Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Флуоресценции основе удлинения праймера методики определения транскрипционных отправных точках и сайты расщепления РНКаз
В этой статье
Резюме
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
Аннотация
Флуоресценции основе удлинения праймера (ФПО) представляет собой молекулярный метод для определения транскрипционных отправные точки или обработки участки молекул РНК. Это достигается с помощью обратной транскрипции РНК интереса с использованием специфических флуоресцентно меченных праймеров и последующий анализ полученных фрагментов кДНК посредством денатурации электрофореза в полиакриламидном геле. Одновременно, традиционный реакции секвенирования Сэнгер запускается на гель для отображения концы фрагментов кДНК с их точными соответствующих баз. В отличие от 5'-RACE (быстрой амплификации концов кДНК), где продукт должен быть клонированной и несколько кандидатов секвенированной, основная часть фрагментов кДНК, порожденных удлинения праймера можно одновременно обнаружить в одной гелевой перспективе. Кроме того, вся процедура (с обратной транскрипцией до конечного анализа результатов) может быть завершена в течение одного рабочего дня. При использовании флуоресцентно меченных праймеров, использование опасных помечены радиоактивным изотопом реагентовможно избежать, и время обработки уменьшается, как продукты могут быть обнаружены во время процедуры электрофореза.
В следующем протоколе, мы опишем люминесцентные праймера метод расширения в естественных условиях, чтобы надежно и быстро обнаружить 5 'концы РНК вывести транскрипции отправные точки и участки для обработки РНК (например, с помощью системных компонентов токсина-антитоксина) в S. стафилококк, Е. палочка и другие бактерии.
Введение
Удлинение праймера 1 представляет собой молекулярный метод для определения 5 'концы конкретных молекул РНК до одной базовой разрешением. Преимущество с другими методами, такими как 5'-RACE (быстрой амплификации концов кДНК) является быстро время обработки и способность легко анализировать смесь различных длин молекул РНК.
Этот метод работает, подвергая молекулы РНК с обратной реакций транскрипции с использованием специфических флуоресцентных праймеров, генерации кДНК фрагменты определенной длины. Эти молекулы кДНК работать параллельно с традиционными Сэнгер секвенирования реакций 2 на денатурирующих полиакриламидных гелей и могут быть обнаружены по их флуоресценции из-за использования флуоресцентно меченных праймеров. Длины фрагментов кДНК затем оценивали по сравнению с секвенирования лестницы, позволяя отображение на концах "5 РНК.
Традиционно праймеров реакции расширения используются в сочетаниис радиоактивными изотопами для обнаружения молекул кДНК на рентгеновских пленок. В связи с опасностями для здоровья, вопросы утилизации отходов и легкостью обработки, новые протоколы используют флуоресценцию для обнаружения удлинения праймера с автоматизированных секвенсоров, хотя их чувствительность несколько ниже. Использование флуоресцентно меченных праймеров, повторяющиеся процедуры радио-маркировка может быть опущен, как люминесцентные грунтовки устойчивы в течение длительного времени (более года в наших руках).
Способ описан здесь использует автоматизированную гель секвенсор, но с небольшими изменениями, капиллярные секвенсеры может быть также использован для разделения и обнаружения кДНК 3. Параллельный характер анализа гель дает возможность обнаружить даже небольшое количество расщепления РНК или обработки. Еще одним преимуществом является высокое разрешение этого метода, как терминал расщепления или обработки даже одной базе могут быть обнаружены.
В связи с обнаружением расщепления РНК или обработки, тypically два различных типа расширений праймеров различают. В одном случае, ферментативная обработка выполняется в пробирке с использованием очищенного РНК и очищенного фермента, в то время как в другом случае, обработка выполняется в естественных условиях, и полученную РНК очищали. В обоих случаях РНК подвергают удлинение праймера проводят в пробирке, однако, в зависимости от источника РНК, либо метод называют в пробирке или в удлинение праймера в естественных условиях. В протоколе приведем здесь, мы сосредоточены только на естественных удлинения праймера в, из-за легкости использования (не очищенных белков необходимо) и возможностью определения транскрипционные отправные точки и обработку, в то же время. Тем не менее, в пробирке расширения праймеров, в принципе, созданной тем же способом и этот протокол может служить в качестве отправной точки.
Способ, показанный здесь, могут быть применены к многих видов бактерий тех пор, пока они поддаются высокойподготовка нуклеиновых кислот чистота и высокодоходным.
Исследования в нашей лаборатории сосредоточена на нормативно рамки токсин-антитоксин (таблица) систем 4,5, поля в которой метод удлинения праймера широко используется. TA-системы представляют собой небольшие генетические элементы, присутствующие в прокариотических геномов, которые состоят из стабильного и активного эндогенно токсичного белка и в основном нестабильной белка или РНК антитоксина, что противодействует токсичности 6,7. Токсин активность иногда оказывает ингибирование репликации, синтеза клеточной стенки или других механизмов, но чаще всего по активности РНКазы 8,9. Как правило, РНКазы специфика определяется путем проведения различных тестов, одним из которых является метод удлинения праймера. Грунтовка реакции удлинительные хорошо подходят для такого применения, как смесь расщепленных фрагментов и полных длины могут быть одновременно анализируют, чтобы определить их 5'-концах. Используя сочетание в пробирке и в естественных расширений праймеров,конкретных токсин РНКазы декольте, например, последовательность специфичность может быть определена 10-13.

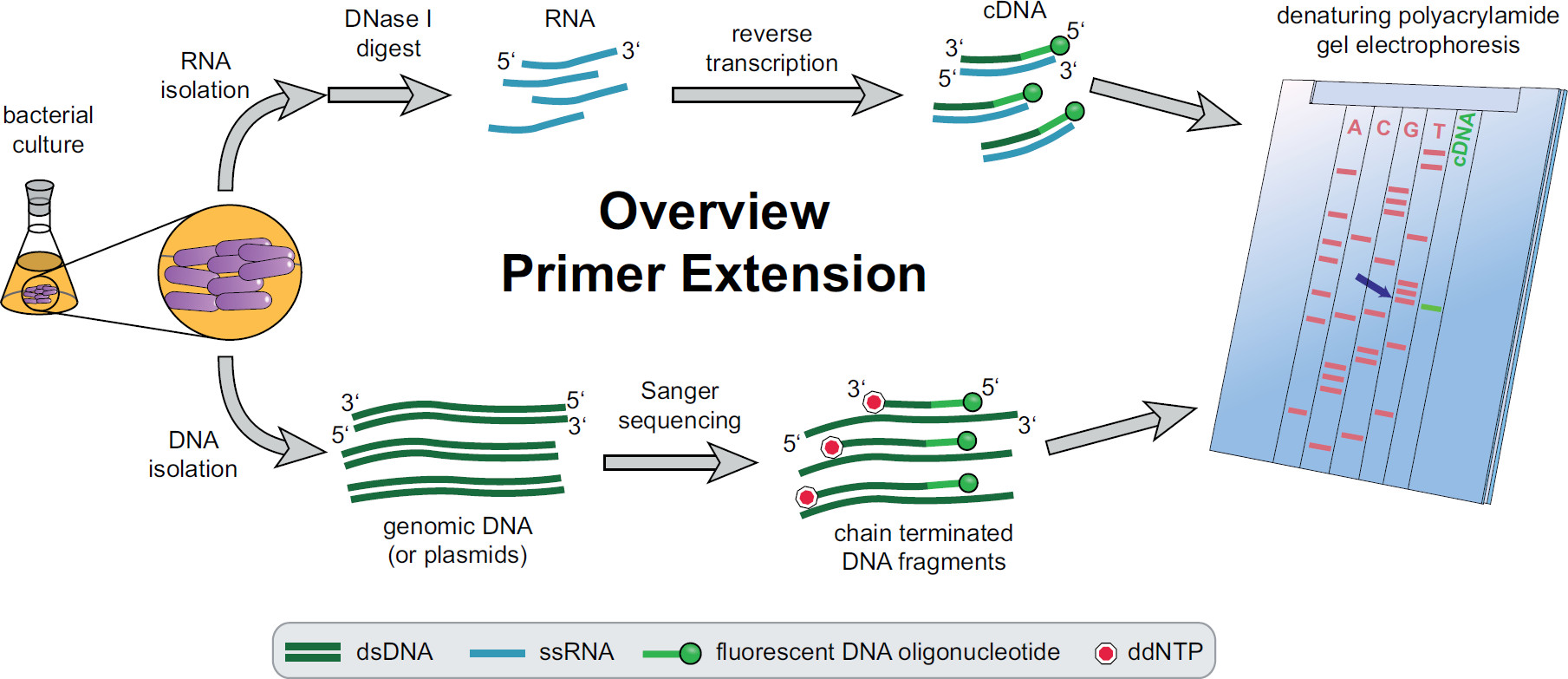
Рисунок 1. Обзор процедуры удлинения праймера. Бактериальные культуры инкубируют и лечение в соответствии с экспериментальными потребностей. Общую РНК экстрагировали из клеток, обработанных ДНКазы I, чтобы удалить следы ДНК и подвергали реакции обратной транскрипции с использованием конкретных целевых флуоресцентных праймеров ДНК, дающие кДНК. Геномную ДНК или плазмиды, извлекаются и впоследствии используется для люминесцентных Сангер секвенирования реакций для сравнения с размером фрагментов кДНК. Продуктов удлинени праймера которые проходят вдоль Сэнгер секвенирования продуктов на денатурирующем полиакриламидном геле мочевины и анализировали с автоматизированной лазерной и микроскопом. Секвенирование база, что линии с полосой кДНК является лаул основание 5'-конца кДНК (синей стрелкой). Более подробная информация в Фекете, и др. 3 Пожалуйста, нажмите здесь, чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
Обзор всей процедуры удлинения праймера можно найти на рисунке 1. Коротко, бактериальные клетки культивируют, собирают, клеточный осадок лизируют и РНК экстрагируют. Очищенная РНК затем обрабатывали ДНКазой I, чтобы удалить следы молекул ДНК, которые могли бы действовать в качестве матриц для обратной транскриптазы. Конкретные флуоресцентные праймеры будут добавлены в РНК, гибридизации в области, представляющей интерес, а затем обратной транскрипции, в результате чего одноцепочечной комплементарной ДНК (кДНК). Секвенирование лестница создается традиционной Sanger секвенирования с использованием флуоресцентных праймеров и разделены на денатурирующего полиакриламидного геля наряду из праймеров фрагментов кДНК расширение. ПолученнуюГель анализируется путем сравнения флуоресцентные полосы, что позволяет идентифицировать 5 'концах интерес. Транскрипции отправные точки и участки обработки Затем оценивается индивидуально по сравнения последовательностей.
протокол
1. High Yield Получение РНК
- Выделение РНК
Примечание: Высокие концентрации суммарной РНК необходимы для реакции удлинения праймера. Наборы столбцов Спин как правило, не дают количество РНК, необходимой (~ 5 - 16 мкг в 5 мкл объем). Поэтому очистка с использованием метода экстракции фенол-тиоцианат-хлороформ кислоты гуанидина рекомендуется, изложены ниже.
ПРИМЕЧАНИЕ: Фенол является канцерогенным, токсичные и коррозионные. Пожалуйста, ознакомьтесь с паспортом безопасности материала и использовать под вытяжкой с соответствующей защитой!- Растут или лечения бактериальных клеток (золотистого стафилококка или кишечной палочки в данном примере), а желательно, и урожай на 10 мин центрифугирования при 4600 × г и 4 ° С. Примечание: Обычно мы собрать полную OD 600 20 - 70. Клеточные осадки могут быть сохранены в течение нескольких недель при температуре -20 ° С.
- Ресуспендируйте осадок клеток в 1 мл кислоты гуанидиния решения тиоцианат-фенол-хлороформ и передать2 мл винт чашку, содержащую 0,5 мл 0,1 мм стекло циркония / оксида кремния бисера.
- Лизиса клеток три раза быстро подготовительной / шарик с насадкой в 6,5 м / с в течение 30 сек в течение трех раундов, охлаждая образцы на льду в течение 5 мин после каждого запуска. Примечание: гомогенизированная образец можно хранить при -80 ° C в течение нескольких недель.
- Инкубируйте лизат в течение 5 мин при комнатной температуре, а затем добавить 200 мкл хлороформа.
- Энергично встряхните или вихрь образец в течение 30 сек, чтобы извлечь РНК.
- Инкубируют при комнатной температуре в течение 3 мин, затем центрифугировать в течение 15 мин при 13000 - 15000 х г и 4 ° С. Примечание: Растворители разделяются на более низкой органической фазы (розовый, содержит белки), межфазного (белый, содержит ДНК) и верхнюю водную фазу (ясно, содержит РНК).
ПРИМЕЧАНИЕ: С этого шага на использовании только РНКазы бесплатно реактивы и пластиковая посуда! - Подготовьте свежий РНКазы 1,5 мл реакционных труб, этикетку, можно добавлять около 500 мкл 100% РНКазы свободной изопропанола каждого (использовать примерно такую же VOЛуме в качестве водной фазы в предыдущем трубки).
- Удерживая трубку под углом и передавать водной фазы (около 500 мкл) на подготовленную труб с использованием РНКазы свободные концы. Не беспокоить межфазной.
- Осадок РНК путем инвертирования несколько раз и инкубации в течение 10 мин при комнатной температуре.
- Центрифуга образцы в течение 15 мин при 13 000 - 15 000 × г и 4 ° С и удалить супернатант с помощью пипетки или аспирации (водоструйного насоса, и искусственное вскармливание). Не беспокоить белую прозрачную РНК гранул в нижней части.
- Добавить 1 мл 70-80% РНКазы свободной этанола (не вихря), чтобы вымыть. Примечание: РНК в этаноле могут быть сохранены в течение нескольких недель при температуре -20 ° С.
- Центрифуга течение 5 мин при 7500 х г и 4 ° С и отбросить супернатант с помощью пипетки или предпочтительно аспирации.
- Воздух сухой осадок РНК в течение 15 - 30 мин под вытяжным шкафом. Не пересушивать, иначе гранулы могут быть трудно растворяться.
- Ресуспендируют гранул в 50 мкл РНКазы свободной DDh 2 O или буфера хранения РНК.
- Измерьте концентрацию РНК с микрообъема UV-VIS спектрофотометр или кварцевую кювету (и обычного фотометр) и перейти к ДНКазы I пищеварения.
- ДНКазы I переваривание РНК, чтобы удалить следы ДНК
Примечание: Так как ДНК может выступать в качестве ложного шаблона в обратной транскрипции (удлинение праймера) реакции, он должен быть удален из образца. Известны различные способы удаления ДНК из РНК решений, которые доступны, как правило, зависят от ДНКазы пищеварения. Простой, но эффективный и экономичный метод для удаления ДНК приводится ниже.- Разогреть на водяной бане до 37 ° С.
- Смешайте соединений, приведенных в таблице 1, в 1,5 мл реакционной трубки.
- Выдержите смесь в течение 1 ч при 37 ° С на водяной бане, а затем перейти непосредственно к экстракции фенолом / хлороформом. Примечание: Тепло инактивация ДНКазы I не рекомендуется, поскольку это может ухудшить РНК.
- Экстракцию фенолом / хлороформом РНК после ДНКазы I пищеварения
Примечание: РНК должны быть очищены для удаления свободных нуклеотидов ДНК-фрагменты и буферных компонентов от ДНКазы I пищеварения. Экстракцию фенолом / хлороформом дает возможность высокой степени извлечения и концентрации образца РНК и, следовательно, описаны ниже. Другие методы очистки РНК также могут быть использованы, если они удовлетворяют этим требованиям.- Сплит 500 мкл ДНКазы I пищеварение смешать в двух 250 мкл образцов в 2 мл реакционных труб.
- Добавить 1 объем (250 мкл) из кислой P / C / I раствора (в водонасыщенного фенола, хлороформа и изопентаноле, соотношение 25: 24: 1, рН 4,5 - 5).
ПРИМЕЧАНИЕ: P решение / C / I является канцерогенным, токсичные и коррозионные. Пожалуйста, ознакомьтесь с паспортом безопасности материала и использовать под вытяжкой с соответствующей защитой! - Энергично вихрь или место в встряхиванием платформы для 1 - 3 мин.
- Центрифуга течение 30 мин при 13000 - 15000 × г и 4 ° С.
- Соберите верхний (водный) фазу и передать чистую пробирку (250 мкл).
- Добавить 1/9 объема (28 мкл) 3 м ацетата натрия рН 5,2.
- Добавить 2,5-3 объемов чистого этанола (700 мкл).
- Смешайте встряхиванием в ближайшее время и место, при -80 ° С в течение 30 мин или при температуре -20 ° С в течение 2 - 3 ч. Если необходимо, хранить РНК O / N при -20 ° С.
- Центрифуга в течение 30 - 60 мин при 13000 - 15000 × г и 4 ° С.
- Удалить супернатант с помощью пипетки или аспирации.
- Промыть осадок путем добавления 1 мл 70% -ного этанола на гранулу. Не вихрь образец.
- Центрифуга образца в течение 5 мин при 13000 - 15000 х г и 4 ° С.
- Удалить супернатант с помощью пипетки или аспирации.
- Воздух сухой осадок под вытяжным шкафом. Храните гранулы при температуре -20 ° CO / N, если необходимо.
- Растворить осадок в 30 мкл обработанной DEPC H 2 O при интенсивном перемешивании в течение 2 мин и использовать это решение, чтобы растворить PELLET соответствующей второй трубы на образец (30 мкл раствора в расчете на одну пару экстракции).
- Измерьте концентрацию РНК, и убедиться, что он превышает 1 мкг / мкл для использования в праймера продлении среднем выраженных мРНК.
- Если необходимо, хранить РНК при -20 ° C в течение нескольких недель до нескольких месяцев.
2. Грунтовка Расширение Реакция
- Грунтовка дизайн
ПРИМЕЧАНИЕ: При проектировании праймеров для расширения эксперимента грунтовки, подчиняются общие руководящие принципы ПЦР дизайна праймеров (см инструкцию на автоматизированную гель секвенсор для получения дополнительной информации и обсуждения раздела в этой статье).- В частности, гарантировать, что праймеры (I) не содержат пробегов оснований, (II) обладают G или С на 3'-конце, (III) имеют сбалансированный GC: AT соотношении, (IV) имеют температуре отжига примерно 55 - 60 ° С, и (v) связывают, по меньшей мере 50 пар оснований, более 100 пар оснований ниже по потоку от области, представляющей интерес, чтобы получить четкое изображение.
- Грунтовка реакция расширение
Примечание: реакции удлинения праймера (синтез кДНК) требуется большое количество матричной РНК. Если суммы РНК материалы выбираются с низким, сигнал может быть слишком низкой для обнаружения! Поэтому мы рекомендуем очистку РНК, как описано выше.
ПРИМЕЧАНИЕ: ВНИМАНИЕ: Использование РНКазы реагентов и пластиковую посуду !!!- Разогреть термо-циклер до температуры 95 ° С и выполнять все дальнейшие шаги инкубации в термо-циклере для простоты использования и воспроизводимости.
- Смешайте соединений из таблицы 2 в ПЦР-пробирку для каждого образца РНК.
- Денатурации образцов в течение 1 мин при 95 ° С.
- Место труб на льду и холод в течение 5 мин для гибридизации РНК и праймеров.
- Установите машину ПЦР до 47 ° С.
- В то же время подготовить обратную транскрипцию мастер-смеси, как описано в таблице 3.
- Добавить 4 мкл обратной транскрипции мастер микс для каждого гибридного РНКобразец.
- Инкубировать пробирки в течение 1 часа при 47 ° С. Примечание: Оптимальная температура для AMV RT 42 ° С, однако более высокие температуры помогают преодолеть вторичные структуры молекул РНК.
- Остановка реакции при нагревании образцов до 95 ° С в течение 2 мин.
ПРИМЕЧАНИЕ: Формамид вызывает коррозию, токсичны и могут быть вредны для неродившегося ребенка. Пожалуйста, ознакомьтесь с паспортом безопасности материала, обращаться с осторожностью и носить соответствующую защиту! - Добавить 6 мкл формамида загрузки красителя (95% (об / об) деионизированной формамида, 10 мМ ЭДТА, 0,05% (вес / объем) бромфенола синего) и хранилище для O / N до двух недель при -20 ° С в темноте.
3. Подготовка секвенирования Лествичник
ПРИМЕЧАНИЕ: реакции секвенирования лестница требует либо умеренное количество плазмид или больших количеств геномной ДНК. Всякий раз, когда это возможно, использование плазмид в реакции последовательности рекомендуется из-за простоты изоляции и высокой сигнал интенсивность. В других случаях, мы обычно используют метод, принятый от Marmur 5,14 подготовить геномной ДНК из E. палочка и С. стафилококк клетки без необходимости использовать фенол. В принципе любой способ, который дает высокое количество и чистоту геномной ДНК может быть использована.
- Геномная Выделение ДНК
- Вырастить 10 мл E. палочка или С. стафилококк клетки O / N в LB, BM 5 или TSB среды.
- Урожай клетки центрифугированием в течение 10 мин при 4600 х г в 15 мл трубки сокола.
- Осадок ресуспендировали в 2 мл буфера Р1, как обнаружено в некоторых мини комплектов подготовки (50 мМ Трис-HCl, рН 8,0, 10 мМ ЭДТА, 100 мкг / мл РНКазы).
- Лизировать клетки для 45 - 60 мин с 20 - 40 мкл лизостафина (0,5 мг / мл, хранение при -20 ° С). Примечание: Для Е. палочки клетки ферментативная предварительная обработка может быть либо опущены или лизоцим, используемые.
- Добавить 100 мкл насыщенного SDS-решения (в 45% этанола) в суспензию и incubatе течение 5 мин при 37 ° С.
- Добавить 650 мкл 5 М NaClO 4 и кратко вихревые клетки.
Примечание: Хлороформ является потенциальным канцерогеном. Пожалуйста, ознакомьтесь с паспортом безопасности материала и использовать под вытяжкой с соответствующей защитой !!! - Добавить 3 мл хлороформа / изопентаноле (24: 1 отношение) к смеси и встряхивают в течение по крайней мере 60 сек. Примечание: Жидкость должна превратиться в однородную белой эмульсии.
- Центрифуга образца в течение 10 мин при 4600 × г и РТ на отдельные фазы.
- Тщательно передачи четкую верхнюю (водный) фазы в новую пробирку. Если раствор мутный, повторить экстракцию хлороформом / изопентаноле. Измеряют объем раствора ДНК и подготовить новую пробирку с 2 объемами этанола (100%).
- Медленно перелить или пипетки раствора ДНК в этаноле, содержащем трубку. Примечание: ДНК должны выпадать в осадок в виде прозрачных, плотные рулоны на дне или при полностью обезвожен в качестве плавучего белого кластера.
- Получить DNA помощью крюков, изготовленных из стекла пипетки Пастера (рис.2) и мыть каждый образец дважды путем погружения в индивидуальной трубки 1 мл 70% -ного этанола.
- Поместите крючки в вертикальном положении в стойке и воздух сухой осадок в течение 60 мин. Если необходимо, хранить высушенный ДНК в течение нескольких дней при комнатной температуре.
- Растворите ДНК путем разрыва ДНК покрыта стеклянные крючки и размещение в 2,0 мл реакционной трубке, содержащей 100 - 500 мкл DDH 2 O. Настройка громкости до конечной концентрации ДНК 1000 - 1500 нг / мкл. С одной реакции секвенирования, используют 10 - 18 мкг геномной ДНК.

Рисунок 2. Инструкция о том, как создать рыбалка ДНК стержень. Удерживая кончик стеклянной пипетки Пастера в пламени горелки Бунзена. Это приводит к тому, стакан, чтобы начать плавления после нескольких секунд, создавая небольшой крюк в тон закончится. Быстро снять с огня и дать остыть в течение 1 мин.
- Плазмиды Изоляция
- Подготовка плазмиды с использованием стандартных наборов мини подготовки и растворить в элюирующего буфера (10 мМ Трис-Cl, рН 8,5). В зависимости от размера плазмиды, используют 100 - 500 нг плазмиды в течение одного секвенирования лестнице.
- Реакция Зангер Секвенирование
Примечание: Найти ниже простой протокол, который использует флуоресцентно меченный праймер для секвенирования с комплектом 7-деаза-дГТФ, который хорошо работает с целью расширения праймеров. Обратитесь к комплект руководстве секвенирования для получения подробной информации. Пожалуйста, обратите внимание, что реакции секвенирования должны использовать один и тот же праймер в качестве праймера реакции удлинения для создания изделий одинаковой длины.- Смешать 12 мкл геномной ДНК (~ 10 - 15 мкг) с 1 мкл ДМСО и 1 мкл флуоресцентно меченого праймера (2 пмоль / мкл).
- Для каждого 1 мкл четырех реакции секвенирования смесей (А, С, G или Т), добавить 3 мкл ДНК / ДМСО / Primer смеси.
- Поместите образцы в машину ПЦР, и выполните следующую программу ПЦР: 95 ° C в течение 2 мин; 35 циклов 95 ° С в течение 20 сек, 54 ° C в течение 20 сек, 70 ° С в течение 30 сек; держать при температуре 4 ° C навсегда.
- После запуска, удалите образцы из машины, добавить 6 мкл красителем и магазина на льду (на короткий срок) или при температуре -20 ° С в течение нескольких дней до нескольких недель.
4. Гель установки и устройство Run
Примечание: Подробная информация о том, как аппарат секвенирования гель собран, гель подготовили и как гель запуска можно найти в протоколе производителя.
- Препараты
- Подготовка 10x TBE как указано в таблице 4.
- В день гелевой перспективе подготовить 1 л 1x TBE буфере сверхчистой DDH 2 O.
- Подготовка 10% (вес / объем) APS. Примечание: Можно хранить в 200 мкл аликвоты при -20 ° С в течение нескольких месяцев, но активность может уменьшаться с течением времени <./ LI>
- Собрание гель литья камеры
- Избегайте пыли и ворсинок между стеклянными пластинами. Поэтому тщательно чистые рабочие поверхности с помощью влажных салфеток.
- Очистить пару 25 см стеклянных пластин с использованием одноразовых бумажных полотенец и дистиллированную воду с обеих сторон, а затем изопропанол для внутренней стороне стеклянных пластин.
- Поместите 0,25 мм проставки на задней стеклянной пластины и снизить зубчатым стеклянную пластину сверху (Рисунок 3).
- Прикрепите гель рельсов с обеих сторон стеклянных пластин с вырезом конце и пилотов железнодорожного въезда, стоящих перед вверх и затяните ручки слегка.

Рисунок 3. разобранном виде электрофореза гель стеклянных пластин. Стеклянных пластин следует использовать направленно. Позаботьтесь, чтобы столкнуться с внутренней стороны стеклянных пластин внутрь и внешнеесторону наружу.

Рисунок 4. вид устройства в сборе гель. После введения раствора геля, карманные прокладка помещена в растворе между стеклянными пластинами. Кастинг пластину затем скользнул между передней стеклянной пластины и гелевых рельсов и обеспечены крепления железнодорожных ручки.
- Кастинг гель
ПРИМЕЧАНИЕ: не полимеризуется акриламида нейротоксичен! Пожалуйста, ознакомьтесь с паспортом безопасности материала и использовать с соответствующей защитой !!!- Добавить соединений, приведенных в таблице 5, в химический стакан и смешать с помощью мешалки и магнитной мешалкой.
- Сразу после добавления APS и TEMED, занимают раствора геля в 50 мл шприца и поместите 0,45 нм фильтр на на кончике.
- Либо провести верхний край стеклянной пластины с одной стороны или разместить сэндвич-ч в кастинге геля стоять создать наклон под углом 10 - 20 °.
- Медленно обойтись раствора геля между стеклянными пластинами при непрерывном перемещении наконечника шприца от одной стороны к другой, и прекратить как только раствор геля соответствует нижний конец.
- Перемещение в сторону или полностью удалить любые образовавшиеся пузыри, используя пузырь крючок.
- Вставьте гель карманные распорку (0,25 мм) между стеклянными пластинами на зубчатый конец, погрузить в раствор геля и исправить путем присоединения литья пластину.
- Закрепите верхний железнодорожные болты слегка (рисунок 4 для полностью собранного устройства).
- Пусть гель набор для 1 - 2 ч.
- Снимите литье пластины и карманные прокладку и очистите карман от соли и геля остатков.
- Промыть DDh 2 O и вытереть излишки раствора с тканей работ.
- Запуск и визуализации геля
ПРИМЕЧАНИЕ: Гели секвенирования непосредственно подвергали электрофорезу в геле томографа, в то время какфлуоресценции одновременно обнаружить с помощью лазерного микроскопа. В отличие от обычной гель-электрофореза, где гель запуска, а затем окрашивали и визуализировали, блок детектирования является фиксированной и сканирует диапазон частот в реальном масштабе времени, когда они проходят лазер. Ниже процедуры для обеспечения сбора ImagIR данных на OS / 2 очерчено, которые могут быть приняты для более поздних версий. Для получения дополнительной информации обратитесь к руководству пользователя.- Авто держатель буферной емкости в гелевых рельсов на передних стеклянных пластин и затяните ручки.
- Поместите гель в геле бака нижнего автоматизированной гель тепловизор с нагревательного элемента и исправить, сдвинув пилота входа железнодорожного в аппарат скобках.
- Заполните 1x буфер TBE в нижних и верхних буферных камер геля, закройте нижнюю буферную камеру и подключите верхнюю палату буфера к власти с помощью кабеля питания.
- Если присутствует, очистите гель карман из соляных остатков по несколько раз пипеткой буфер в карман.
- Закройте верхнюю камеру буферной емкости с помощью верхнюю крышку буфера.
- Закройте дверцу машины и переключиться на тепловизор и компьютер и запустите программу сбора База ImagIR данных.
- Создайте новый файл проекта (File> New ...), ввести имя файла проекта, выбрать соответствующие лазерные диапазоны (700 или 800 нм) и подтвердить кнопкой ОК.
- Выберите Настройки-> Автоусиление ... из меню изображения в верхней, нажмите Auto, чтобы начать автоматический измерение усиления и принять настройки, нажав кнопку ОК.
- Фокус лазер, выбрав Настройки-> Фокус ... из меню управления сканером, нажав на кнопку Auto и принятия настройки, нажав кнопку ОК.
- Повторите процедуру автоматического усиления, чтобы приспособиться к новой ориентированной области.
- Настройка управления сканером соответствии с этими настройками: 2000 V, 35 мА, 45 Вт, 45 ° C, Scan фильтр: 3, Скорость сканирования: 3.
- Prerun пустой гель в течение 20 мин (выбран напряжения и нажмите ).
- В то же время, нагревать секвенирования лестнице и продуктов удлинени праймера в машине ПЦР в течение 2 мин до 90 ° С, затем охлаждают на льду.
- Остановите электрофорез, открыть автоматизированный гель секвенсор и снимите верхнюю крышку буферной емкости.
- Вставьте акулы зубную гребенку между стеклянными пластинами и слегка прокалывают гель с зубов акулы (рисунок 5).

Рисунок 5. Крупным планом геля с акулой зуба гребенки. Образец (фиолетовый) применяется между зубами акул.
- Пипетка либо 1 - 2 мкл праймера продукты удлинения или секвенирование лестничные реакции в каждом кармане гель (образованной из акульих зубов).
- Если не все карманы нужны, заполнить пустые карманы с красителем, чтобы предотвратить впоследовательное поведение бег.
- Закройте буферную емкость и дверь гель секвенсор.
- Начните электрофорез и включить лазерный (Select напряжение на и лазер на и нажмите ).
- Стоп-электрофореза раз область интереса прошел лазер.
Результаты
Как показано на рисунке 6, праймер реакции расширение может использоваться для определения транскрипционные начальные точки транскриптов, представляющим интерес, и может помочь вывести промоторные области (обычно идентифицируется -10 и -35 элементов). Самый верхний (длинный) Фр...
Обсуждение
Расширение Люминесцентная грунтовка представляет собой простой и быстрый способ определения 5 'концы РНК, либо для TSP- или вторичной идентификации обработки РНК. Благодаря использованию флуоресцентных праймеров, реакции могут быть созданы и работают без дополнительных мер безопасн...
Раскрытие информации
The authors have nothing to disclose that would present a conflict of interest.
Благодарности
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
Материалы
| Name | Company | Catalog Number | Comments |
| Name | Supplier | Catalog Number(s) | Comment |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
Ссылки
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены