためにサーモサイクラー内のすべてのさらなるインキュベーション工程を行う。
- 各RNAサンプルのPCRチューブに、表2からの化合物を混合する。
- 95℃で1分間、サンプルを変性。
- RNAおよびプライマーをハイブリダイズさせるために5分間、氷と寒さにチューブを置きます。
- 47℃にPCR装置を設定してください。
- 一方で、表3に記載したように逆転写マスターミックスを調製する。
- 各ハイブリダイズされたRNAに逆転写マスターミックス4μlを添加するサンプル。
- 47℃で1時間のためのチューブをインキュベートします。注:AMV RTのための最適温度は42℃である、しかし、より高い温度は、RNA分子の二次構造を克服するのに役立つ。
- 2分間95℃にサンプルを加熱することによって反応を停止する。
注:ホルムアミドは有毒で、腐食性で、胎児に有害であることができます。 、材料安全データシートを読んで取り扱いに注意して、適切な保護具を着用してください! - 暗闇の中で-20℃で2週間ホルムアミドローディング色素(95%(v / v)の脱イオン化ホルムアミド、(w / v)のブロモフェノールブルー10mMのEDTA、0.05%)とO / Nのための店の6μlを添加する。
シーケンシングラダーの調製
注:シークエンスラダー反応は、プラスミドまたはゲノムDNAの大量の中程度の量のいずれかが必要です。可能な限り、シーケンス反応におけるプラスミドの使用は、分離のしやすさと高SIGに推奨されますNAL強度。他のケースでは、我々は日常的にE.からゲノムDNAを調製するためにMarmur 5,14から採用する方法を使用し大腸菌およびS.フェノールを使用する必要なく、 黄色ブドウ球菌細胞。原理的に多量のゲノムDNAの純度を生じる任意の方法を用いることができる。
- ゲノムDNAの単離
- Eの10ミリリットルを育てるcoliまたはS. LB、BM 5またはTSB培地中で黄色ブドウ球菌細胞のO / N。
- 15ミリリットルファルコンチューブ中で4600×gで10分間の遠心分離によって細胞を回収する。
- いくつかのミニ調製キット(50mMのトリス-HCl pH8.0で、10mMのEDTA、100μg/ mlのRNアーゼA)に見られるように、2ミリリットルの緩衝P1で再懸濁したペレット。
- 40μlのリソスタフィン(-20°Cで0.5 mg / mlの、ストレージ) - 20と60分 - 45溶解細胞。注意:E.について大腸菌細胞は、酵素前処理を省略することができるいずれか、またはリゾチームを使用した。
- サスペンションとincubatに飽和SDS溶液(45%エタノール中)を100μlを追加します。37℃で5分間の電子。
- 650μlの5 MのNaClO 4と簡潔に渦セルを追加。
注:クロロホルムが潜在的な発癌物質である。化学物質等安全データシートを読み、適切な保護とヒュームフードの下で使用してください!!! - 混合物には、(1比24)と少なくとも60秒間振るクロロホルム/イソペンタノールの3ミリリットルを追加します。注:液体は均質な白色エマルジョンに変わるはずです。
- 相を分離する4600×gで、室温で10分間遠心分離サンプル。
- 慎重に新しいチューブに明確な上限(水性)相を転送します。解が濁っている場合は、クロロホルム/イソペンタノール抽出を繰り返す。 DNA溶液の体積を測定し、エタノール(100%)の2倍量の新鮮なチューブを準備する。
- ゆっくりとチューブを含むエタノールにDNA溶液をデカント又はピペット。注:DNAはボトム時や白浮きクラスタとして完全に脱水上のような透明な、密なコイルを沈殿させる必要があります。
- Dを取得NAガラスパスツールピペット( 図2)から作られたフックを使用し、1mlの70%エタノール中の個々の管の中に浸漬することにより、二回、各サンプルを洗う。
- ラックに直立フックを置き、空気が60分間、ペレットを乾燥させます。必要であれば、室温で数日間乾燥させたDNAを保存する。
- のddH 2 O500μlの- DNAは、100を含む2.0ミリリットル反応管にガラスフック配置をカバー切り離すことによりDNAを溶解する1500 ng /μLで - 千の最終DNA濃度に音量を調整します。ゲノムDNAの18μgの - 1の配列決定反応のために、10を使用しています。

図のDNA釣り竿を作成する方法について2.命令。ブンゼンバーナーの炎にガラスパスツールピペットの先端を持ってください。これは、tにおける小さなフックを作成、ガラスが数秒後に溶融を開始させる彼が終わる。すばやく炎から削除して、1分間冷ます。
- プラスミド分離
- 標準ミニ調製キットを用いてプラスミドを調製し、溶出緩衝液(10mMのトリス-Cl、pH8.5)中に溶解する。 1シークエンシングラダー用プラスミド500ngの - プラスミドの大きさに応じて、100を使用しています。
- サンガー配列決定反応
注:プライマーエクステンションの目的のためにうまく機能7-デアザdGTPを持つ蛍光標識されたプライマー配列決定キットを使用して単純なプロトコルを下回って下さい。詳細については、シーケンシングキットのマニュアルを参照してください。シークエンシング反応が同じ長さの製品を作成するために、プライマー伸長反応と同じプライマーを使用しなければならないことに注意してください。- 1μlのDMSOおよび1μlの蛍光標識したプライマー(2ピコモル/μL)と - (15μgの〜10)のゲノムDNAを12μlを混ぜる。
- 4配列決定反応ミックス(A、C、GまたはT)の各1μlに、DNA / DMSO /プライマーミックスの3μlを添加する。
- PCR装置にサンプルを置き、以下のPCRプログラムを実行します。2分間95℃。 20秒、20秒、54℃、30秒間70℃、95℃の35サイクル。 4℃で永遠に保つ。
- 実行後に、マシンからサンプルを削除する数日から数週間のために氷(短期)または-20℃で上のローディング色素と店の6μlを添加する。
4.ゲルのセットアップおよび装置ラン
注:配列決定ゲル装置を組み立てる方法の詳細は、ゲルを調製する方法、およびゲルが実行され、製造業者のプロトコルに記載されています。
- 調剤
- 表4に示したように10倍TBEを準備します。
- ゲルランの日に、超純をddH 2 Oで1×TBEバッファー1リットルを準備
- 10%(w / v)のAPSを準備します。注:数ヶ月間-20℃で200μlのアリコートで保存することができますが、アクティビティは、時間の経過とともに低下することがあり<。/ LI>
- ゲルキャスティングチャンバアセンブリ
- ガラス板の間ほこりや糸くずを避けてください。そこで徹底的にウェットティッシュを使用して作業面をクリーニングします。
- ガラス板の内側にイソプロパノール、次に両側に使い捨ての紙タオル、蒸留水を用いて、25センチメートルのガラス板の対をきれい。
- 背面ガラス板上の0.25mmのスペーサを配置し、上部にノッチガラス板( 図3)を下げる。
- 切り欠きのある端を持つガラス板と上向きにレールエントリーパイロットの両側にゲルレールを取り付け、軽くノブを締めます。

図3は、ゲル電気泳動のガラス板の分解斜視図。ガラスプレートは一方向に使用されるべきである。内方ガラス板の内側と外側を向くように注意してくださいサイド外側に。

組み立てられたゲル装置4の表示図。ゲル溶液を注入した後、ポケットスペーサはガラス板の間溶液中に置かれる。鋳造板は、フロントガラス板とゲルレールの間にスライドレールノブを締め付けることにより固定されている。
- ゲルキャスティング
注:非重合したアクリルアミドは神経毒です!化学物質等安全データシートを読み、適切な保護を使用してください!!!- ビーカーに、表5に列挙した化合物を加え、攪拌棒及びマグネチックスターラーを用いて混合する。
- すぐにAPSとTEMEDを追加した後、50ミリリットルのシリンジ内のゲル溶液を取るチップを0.45 nmフィルターを配置。
- どちらか一方の手でガラス板の上端を保持またはsandwicを配置20° - ゲルキャスティングにおけるhは傾き角度の付いた10を作成するために立っている。
- ゆっくりと連続的に一方の側から他方の側のシリンジ先端を移動させながらガラス板の間ゲル溶液を分配し、ゲル溶液は、底端部を満たしたら止める。
- 側に移動したり、完全にバブルフックを使用して、任意の形成された気泡を除去。
- 切り欠きのある端でガラスプレート間のゲルポケットスペーサー(0.25 mm)をスライドさせて、ゲル溶液中に沈め、鋳造板を取り付けることにより固定する。
- (完全に組み立てられた装置については、図4を参照してください)軽く上レールのネジを締めます。
- 2時間 - 1のためのジェルセットをしてみましょう。
- キャストプレートとポケットスペーサーを外し、塩とゲル残留物からポケットを掃除。
- のddH 2 Oですすぎ、ティッシュペーパーで余分な溶液を拭き取る。
- ゲルの実行と可視化
注:配列決定用ゲルに直接ゲルイメージャーで電気泳動され、一方蛍光を同時にレーザー顕微鏡によって検出される。ゲルが最初に実行し、染色し、可視化される従来のゲル電気泳動とは対照的に、検出部を固定し、それらがレーザーを渡すようにリアルタイムでバンドをスキャンする。より最近のバージョンに採用することが可能で概説されているOS / 2上でImagIRデータ収集ソフトウェアのための手順は、以下。詳細については、ユーザーマニュアルを参照してください。- フロントガラス板上でのゲルレールにバッファタンクホルダーをスライドさせ、ノブを締めます。
- 加熱板に対して自動化されたゲルイメージャの下ゲル槽にゲルを置き、装置のブラケットにレールエントリパイロットをスライドさせて固定します。
- 電源コードを使用して電源に上部バッファー槽を下バッファ室を閉じ、接続し、下方および上方ゲル緩衝室に1×TBEバッファーを埋める。
- 存在する場合、繰り返しポケットにバッファをピペッティングすることにより、塩残留物からゲルポケットを清掃してください。
- トップバッファの蓋を用いたトップバッファタンク室を閉じてください。
- 機械のドアを閉じ、イメージャ及びコンピュータ上で切り替えて、ベースImagIRデータ収集ソフトウェアを起動します。
- 新しいプロジェクトファイル( ファイル- >新規...)を作成し、プロジェクト·ファイル名を入力し、適切なレーザーレンジ(700または800 nm)を選択し、[OK]で確定します。
- [オプション] - > [オートゲイン...トップの画像メニューからを選択し、オートゲイン測定を開始し、[OK]をクリックして設定を受け入れるには、[自動]をクリックします。
- フォーカス...スキャナ制御メニューから、[自動]ボタンをクリックして、[OK]をクリックして設定を受け入れる> [オプション]を選択してレーザーの焦点を合わせる。
- 新たに焦点を当てた領域に調整するオートゲイン手順を繰り返します。
- セットアップこれらの設定に応じてスキャナ制御:2000 V、35ミリアンペア、45 W、45℃、スキャンフィルター:3、スキャン速度:3。
- 20分間の空のゲルをプレラン(電圧オンを選択してEnterを押します)。
- 一方、その後、90℃まで2分間、PCR機におけるシークエンシングラダーおよびプライマー伸長生成物を加熱し、氷上で冷却する。
- 、電気泳動を停止し、自動化ゲルシーケンサーを開き、上部バッファータンクのふたを取り外します。
- ガラス板の間にサメの歯のコームを差し込み、少しサメの歯を有するゲルを貫通します( 図5を参照)。

図5.クローズアップサメの歯のコームを有するゲルの眺め。サンプル(紫色)は、サメの歯の間に適用されます。
- (サメの歯によって形成される)各ゲルのポケットにプライマー伸長産物または配列決定ラダー反応の2μlの - ピペットどちらか1。
- すべてのポケットが必要な場合は、内防ぐためにローディング色素と空のポケットを埋める一貫性のあるランニング振る舞い。
- ゲルシーケンサーのバッファタンクとドアを閉じます。
- (ON電圧とレーザーONを選択し、を押し、電気泳動を起動して、レーザーをオンにする )。
- 関心領域は、レーザーを通過した後に電気泳動を停止します。
結果
図6に示されるように、プライマー伸長反応は、目的の転写物の転写開始点を決定するために用いることができる(通常-10および-35要素によって識別される)のプロモーター領域を推定するのを助けることができる。一番上の(最長)のcDNA断片は、mRNAの5 '末端を表し、従って、配列決定ラダーと比較した場合に容易にマッピングすることができる。
ディスカッション
蛍光プライマー伸長はTSP-または二次RNA処理識別のためのいずれか、RNAの5 '末端を決定するための簡単かつ迅速な方法である。原因蛍光プライマーの使用のために、反応が設定することができ、(放射性標識したプライマーの場合と異なり)追加のセキュリティ対策なしで実行されます。サンプルは、蛍光によって検出されるように、電気泳動、X線フィルムが一般的に使用される放射?...
開示事項
The authors have nothing to disclose that would present a conflict of interest.
謝辞
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
資料
| Name | Company | Catalog Number | Comments |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
参考文献
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved