
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Fluoreszenzbasierten Primer Extension-Technik, um Transkriptionsstartpunkte und Schnittstellen von RNasen Bestimmen
In diesem Artikel
Zusammenfassung
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
Zusammenfassung
Fluoreszenzbasierten Primer-Extension (FPE) ist ein Molekular Methode, um Transkriptionsstartpunkte oder Verarbeitungsstätten von RNA-Molekülen zu bestimmen. Dies wird durch die reverse Transkription der RNA von Interesse mit spezifischen fluoreszenzmarkierten Primern und anschließender Analyse der resultierenden cDNA-Fragmente durch denaturierende Polyacrylamid-Gelelektrophorese erreicht. Gleichzeitig wird eine traditionelle Sanger-Sequenzierungsreaktion auf dem Gel laufen gelassen, um die Enden der cDNA-Fragmente anzeigen, um deren genaue entsprechenden Basen. Im Gegensatz zu 5'-RACE (Rapid Amplification von cDNA-Enden), wobei das Produkt muss kloniert und sequenziert mehreren Kandidaten kann die Masse der cDNA-Fragmente durch Primerverlängerung erzeugt werden gleichzeitig in einem Gel laufen detektiert werden. Zusätzlich kann das gesamte Verfahren (von der reversen Transkription der endgültigen Analyse der Ergebnisse) an einem Arbeitstag abgeschlossen sein. Durch die Verwendung fluoreszierend markierter Primer, die Verwendung von gefährlichen radioaktiven Isotop markierten Reagenzienvermieden werden und die Bearbeitungszeit verringert werden als Produkte können während der Durchführung der Elektrophorese nachgewiesen werden.
In dem folgenden Protokoll beschreiben wir ein in vivo Fluoreszenz Primer-Extension-Methode auf das 5'-Ende der RNA schnell und zuverlässig zu erfassen, abzuleiten Transkriptionsstartpunkte und RNA-Verarbeitungsstellen (zB durch Toxin-Antitoxin-Systemkomponenten) in S. aureus, E. coli und anderen Bakterien.
Einleitung
Primerverlängerungs 1 ist ein Molekular Methode, um die 5'-Enden der spezifischen RNA-Moleküle bis zu einer eine Basisauflösung bestimmen. Der Vorteil gegenüber anderen Verfahren, wie 5'-RACE (schnelle Amplifikation von cDNA-Enden) ist die schnelle Durchlaufzeit und die Fähigkeit, leicht zu analysieren, die eine Mischung von verschiedenen Längen von RNA-Molekülen.
Diese Methode funktioniert, indem RNA-Moleküle, die Transkription Reaktionen Reverse mit spezifischen fluoreszierenden Primer, Erzeugung cDNA-Fragmente bestimmter Längen. Diese cDNA-Moleküle werden neben der traditionellen Sanger-Sequenzierungsreaktionen 2 auf denaturierenden Polyacrylamid-Gelen und durch ihre Fluoreszenz aufgrund der Verwendung von fluoreszenzmarkierten Primern nachgewiesen werden. Die Längen der cDNA-Fragmente werden dann durch Vergleich mit der Sequenzierungsleiter beurteilt, so dass die Zuordnung der 5'-RNA-Ende.
Traditionell werden Primer-Verlängerungsreaktionen in Verbindung verwendet werdenmit radioaktiven Isotopen, um cDNA-Moleküle auf Röntgenfilmen zu erkennen. Wegen der Gesundheitsrisiken, Entsorgungsprobleme und der einfachen Handhabung, neuere Protokolle zu nutzen Fluoreszenz zum Nachweis der Primer-Extension mit automatisierten Sequenzer, wenn ihre Empfindlichkeit ist etwas niedriger. Mit fluoreszenzmarkierten Primern kann die wiederkehrende Radiomarkierungsverfahren verzichtet werden, als fluoreszierende Primer sind für eine lange Zeit (mehr als ein Jahr in unseren Händen) stabil.
Die Methode, die wir hier beschreiben, nutzt ein automatisiertes Gel Sequenzer, aber mit leichten Modifikationen können Kapillare Sequenzern auch für die cDNA Trennung und Detektion 3 verwendet werden. Die Parallelität der Gel-Analyse ermöglicht es, auch eine kleine Menge der RNA-Spaltung oder Verarbeitung zu erfassen. Ein weiterer Vorteil ist die hohe Auflösung dieses Verfahrens als terminale Spaltung oder Verarbeitung von nur einer Basis detektiert werden.
In Bezug auf den Nachweis von RNA-Spaltung oder Verarbeitung, typically zwei verschiedene Arten von Primer-Extensions unterschieden. In einem Fall wird die enzymatische Behandlung in vitro durchgeführt unter Verwendung gereinigter RNA und gereinigtes Enzym, wohingegen in dem anderen Fall, wird die Verarbeitung in vivo durchgeführt und die resultierende RNA wird gereinigt. In beiden Fällen wird die RNA unterzogen, um eine Primer-Extension durchgeführt in vitro, jedoch in Abhängigkeit von der Quelle der RNA wird das Verfahren entweder bezeichnet eine in vitro oder in vivo Primerverlängerung. In dem Protokoll wir hier konzentrieren wir uns lediglich auf der in-vivo-Primerverlängerung, aufgrund der Einfachheit der Nutzung (keine gereinigten Proteine notwendig) und die Möglichkeit zur Transkriptionsstartpunkte und die Verarbeitung in der gleichen Zeit zu bestimmen. Allerdings sind in vitro Primerverlängerungen im Prinzip die gleiche Weise eingestellt und dieses Protokoll kann als Ausgangspunkt dienen.
Das hier dargestellte Verfahren kann auf viele Bakterienarten, solange sie zugänglich sind hoch angewendetReinheit und High-Yield-Vorbereitung von Nukleinsäuren.
Die Forschung in unserem Labor konzentriert sich auf den Regelungsbereich von Toxin-Antitoxin (TA) Systeme 4,5, einem Bereich, in dem die Primer-Extension-Methode wird intensiv genutzt. TA-Systeme sind kleiner in prokaryotischen Genom, das aus einem stabilen und endogen aktive toxische Protein und einer meist instabil Protein oder RNA-Antitoxin, die Toxizität entgegen 6,7 bestehen vorliegenden genetischen Elemente. Toxinaktivität wird manchmal durch Hemmung der Replikation, der Zellwandsynthese oder andere Mechanismen, die von RNase-Aktivität 8,9 ausgeübt wird, aber meistens. Typischerweise wird RNase Spezifität durch die Durchführung verschiedener Tests, von denen der eine der Primer-Extension-Methode bestimmt. Primer-Verlängerungsreaktionen sind für diese Anwendung gut geeignet, da eine Mischung aus gespaltenen und in voller Länge Fragmente können gleichzeitig analysiert werden, um festzustellen ihren 5'-Enden. Verwendung einer Mischung von in vitro und in vivo Primerverlängerungen, diespezifische Toxin RNase-Spaltung, zB Sequenzspezifität bestimmt 10-13 werden.

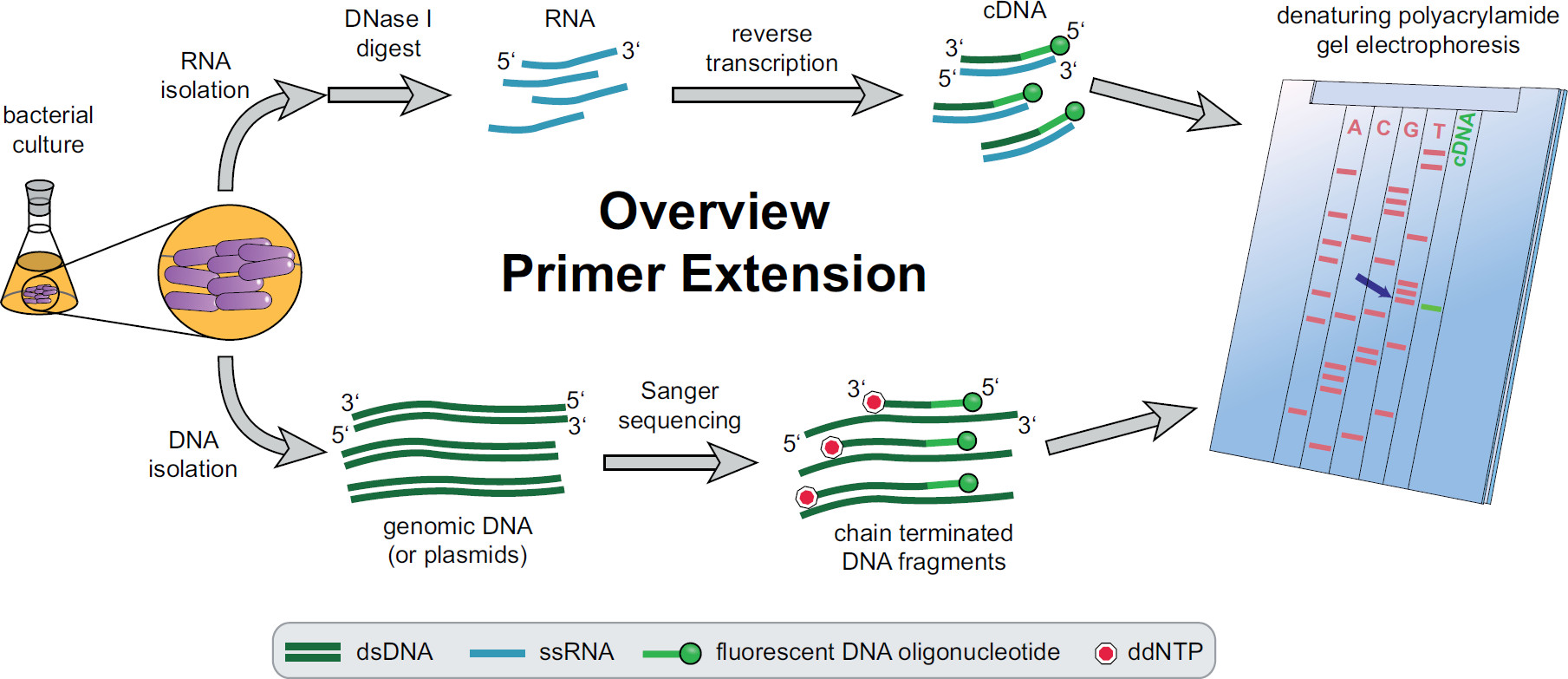
Abbildung 1. Übersicht der Primerverlängerungsverfahren. Bakterielle Kulturen werden nach den experimentellen Anforderungen inkubiert und behandelt. Gesamt-RNA wird aus den Zellen mit DNase I behandelt, um DNA-Spuren zu entfernen, und einer Umkehrtranskriptionsreaktion unter Verwendung von Ziel-DNA spezifischen Fluoreszenz Primer wodurch cDNA unterworfen extrahiert. Genomische DNA oder Plasmide extrahiert und anschließend zur Fluoreszenz Sanger-Sequenzierungsreaktionen zum Größenvergleich mit den cDNA-Fragmenten verwendet. Primerverlängerungsprodukte sind neben Sanger-Sequenzierungsprodukte auf einem denaturierenden Harnstoff-Polyacrylamidgel laufen und mit einem automatisierten Laser und Mikroskop analysiert. Die Sequenzierung Basis, die Linie mit dem cDNA-Band ist die last Basis des 5 'cDNA Ende (blauer Pfeil). Weitere Informationen im Fekete et al. 3 Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
Eine Übersicht über den gesamten Primerverlängerungsverfahren kann in 1 gefunden werden. Kurz gesagt, kultiviert, geerntet sind bakterielle Zellen, das Zellpellet lysiert und die RNA extrahiert. Gereinigte RNA wird dann mit DNase I behandelt, um Spuren von DNA-Molekülen, die als Matrizen für die reverse Transkriptase wirken könnten, zu entfernen. Spezifische fluoreszierende Primer an die RNA gegeben, um die Region von Interesse hybridisiert und anschließend revers transkribiert, wodurch einzelsträngige komplementäre DNA (cDNA). Ein Sequenzierleiter wird von traditionellen Sanger-Sequenzierung erstellt die Verwendung von Fluoreszenz Primer und auf einem denaturierenden Polyacrylamid-Gel neben der Primerverlängerung cDNA-Fragmente getrennt. Das resultierendeGel wird durch den Vergleich der Fluoreszenzbanden, die eine Identifizierung des 5'-Enden von Interesse analysiert. Transkriptionsstartpunkte und Verarbeitungsstellen werden dann einzeln durch Sequenzvergleiche geprüft.
Protokoll
1. High Yield RNA-Präparation
- RNA-Isolierung
HINWEIS: Hohe Konzentrationen von Gesamt-RNA werden für die Primer-Verlängerungsreaktion benötigt. Spinsäule Kits in der Regel nicht die Menge der RNA benötigt Ausbeute (~ 5 - 16 & mgr; g in 5 & mgr; l Volumen). Daher Reinigung unter Verwendung der Phenol-Chloroform-Extraktion Methode empfohlen, unten beschrieben.
HINWEIS: Phenol ist krebserregend, giftig und ätzend. Bitte lesen Sie die Sicherheitsdatenblätter und verwenden Sie unter einer Abzugshaube mit entsprechenden Schutz!- Wachsen oder Behandlung der Bakterienzellen (S. aureus oder E. coli in diesem Beispiel), wie gewünscht und die Ernte um 10 min Zentrifugation bei 4600 × g und 4 ° C. Anmerkung: In der Regel ernten wir insgesamt OD 600 von 20 - 70. Die Zellpellets können für mehrere Wochen bei -20 ° C gelagert werden.
- Zellpellet in 1 ml saure Guanidiniumthiocyanat-Phenol-Chloroform-Lösung und Transfer zu einem2 ml Schraube Tasse mit 0,5 ml 0,1 mm Glas Zirkonium / Silica-Kügelchen.
- Lyse der Zellen dreimal in einer schnellen prep / Kugelmühle mit 6,5 m / sec für 30 sec für drei Runden, die Kühlung der Proben auf Eis für 5 min nach jedem Lauf. Hinweis: homogenisierte Probe bei -80 ° C für mehrere Wochen gelagert werden.
- Inkubieren Lysat 5 min bei RT und dann mit 200 ul Chloroform.
- Kräftig schütteln oder vortexen die Probe für 30 s bis RNA zu extrahieren.
- Inkubieren bei Raumtemperatur für 3 min, dann für 15 min bei 13000 zentrifugieren - 15.000 × g und 4 ° C. Hinweis: Die Lösungsmittel werden in eine untere organische Phase abgetrennt (rosa, enthält Proteine), eine Interphase (weiß, enthält DNA) und eine obere wässrige Phase (klar, enthält RNA).
HINWEIS: Von diesem Schritt auf die Verwendung nur RNase freie Reagenzien und Plastikwaren! - Bereiten Sie frischen RNase-freies 1,5 ml Reaktionsgefäße, Label angemessen und etwa 500 ul 100% RNase frei Isopropanol jeder (verwenden Sie etwa die gleiche volumen als wässrige Phase in der vorhergehenden Rohr).
- Halten Sie das Röhrchen in einem Winkel und übertragen die wässrige Phase (etwa 500 ul) auf die vorbereiteten Röhrchen mit RNase kostenlose Tipps. Bitte nicht stören die Interphase.
- Ausfällung der RNA durch mehrmaliges Wenden und Inkubation für 10 min bei RT.
- Zentrifugieren Sie die Proben für 15 min bei 13.000 - 15.000 × g und 4 ° C und entfernen Überstand mit einer Pipette oder Aspiration (Wasserstrahlpumpe und Babyflasche). Bitte nicht stören die weiße transparente RNA Pellet am Boden.
- 1 ml der 70-80% RNase freiem Ethanol (nicht vortexen) zu waschen. Hinweis: RNA in Ethanol kann für mehrere Wochen bei -20 ° C gelagert werden.
- Zentrifuge für 5 min bei 7500 × g und 4 ° C und Überstand verwerfen durch Pipettieren oder vorzugsweise Aspiration.
- Luft trocknen RNA-Pellet für 15 - 30 min unter dem Abzug. Nicht übertrocknen, sonst Pellets schwer wieder zu lösen sein.
- Das Pellet in 50 ul RNase freies ddH2O oder RNA Speicherpuffer.
- Messen RNA-Konzentration mit einem Mikrovolumen UV-Vis-Spektrophotometer oder einer Quarzküvette (und konventionellen Photometer) und fahren Sie mit DNase I Verdauung.
- DNase I-Verdau der RNA zu DNA-Spuren zu entfernen
Hinweis: Da die DNA kann als eine Matrize in der falschen Reverse Transkription (primer extension) Reaktion wirken, sollte es aus der Probe entfernt werden. Verschiedene Verfahren zur Entfernung von DNA-RNA-Lösungen sind in der Regel auf DNase-Verdau verlassen. Eine einfache, aber effektive und kostengünstige Methode zur DNA Entfernung wird im Folgenden erläutert.- Vorheizen Wasserbad auf 37 ° C.
- Mischen der in Tabelle 1 aufgeführten Verbindungen in einem 1,5 ml Reaktionsröhrchen.
- Inkubieren der Mischung für 1 h bei 37 ° C in einem Wasserbad und anschließend unverzüglich um das Phenol / Chloroform-Extraktion. Hinweis: Die Hitzeinaktivierung der DNase I wird nicht empfohlen, da dies die RNA abzubauen.
- Phenol / Chloroform-Extraktion von RNA nach der DNase I-Verdau
HINWEIS: Die RNA muss gereinigt, freien Nukleotiden, DNA-Fragmente und Pufferkomponenten aus dem DNase I zu entfernen. Phenol / Chloroform-Extraktion erlaubt eine hohe Rückgewinnung und Konzentration der RNA-Probe und wird daher nachfolgend beschrieben. Andere Verfahren zur RNA-Reinigung können auch verwendet werden, wenn sie diese Anforderungen erfüllen.- Teilen Sie die 500 ul DNase I in zwei 250 ul Proben mischen in 2 ml Reaktionsgefäße.
- 1 Volumen (250 ul) von sauren P / C / I-Lösung (in Wasser gesättigtem Phenol, Chloroform und Isopentanol, Verhältnis von 25: 24: 1, pH 4,5-5).
HINWEIS: P / C / I-Lösung ist krebserregend, giftig und ätzend. Bitte lesen Sie die Sicherheitsdatenblätter und verwenden Sie unter einer Abzugshaube mit entsprechenden Schutz! - Kräftig Wirbel oder in einem Vortex-Plattform für 1-3 min.
- Zentrifuge für 30 min bei 13.000 - 15.000 × g und 4 ° C.
- Sammeln Sie die obere (wässrige) Phase und Transfer zum frisches Röhrchen (250 ul).
- Hinzufügen 1/9 Volumen (28 ul) von 3 M Natriumacetat pH 5,2.
- In 2,5-3 Volumen reinem Ethanol (700 ul).
- Vortexen kurz und Ort bei -80 ° C für 30 min bei -20 ° C für 2 - 3 Std. Falls erforderlich, speichern Sie die RNA O / N bei -20 ° C.
- Zentrifuge ca. 30 - 60 min bei 13.000 - 15.000 × g und 4 ° C.
- Überstand entfernen durch Pipettieren oder Aspiration.
- Waschen Sie das Pellet durch Zugabe von 1 ml 70% igem Ethanol auf den Pellets. Nicht mit dem Vortex Probe.
- Zentrifuge Proben für 5 min bei 13.000 - 15.000 × g und 4 ° C.
- Überstand entfernen durch Pipettieren oder Aspiration.
- Luft trocknen Pellet unter dem Abzug. Bewahren Sie das Pellet bei -20 ° CO / N, wenn nötig.
- Das Pellet in 30 ul DEPC behandeltem H 2 O durch Vortexen für 2 min und die Nutzung dieser Lösung an die PE auflösenllet des entsprechenden zweiten Rohr pro Probe (30 ul Lösung pro Extraktions pair).
- RNA-Konzentration zu messen, und sicherzustellen, dass 1 & mgr; g / & mgr; l für den Einsatz in Primerverlängerungs von durchschnittlich exprimierten mRNAs überschreitet.
- Falls erforderlich, speichern die RNA bei -20 ° C mehrere Wochen bis zu einigen Monaten.
2. Primer-Extension-Reaktion
- Primerdesign
HINWEIS: Bei der Gestaltung von Primern für eine Primer-Extension-Experiment, gehorchen allgemeinen Leitlinien der PCR-Primer-Design (siehe die Begleitliteratur der automatisierten Gel Sequenzer für weitere Informationen und Diskussionsteil in diesem Papier).- Insbesondere ist sicherzustellen, dass die Primer (i) Sie verläuft von Basen enthalten, (ii) über ein G oder C am 3'-Ende, (iii) eine ausgewogene GC: AT-Verhältnis, (iv) eine Glühtemperatur von etwa 55-60 ° C und (v) bind mindestens 50 bp, besser 100 bp stromabwärts von der Region von Interesse, um klare Bilder zu erhalten.
- Primer-Extension-Reaktion
HINWEIS: Die Primer-Verlängerungsreaktion (cDNA-Synthese) erfordert hohe Mengen an Matrizen-RNA. Wenn die Mengen der RNA verwendet werden, um niedrig gewählt wird, kann das Signal zu niedrig ist, um zu erfassen! Wir empfehlen daher die Reinigung der RNA, wie oben beschrieben.
HINWEIS: VORSICHT: RNase freie Reagenzien und Plastikwaren !!!- Heizen Sie den Thermocycler auf eine Temperatur von 95 ° C und die Durchführung aller weiteren Inkubation Schritte in einem Thermocycler für Benutzerfreundlichkeit und Reproduzierbarkeit.
- Mischen Sie die Verbindungen aus Tabelle 2 in einem PCR-Röhrchen für jede RNA-Probe.
- Denaturierung der Proben für 1 min bei 95 ° C.
- Die Röhrchen auf Eis und Kälte für 5 min auf RNAs und Primer hybridisieren.
- Stellen Sie die PCR-Maschine bis 47 ° C.
- In der Zwischenzeit bereiten die reverse Transkription Master-Mix, wie in Tabelle 3 beschrieben.
- In 4 ul reverse Transkription Mastermix jedem hybridisierten RNAProbe.
- Die Röhrchen für 1 h bei 47 ° C inkubieren. Hinweis: Die optimale Temperatur für AMV RT 42 ° C, helfen jedoch höhere Temperaturen, um Sekundärstrukturen der RNA-Moleküle zu überwinden.
- Stoppen der Reaktion durch Erhitzen der Proben auf 95 ° C für 2 min.
HINWEIS: Formamid ist ätzend, giftig und kann das Kind im Mutter schädlich sein. Bitte lesen Sie die Sicherheitsdatenblätter, mit Vorsicht hand und tragen Sie geeignete Schutz! - Hinzufügen 6 ul Formamid-Ladungsfarbstoff (95% (v / v) deionisiertes Formamid, 10 mM EDTA, 0,05% (w / v) Bromphenolblau) und Speicher für O / N bis zu zwei Wochen bei -20 ° C im Dunkeln.
3. Vorbereitung der Sequenzierleiter
HINWEIS: Die Sequenzierleiter Reaktion entweder moderate Mengen von Plasmiden oder hohe Mengen an genomischer DNA benötigt. Wenn möglich, wird die Verwendung von Plasmiden in der Sequenzreaktion aufgrund der Leichtigkeit der Isolierung und hohe sig empfohlennal Intensität. In anderen Fällen, die wir routinemäßig eine von Marmur 5,14 angewandte Methode, um genomische DNA aus E. vorbereiten coli und S. aureus-Zellen ohne die Notwendigkeit, Phenol verwenden. Grundsätzlich kann jedes Verfahren, das hohe Mengen und Reinheit der genomischen DNA ergibt, verwendet werden.
- Isolierung genomischer DNA
- Wachsen 10 ml E. coli oder S. aureus-Zellen O / N in LB, BM 5 oder TSB-Medium.
- Ernten der Zellen durch Zentrifugation für 10 min bei 4.600 × g in einem 15 ml Falcon-Röhrchen.
- Resuspendierte Pellet in 2 ml Puffer P1, wie in einigen Minivorbereitung Kits gefunden (50 mM Tris-HCl pH 8,0, 10 mM EDTA, 100 ug / ml RNase A).
- Zellen lysieren für 45 - 60 min bei 20 bis 40 & mgr; l Lysostaphin (0,5 mg / ml, Aufbewahrung bei -20 ° C). Hinweis: Für E. coli-Zellen die enzymatische Vorbehandlung kann entweder weggelassen werden oder Lysozym verwendet.
- Füge 100 & mgr; l gesättigter SDS-Lösung (in 45% Ethanol) zu der Suspension und INCUBATe für 5 min bei 37 ° C.
- In 650 ul 5 M NaClO 4 und die kurz Wirbelzellen.
HINWEIS: Chloroform ist ein potenzielles Karzinogen. Bitte lesen Sie die Sicherheitsdatenblätter und verwenden Sie unter einer Abzugshaube mit der entsprechenden Schutz !!! - 3 ml Chloroform / Pentanol (24: 1-Verhältnis) zu dem Gemisch und schüttelt für mindestens 60 sec. Hinweis: Die Flüssigkeit sollte in eine homogene weiße Emulsion verwandeln.
- Zentrifuge Probe für 10 min bei 4.600 × g und RT Phasen trennen.
- Die klare obere (wässrige) Phase in ein frisches Röhrchen vorsichtig zu übertragen. Wenn Lösung trüb, wiederholen Sie das Chloroform / Isopentanol Extraktion. Messen Sie das Volumen der DNA-Lösung und bereiten ein frisches Röhrchen mit 2 Volumina Ethanol (100%).
- Langsam dekantieren oder pipettieren die DNA-Lösung in das Röhrchen mit Ethanol. Hinweis: DNA als transparent, dichte Spulen am Boden oder bei vollständig entwässert als schwimmenden weißen Cluster auszufällen.
- Rufen Sie die DNA mit Haken vom Pasteur-Glaspipetten (Figur 2) gestellt und zweimal waschen jede Probe durch Eintauchen in eine einzelne Röhre von 1 ml 70% Ethanol.
- Setzen Sie die Haken aufrecht in einem Ständer und Luft trocknen das Pellet für 60 min. Falls erforderlich, speichern die getrocknete DNA mehrere Tage bei RT.
- Man löst DNA durch Abbrechen die verdeckten DNA Glashaken und Platzierung in einem 2,0 ml-Reaktionsgefäß mit 100 bis 500 & mgr; l ddH 2 O. Stellen Sie die Lautstärke auf eine endgültige DNA-Konzentration von 1,000 - 1,500 ng / ul. Zum einen Sequenzierungsreaktion, verwenden 10 - 18 ug genomische DNA.

Abbildung 2. Instruction, wie man eine DNA Angelrute erstellen. Halten Sie die Spitze einer Glaspasteurpipette in die Flamme eines Bunsenbrenners. Dies bewirkt, dass das Glas an zu schmelzen beginnen nach einigen Sekunden, die Schaffung eines kleinen Haken bei ter beenden. Schnell zu entfernen, von der Flamme nehmen und abkühlen lassen für 1 min.
- Plasmid Isolation
- Vorbereitung Plasmide unter Verwendung von Standardminipräparation Kits und in Elutionspuffer (10 mM Tris-Cl, pH 8,5) zu lösen. Je nach Plasmid Größe, verwenden 100-500 ng Plasmid für eine Sequenzierleiter.
- Sanger-Sequenzierungsreaktion
HINWEIS: Nachstehend eine einfaches Protokoll, das eine fluoreszenzmarkierte Primer Sequenzier-Kits mit 7-deaza-dGTP, die gut für den Zweck der Primerverlängerungen arbeitet verwendet. Siehe den Sequenzierungs-Kit Hand für detaillierte Informationen. Bitte beachten Sie, dass die Sequenzierungsreaktion müssen die gleiche Primer als Primer-Extension-Reaktion zu verwenden, um Produkte mit der gleichen Länge zu erstellen.- Mischungs 12 & mgr; l genomische DNA (~ 10 bis 15 & mgr; g) mit 1 & mgr; l DMSO und 1 ul fluoreszenzmarkierten Primer (2 pmol / & mgr; l).
- Zu jeweils 1 ul der vier Sequenzierungsreaktion Mischungen (A, C, G oder T), fügen Sie 3 ul der DNA / DMSO / Primer-Mix.
- Legen Sie die Proben in einer PCR-Maschine, und führen Sie den folgenden PCR-Programm: 95 ° C für 2 Minuten; 35 Zyklen von 95 ° C für 20 sec, 54 ° C für 20 sec, 70 ° C für 30 sec; halten bei 4 ° C für immer.
- Nach dem Lauf, entfernen Sie die Proben aus der Maschine, fügen 6 ul Ladefarbstoff und auf Eis aufbewahren (kurzfristig) oder bei -20 ° C für mehrere Tage bis Wochen.
4. Gel-Setup und Apparatebau Run
HINWEIS: Detaillierte Informationen darüber, wie die Sequenzierung Gel-Apparatur zusammengebaut ist, wird das Gel hergestellt und wie das Gel laufen kann in der Hersteller-Protokoll gefunden werden.
- Vorbereitungen
- Vorbereitung 10x TBE, wie in Tabelle 4 angegeben.
- Am Tag des Gels Lauf vorzubereiten 1 l 1x TBE-Puffer mit Reinstwasser ddH 2 O.
- Herstellung von 10% (w / v) APS. Anmerkung: Dies kann in 200 & mgr; l-Aliquots bei -20 ° C mehrere Monate gelagert werden kann, aber die Aktivität mit der Zeit nachlassen <./ Li>
- Versammlung der Gelgießen Kammer
- Staub und Flusen zwischen den Glasplatten zu vermeiden. Daher gründlich reinigen Arbeitsflächen mit Feuchttüchern.
- Reinigen ein Paar von 25 cm Glasplatten unter Verwendung von Einweg-Papierhandtücher und destilliertem Wasser auf beiden Seiten und Isopropanol für die Innenseite der Glasplatten.
- Zeigen 0,25 mm-Distanzscheiben an der Hinterglasplatte und senken Sie die gekerbten Glasplatte auf der Oberseite (Abbildung 3).
- Bringen Sie die Gel-Schienen an beiden Seiten der Glasplatten mit dem gekerbten Ende und den Schienen Eintrag Piloten nach oben und ziehen Knöpfe leicht.

Abbildung 3. Explosionsansicht der Gelelektrophorese Glasplatten. Glasplatten sollte richtungs verwendet werden. Achten Sie auf die Innenseite der Glasplatten nach innen und die äußere FlächeSeite nach außen.

Abbildung 4. Ansicht eines zusammengebauten Gel-Apparatur. Nach Injektion der Gel-Lösung, die Taschenabstandshalter ist in der Lösung zwischen den Glasplatten gelegt. Das Gussplatte wird dann zwischen der vorderen Glasplatte und das Gel Schienen aufgeschoben und durch Anziehen der Schienen Noppen befestigt.
- Gießen des Gels
HINWEIS: Nicht polymerisierte Acrylamid ist neurotoxisch! Bitte lesen Sie die Sicherheitsdatenblätter und die Verwendung mit geeigneten Schutz !!!- Fügen Sie die in der Tabelle 5 aufgeführten Verbindungen in ein Becherglas und mischen mit einem Rührstab und einem Magnetrührer.
- Unmittelbar nach der Zugabe von APS und TEMED, nehmen die Gel-Lösung in einem 50-ml-Spritze und setzen Sie ein 0,45-nm-Filter an der Spitze.
- Entweder halten Sie die obere Kante der Glasplatte mit einer Hand oder legen Sie das sandwich in einem Gelgießen stehen, um eine Neigung Neigung 10 zu schaffen - 20 °.
- Langsam abzug die Gellösung zwischen den Glasplatten während kontinuierlich Bewegen des Spritzenspitze von einer Seite zur anderen und zu stoppen, wenn die Gel-Lösung am unteren Ende trifft.
- Bewegen zur Seite oder vollständig zu entfernen alle gebildeten Blasen mit einem Blasen Haken.
- Schieben Sie die Geltasche Abstandshalter (0,25 mm) zwischen den Glasplatten an der eingekerbte Ende, tauchen in die Gel-Lösung und fixieren durch das Anbringen der Gussplatte.
- Befestigen oberen Schiene Schrauben leicht (siehe Abbildung 4 für komplett montierte Gerät).
- Lassen Sie Gel-Set für 1-2 Std.
- Entfernen Sie die Gussplatte und Taschenabstandshalter und reinigen Sie die Tasche aus Salz und Gel-Resten.
- Spülen Sie mit ddH 2 O und aufwischen überschüssige Lösung mit Tissue-Papiere.
- Laufen und Visualisierung des Gels
HINWEIS: Die Sequenzierung Gele direkt einer Elektrophorese in dem Gel Imager unterworfen, währenddie Fluoreszenz wird gleichzeitig durch ein Lasermikroskop nachgewiesen. Im Gegensatz zu herkömmlichen Gelelektrophorese, wobei das Gel wird zuerst ausgeführt und dann angefärbt und sichtbar gemacht wird die Erfassungseinheit befestigt und tastet die Bänder in Echtzeit, während sie die Lasergeben. Unterhalb eines Verfahrens zur Datenerfassung ImagIR Software auf OS / 2 skizziert, die neuere Versionen übernommen werden kann. Weitere Informationen finden Sie in der Bedienungsanleitung.- Schieben Sie den Puffertank Halter in die Gelt Schienen auf den vorderen Glasplatten und ziehen Sie die Knöpfe.
- Zeigen Gel in den unteren Geltank des automatisierten Gel Imager gegen die Heizplatte und fixieren, indem die Schiene Eintrag Pilot in das Gerät Klammern.
- Füllen 1x TBE-Puffer in der unteren und oberen Gel Pufferkammern, schließen Sie die untere Pufferkammer und schließen Sie die obere Pufferkammer mit der Stromversorgung mit dem Netzkabel.
- Falls vorhanden, reinigen Sie die Geltasche aus Salzrückstände durch mehrmaliges Pipettieren Puffer in die Tasche.
- Schließen Sie die obere Puffertankkammer mit der oberen Puffer Deckel.
- Schließen Sie die Gerätetür und schalten Sie den Imager und Computer ein und starten Sie die Basis ImagIR Software zur Datenerfassung.
- Erstellen Sie eine neue Projektdatei (Datei-> Neu ...), geben Sie einen Projektdateinamen, wählen Sie die entsprechenden Laserbereiche (700 oder 800 nm) und mit OK bestätigen.
- Wählen Sie Optionen-> Auto gain ... aus dem Bild-Menü oben, klicken Sie auf Auto, um die automatische Verstärkungsmessung beginnen und akzeptieren die Einstellungen mit OK.
- Fokus des Lasers durch die Auswahl Optionen-> Fokus ... aus dem Scanner-Steuerungsmenü, indem Sie auf die Schaltfläche Auto und akzeptieren Sie die Einstellungen mit OK.
- Wiederholen Sie den Auto-Gain-Verfahren, um auf die neu fokussiert Region einzustellen.
- Richten Sie die Scannersteuerung entsprechend diesen Einstellungen: 2000 V, 35 mA, 45 W, 45 ° C, Scan-Filter: 3, Scangeschwindigkeit: 3.
- Vorlauf den leeren Gel für 20 min (Auswahlspannung ON und drücken Sie ).
- In der Zwischenzeit erhitzen Sequenzierungsleiter und der Primer-Verlängerungsprodukte in einer PCR-Maschine für 2 min auf 90 ° C, kühlt dann auf Eis.
- Stoppen Sie die Elektrophorese, öffnen Sie die automatisierte Gel Sequenzer und entfernen Sie die obere Puffertank Deckel.
- Legen Sie die Haifischzahn-Kamm zwischen die Glasplatten und leicht durchbohren das Gel mit dem Haifischzähne (siehe Abbildung 5).

Abbildung 5. Nahaufnahme von Gel mit Haifischzahn Kamm. Probe (violett) liegt zwischen den Hai-Zähne aufgetragen.
- Pipette entweder 1-2 ul der Primerverlängerungsprodukte oder Sequenzierleiter Reaktionen in jede Geltasche (von der Haifischzähne gebildet).
- Wenn nicht alle Taschen benötigt werden, füllen leere Taschen mit Ladefarbstoff zu verhindern inkonsistente Laufverhalten.
- Schließen der Pufferbehälter und Tür des Gels Sequenzer.
- Starten Elektrophorese und schalten Sie Laser (Select Spannung EIN und Laser ON, und drücken Sie ).
- Stoppen Elektrophorese einmal interessierende Region hat den Laser übergeben.
Ergebnisse
Wie in 6 dargestellt, kann ein Primer-Verlängerungsreaktion verwendet werden, um die Transkriptionsstartpunkte von Transkripten von Interesse zu bestimmen, und kann helfen, Promotorregionen ableiten (typischerweise -10 bis -35 Elemente identifiziert). Die oberste (längste) cDNA Fragment stellt das 5'-Ende der mRNA und somit leicht zugeordnet werden, wenn im Vergleich zu der Sequenzierungsleiter werden.
Diskussion
Fluoreszierende Primerverlängerung ist ein einfaches und schnelles Verfahren zur Bestimmung der 5'-Enden der RNAs, entweder für TSP- oder sekundäre RNA-Prozessierung Identifikation. Durch die Verwendung von fluoreszierenden Primern können die Umsetzungen eingerichtet werden und ausgeführt werden, ohne zusätzliche Sicherheitseinrichtungen (anders als im Fall von radioaktiv markierten Primern). Da die Proben durch Fluoreszenz detektiert, können sie abgebildet werden, während der Elektrophorese durchgeführt wi...
Offenlegungen
The authors have nothing to disclose that would present a conflict of interest.
Danksagungen
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
Materialien
| Name | Company | Catalog Number | Comments |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
Referenzen
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten