
Method Article
ポリ(εカプロラクトン)エレクトロ糸にヒト間葉系幹細胞の添付ファイルの最適化
要約
This article describes a range of set-ups for seeding human mesenchymal stem cells onto materials, in this case electrospun yarns, that do not cover the base of standard culture well plates in order to maximize and quantify the number of cells that initially attach compared to the known seeding density.
要約
生体材料および組織工学の研究は、多くの場合、出発細胞数の初期の知識を必要とする細胞ベースのin vitroでの研究を含む。研究者は、一般的に彼らの播種密度を参照するが、これは必ずしも問題の物質に付着した細胞の実際の数を示すものではありません。これは、特に、標準的な細胞培養ウェルプレートの底部をカバーしていない材料、または足場の場合である。この研究は、培養中の4時間後にエレクトロスピニングポリ(εカプロラクトン)糸の上に既知の数で播種ヒト間葉系幹細胞の初期の添付ファイルを調査する。電界紡糸糸9 rpmで、ペトリ皿内に配置低い結合ウェルプレート、ポリテトラフルオロエチレン(PTFE)のトラフ内に配置細胞培養インサートで回転バイオリアクター容器を含む、いくつかの異なるセットアップ内に保持した。後者の二つは、シェーカープレート上で静的な条件のどちらかにさらさまたは配置した(30 37°Cの 、5%CO 2で4時間インキュベートした後、播種した細胞の位置を、細胞DNAアッセイによって決定した。足場は、その容器から除去し、溶解緩衝液に入れた。メディア画分も同様に除去し、遠心分離し、 - 上清を捨て、ペレットを溶解緩衝液で壊れ。溶解緩衝液を各容器に添加し、またはウェル、および存在し得る任意の細胞を解放するために削り取られた。細胞DNAアッセイは足場、メディアよく画分内に存在する細胞の割合を決定した。細胞接着は、糸細胞培養インサート内に保持され、運動を振とうするための最大のアタッチメント(30%)を、全ての実験セットアップのために低かった。この研究は、関係なく、記載された細胞播種密度の足場への添付細胞の実際の数に意識を発生させます。
概要
足場は、日常的に開発され、生体材料および組織工学用途のために研究されている。そのようなものとして、それらは一般に、例えば、細胞増殖および細胞数を決定するアッセイによって特徴付けられる細胞およびそれらのin vitro挙動を播種する。このような実験のために、初期細胞数が既知であることが必須であると研究者は、多くの場合、溶液または1cm 2あたりの細胞の数の点で播種濃度を述べる。これは特にスケールアップの目的のために、良い方法ですが、それは足場の表面に付着する細胞の実際の数(ま た生体材料表面1の接着特性に依存する)を考慮していない。細胞が構築物から離れて落下する可能性があると、により、実験の多くの場合、静的な性質のために、INTEの材料と接触戻ってこないかもしれないので、これは、細胞培養ウェルプレートの全塩基をカバーしていない足場では特に顕著です残り。電界紡糸繊維糸は、ウェル( 図1A)のベースをカバーしていない足場の良い例である。この場合には、表面処理されていない低結合ウェルプレートは、プレートの表面に付着し、従って、任意のウェルベースのアッセイの結果を歪めるから細胞を防ぐために使用されるべきである。
ウェルプレートは、容易に足場上に細胞播種のために使用されているが、これらは利用可能な唯一の方法ではありません。ロータリー細胞培養系、1980年代後半にNASAでのライフサイエンス部門によって開発されたバイオリアクターのタイプは、同様に、模擬微小重力を有する三次元(3D)環境内で足場を播種するために使用することができる。バイオリアクターのこのタイプは、世界中の研究者との人気のある選択肢のままで、セル4,5および組織工学6,7幹細胞は2,3をシグナリングするための研究に組み込まれている。どのようなウェルプレートに好適な回転式バイオリアクターを作ることは、メンテナンスですしばしばそうであるように、脱分化から分化細胞を防ぐことができ、3D環境のとき従来の2D条件8内で培養した。
本論文では、4時間の期間内にこれらの足場に付着した細胞の初期数を最大にするためにボスワースら 、9で作製したように電界紡糸ポリ(εカプロラクトン)繊維糸にヒト間葉系幹細胞を播種するための異なる技術を調査する。 2D培養のために、糸を確実にウェルプレートまたはカスタムメイドのポリ(テトラフルオロエチレン)(PTFE)のトラフ内に保持され、静的な条件下で飼育し、30 rpmで振とうした。 3D培養のために、糸と細胞が9で回転バイオリアクター容器内に保持された。
プロトコル
1.Scaffold作製と滅菌
- 10%のw / vの濃度を得1,1,1,3,3,3-ヘキサフルオロイソプロパノールでPCLを溶解する。 。ポリマー溶液(パラメータ:20 kVの、1ミリリットル/時、20cm)にエレクトロスピニングボスワースら 、9で説明したように、回転するマンドレル(600回転)の端に繊維を整列収集します。メスで収集された繊維のリボンを削除してから、短い長さに切断 - (細胞培養インサート用)(谷と回転船舶用)3センチメートルと4センチメートル長さ。
- 細かい鉗子を使用すると、蒸留水に個々のストリップを水没して削除する。
- 親指と人差し指の間の両端を保持する。それは、スレッドに似ているまで、手動でストリップをねじる。
- 簡単に説明すると乾燥するために、クリーン、非繊維状カードに蒸留水と所定の位置に、このスレッドのような足場を沈める。
- きれいな微量遠心チューブ中で個別に乾燥したら、場所、蒸留水中50%v / vのエタノールの1ミリリットルを追加します。蓋を閉じて、2に向けて出発4時間。
注:層流の下で次の手順を実行します。 - クリーンベンチでマイクロチューブを配置し、50%のV / v溶液を吸引。 1ミリリットルの蒸留水中70%v / vのエタノール、近くに蓋を交換し、24時間に向けて出発。
- 90%を繰り返し、蒸留水100%(v / v)のエタノール(1ミリリットル容量)。
- 洗浄液は、リン酸緩衝生理食塩水(PBS)、洗浄あたり24時間(2×1ml)で二回足場。
- PBSを除去し、1ミリリットル細胞培養培地と交換してください。注:足場は、使用後の24の時間の準備ができている。
2。足場の表面積と細胞数を測定
- 光学顕微鏡および画像化ソフトウェアを使用して、平均値を決定するために、その長さに沿って電界紡糸ヤーンの直径を測定する。
- 円筒状のロッドであることが糸を想定し、使用して表面積を近似する。
- A =表面積あり、r =半径であり、h =長さ
注:表面積が18902と計算された、800μmの2。さらに、糸は、表面積を増加させる微細繊維、数百から構成されているように、実際の表面積が、この計算よりも大きくなることに留意すべきである。その結果、細胞の数が多いほど、足場に取り付けることができるはずです。しかし、これは行われた試験群間の直接比較に影響を与えない。 - によって公開さ足場表面に付着できたセルの最大数を決定します。
注:光学顕微鏡および画像化ソフトウェアを使用して、ヒト間葉系幹細胞の直径が20μmであると決定された(細胞が丸くと仮定して)、細胞= 60200のため、この場合には、数。
3.足場セットアップ - セルカルチャーインサート(図1A)
- 層流下、無菌の6ウェル細胞培養インサートを開いて、より広い環状体から歯に短いリングを分離する。
- 上向き歯とリングを取る。ドレープ1確認は、両側に重なる作るリングの中心上に4cmの足場。歯付きリングと、足場の上に環状体と位置を取り、足場位置に留まり、細胞培養インサートの中心を通ってある確認して下方に押し込む。
- 6ウェル、低結合プレートのウェルに足場を持つ細胞培養インサートを置きます。
- 足場に培養培地の10ミリリットルを追加します。
4.足場セットアップ - トラフ(図1B)
- 層流の下では、場所のポリ(テトラフルオロエチレン)(PTFE)は、個々のペトリ皿に谷と谷に培養培地の10ミリリットルを追加します。
- 鉗子を使用すると、必ず、その長さは、トラフの長辺に平行に位置作るトラフに3cmの足場のいずれかをドレープ。
5.足場セットアップ - バイオリアクター容器(図1C)
- 層流の下で、バイオリアクター容器のを通して滅菌PBSを10ml分配メインポートと10分間のまま。
- PBSを除去し、8ミリリットル培養培地と交換してください。
- 鉗子を使用して、メインポートを介して容器に3cmの足場の1を挿入します。
- メインポートを閉じる。
6.細胞の計数
- 最大収穫前の通路4に、製造業者のプロトコルに従って、骨髄由来の培養ヒト間葉系幹細胞(hMSCを)。
- 骨髄(通路4、80%のコンフルエンシー)に由来したhMSCを含む75cm 2のフラスコから培地を吸引除去する。
- 10ミリリットル滅菌PBSと吸引で細胞を洗浄。
- 3ミリリットルのトリプシンを添加し、細胞がフラスコ表面から除去されるまで、37℃、5%CO 2でフラスコをインキュベートする。
- 酵素を不活性化し、遠心チューブにこの総容量(10ml)に転送するために7ミリリットルの培地を加える。
- 均質内の細胞を分散させるために数回上下にピペッティングすることにより、この細胞懸濁液を再懸濁メディアリミット細胞凝集体(10 mlピペットを用いて、)。
- 細胞懸濁液の20μlのを削除し、血球計数器に転送。
- X10の目的で、光学顕微鏡と画像の下に血球計数器を配置します。
- グリッド線を集中し、グリッドの各隅に4×4の正方形のセルの数をカウントし、その正方形、右手または下部境界線と交差しているものに含まれる。 4×4の正方形(グリッドあたり4カウント)のセットごとにカウントします。
- 再懸濁し、細胞数を3回(6.6から6.9ステップ)繰り返します。
- 平均細胞数を計算し、細胞の再懸濁(200μl中の細胞濃度60200)に必要なメディアの量を決定する。例えば、血球計数器からの平均細胞数を決定した後、3つの別々のカウントからの細胞の全体的な平均数を決定する。 1ml当たりの細胞数を与え、その後に与える細胞懸濁液の全体積を掛けるために、1×10 4によって、これを掛ける細胞のTAL番号。再懸濁のために必要なメディアの容量を決定するために、次の式を使用します。
- 5分間、241×gで細胞懸濁液を遠心。
7.細胞播種
- 細胞ペレットを残して遠心分離チューブから培地を吸引除去し、計算されたメディアボリュームと交換します。
- でも混ぜるための細胞および培地を懸濁します。
- P200ギルソンピペットを使用して、ゆっくりと足場の長さに沿って媒体の液面下にピペットの先端を実行することによって、各足場上の細胞懸濁液200μlを分注する。 20分間邪魔されずにしておきます。
- バイオリアクター容器の場合は、メインポートを介して細胞懸濁液200μlを分注する。注射器ポートを介してトップアップ残りの2mlの培地を、10mlの全容積を得た。
8.実験スタート
- RCCS-4DQバイオリアクターに、バイオリアクター容器を移し、9 rpmで回転するように設定。
- トランシェーカープレートに細胞培養インサートおよび谷を有するウェルプレートをsferおよび30rpmで回転するように設定。
- 37℃、5%CO 2(静置培養)に設定細胞インキュベーターの棚に細胞培養インサートおよび谷を有するウェルプレートに移す。
9. DNAアッセイ
- 溶解バッファー、1×TEバッファー、DNA標準と細胞DNAの作業溶液 - - 製造業者の指示に従って溶液を調製する。
- 4時間後、インキュベーターからサンプルを除去し、層流の下に置く。
- 別途ラベル遠心管の全サンプルと場所からメディア画分を削除します。チューブ241 XGを遠心。上清を除去し、3ミリリットルの溶解バッファーを追加します。ブレークアップするために細胞ペレットをソリューションを懸濁します。
- 鉗子は3ミリリットルの溶解緩衝液を含む遠心管における足場と場所を削除]を使用して(バイオリアクター血管内足場のために、前に私を吸引する足場を削除DIA)。細胞培養インサート内に保持された足場のために、最初のメスを用いて、挿入物の端部の近くに足場を切断して足場を解放する。
- 各足場コンセントに溶解バッファーの3ミリリットルを追加し、表面をこすり(精力的にバイオリアクター船舶の攪拌)。溶解バッファーを外し、別途ラベル遠心管を内の場所。
- ボルテックスを約1分間、各遠心管細胞の十分な攪拌を確保し、バッファリングし、細胞膜の溶解を促進するため。
- 足場、メディアとうまく(複製) - 黒の96ウェルプレートでは、各サンプル画分についての溶解バッファー100μlを添加する。
- 暗闇の中で、溶解緩衝液を含む全てのウェルに、細胞DNA溶液100μlを追加し、穏やかに混合する。
- ウェルプレートのための陰性および陽性対照を提供することなくDNAおよび細胞DNA溶液を含まない溶解緩衝液でウェルを含む。
- 蛍光プレートリーダーを使用して、ウェルの吸光度を測定する485nm励起および520nmの発光を用いだ。
- メーカーの説明書に従ってDNA標準から生成された標準曲線とのデータを比較してください。
10.走査型電子顕微鏡(SEM)固定
- 層流の下で次の手順を実行します。4時間後、新たな6ウェルプレート内でのレセプタクルと場所(別々のウェル)からすべての足場を削除する。
- PBSで2回足場を洗ってください。
- 各ウェルに、足場の完全なカバレッジを確保するために、PBS中の1.5%(v / v)のグルタルアルデヒドの2ミリリットルを追加します。オープンベンチで次の手順を実行します。
- 細胞固定のために、4℃で、最低30分間プレートのままにしておきます。
- 固定液を除去し、PBSで2回足場を洗う。
- 70%v / vの90%v / vで、続いて、50%v / vで始まる蒸留水にエタノールの濃度を増加させて足場を脱水する。各濃度について、完全にsolutioで足場を沈めるN(2ミリリットル)と3分間のまま。液を捨て、繰り返します。
- 完全に溶液中で足場(2ml)中に浸すと、5分間放置することにより、100%エタノールで脱水する。液を捨て、繰り返します。
- 化学的にヒュームフード内でヘキサメチルジシラザン(HMDS)を使用して足場を乾燥させてください。 HMDSで足場(2ミリリットル)を浸し、5分間のまま。 HMDSを取り外して、繰り返します。
- HMDSを削除し、足場を乾燥させる。 (この場合は接着剤のカーボンタブを持つステンレス鋼のスタブで)市販のSEMスタブ上に足場をマウントします。
- SEM内に表示容易にするために、2分間の金スパッタコーターを使用してコート試料は、薄くて均一な被覆を確実にする。
- 場所のSEM内のサンプルと5ドーズの電子ビームを用いて、細胞播種足場を視覚化する。
結果
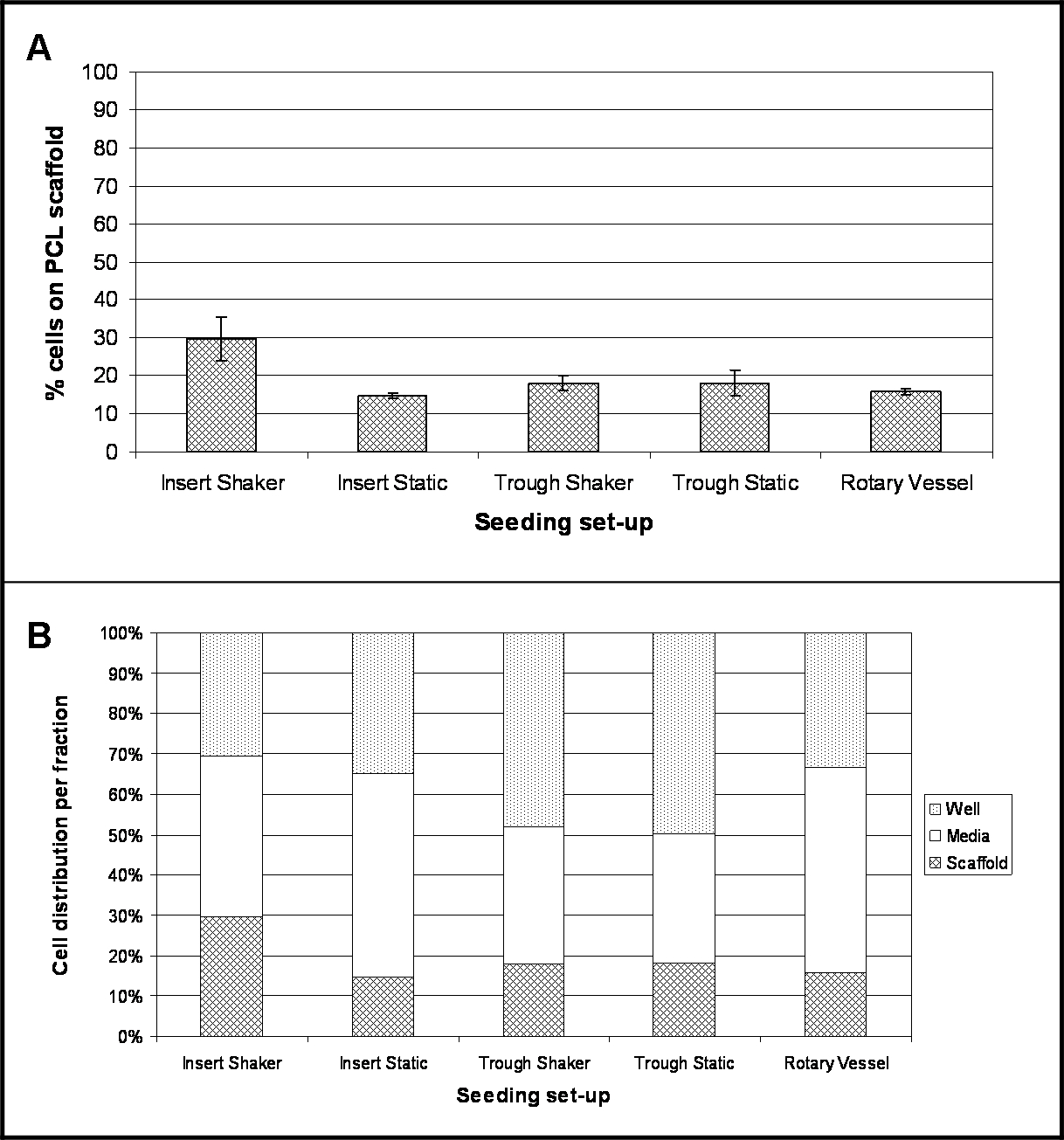
結果を調べ、各実験設定のための4時間後、播種後の細胞の位置を強調している。 図2Aは、この時間の間、足場の表面に付着した細胞のパーセンテージを示す。 8.5 pgの/細胞の変換係数は、細胞数に測定されたDNAの内容を変換し、したがって、セル10の割合を決定するために使用された。全て播種セットアップを調査するために、細胞接着の割合は、細胞培養インサート内に保持し、30rpmで(インサートシェーカー)で振とうした足場のための最大の細胞接着(30%)で、比較的低い。最も低い順守(15%)が細胞培養インサート内に保持された足場のためであり、静止状態(挿入静的)下に維持した。
多数のセルは、メディア画分( 図2B)内に存在し、最も顕著には、50%および51%である低結合プレート(挿入静的)及び回転血管内に保持された細胞培養インサートのためのそれぞれ。トラフシェーカーのための細胞の48%、トラフ静的50% - のトラフ内に保持された足場は、ホルダー自体の中に存在する細胞の隆起数を示した。
走査電子顕微鏡は、細胞播種足場( 図3)の視覚的評価を可能にした。代表的な画像にかかわらず播種セットアップの、繊維状の表面に細胞の限られた存在を強調した。しかし、細胞および細胞凝集体の数が多いほど、細胞培養インサート内に保持された足場上に存在し、30rpmで(インサートシェーカー)で振とうした。

エレクトロ糸が内に保持されている。図1。実験セットアップ、。 (A)細胞培養インサート及び低結合ウェルプレート;(B)ポリ(テトラフルオロエチレン)(PTFE)トラフシャーレ。と(C)バイオリアクター容器。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

異なる播種セットアップを使用してPCLエレクトロ糸の上に細胞を4時間後の播種の図2.場所。よくメディア、および足場た(n = 4、として提示されたデータ- 3つの画分内の細胞の位置のパーセンテージの広がりを強調し(B)、(A)は、(平均±標準偏差)PCL足場に付着した細胞のパーセンテージを実証平均値)。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図3.代表走査型電子顕微鏡写真をヒト間葉系幹細胞とPCLエレクトロ糸、4最初の播種は異なる実験セットアップを使用した後の時間。用 (1,000倍の倍率でのすべての画像、スケールバー=130μmの。) 表示するには、こちらをクリックしてくださいこの図の拡大版。
{kind=link}
ディスカッション
バイオポリマーから製造された電界紡糸繊維のマ トリックスは、定期的にバイオマテリアルおよび/ または組織工学アプリケーション11,12のための細胞の付着および増殖を支持するために使用される。これらのケースでは、マトリックスは、容易に細胞培養ウェルプレートの全塩基を覆い、したがって、細胞の付着を改善播種された細胞と完全に接触している繊維の薄いシートであることが多い。しかしながら、生体材料足場が完全にウェルプレートの底をカバーしていない場合には、播種された細胞の大部分は足場に接触して留まらないであろうし、最終的に取り付けることができないという高い可能性がある。この研究は、将来の細胞ベースの実験のために推奨されることができ、最適化手法を決定するために、ウェルプレートの底部をカバーしていない足場上に細胞を播種するためのいくつかの異なる方法を検討した。
ファイブ異なるセットアップが( 図1)を調査した:scaffoLDS(エレクトロ糸)低結合ウェルプレート内の細胞培養インサートを使用して保持され、静的な条件下で飼育し、30 rpmで振とうし;足場は狭いPTFEトラフ内に配置し、静的開催または30 rpmで振とう。そして9で回転バイオリアクター容器の内部に収容された足場。全て播種セットアップ( 図2)のためのアタッチメントの低い割合を示したDNAアッセイによってエレクトロスパン糸に付着した細胞の数を決定する。これは、さらに電子顕微鏡写真(SEM)( 図3)をスキャンすることを確認した。最大の細胞付着 - 30%〜18060細胞を細胞培養インサート内に保持され、連続した動きを行った糸について観察-was。興味深いことに、最も低い細胞付着(15%)は、半径方向の運動を含むことが足場と接触した細胞を維持する上でプラスの効果を有することを示唆している細胞培養インサートに保持されたが、静的条件下で飼育糸、達成された。しかし、それは、メディアの流れの連続的な旋回は、SEM像から観察された細胞凝集に関与するかもしれないことに留意すべきである。 30回転 - - このセットアップに制限することができるシェーカープレートは最も低い設定に設定した。より低速ラジアル運動を使用して、細胞凝集を防止または低減するのを助けることができ、細胞がより少ない力を経験するように、細胞の付着を改善することができる。今後の実験では、改善された細胞付着のための理想的なシェーカー速度を最適化することに焦点を当てるべきである。両方のシナリオでは18%の添付ファイル(〜10836細胞)を得て、同様の傾向をもたらさなかったトラフ内に保持された糸のために運動を組み込む。これは、トラフ内の足場の部分的な浮上によるものかもしれないが、彼らはベースに固定されていなかったとして(シェーカープレート上に置い谷のために観察された)。足場の部分的浮動は、材料と接触すると付着するの谷の底に沈んでいるすべてのセルを防ぐことができます。 tについて彼の特定のセットアップ、トラフは、ペトリ皿内に収容され、媒体の全体積を10mlを加えた。トラフの小さな寸法は、メディアの大部分は、シャーレ内に存在する任意の動きがある場合、細胞はシャーレに離れてトラフからドリフト足場の手の届かないところに完全に残ることを意味する。これはにさらされる足場のための彼らの浮上、特に動きを(防ぐ必要があるとして、これらの制限を克服するために、さらなる実験は、足場の端部が無菌微針を使用して、トラフのベースに固定されていることにより、プロトコル、中に余分なステップを含める必要があります最終的に、足場に付着する細胞数の増加をもたらすはずである半径方向の動き)。細胞の16%は、回転血管内に存在する糸に付着していた。 3D培養のためのよく確立された技術であるにもかかわらず、問題が緩くATTACをもたらしている可能性がある、船舶の主要港から足場の除去を生じなかったHED細胞が失われている。完全に開くことができる容器は、この問題を排除するであろう。これらは、購入可能であるが、相当に、この研究で使用される使い捨ての容器よりも高価である。
本研究では、標準的な細胞培養ウェルプレートの全塩基をカバーしていない足場に播種すると、現在の問題を示しています。既知の細胞数を播種すると、すべての細胞が接着することを可能に足場の表面積にもかかわらず、足場に第三取付より小さいとなった。これは、潜在的な将来の医療機器等の足場と生体適合性及び細胞物質/細胞 - 細胞の挙動との相互作用を評価することができる他の細胞ベースのアッセイにおける有害な結果を有することができる。研究のさらなる制限は、4時間の時点を含むこと-十分な長さ(細胞がしっかりと30分13,14,15内の基板に取り付けることが示されている)最初の細胞播種を確実にするためであるにもかかわらず、それが内に合理的であることこれはそうでなければ出発細胞数をスキューと同じように、細胞がより長い時間枠の間に増殖しない提供し、後の時点をvestigate。この場合には、10ミリリットルの培地の容積を減少させる、また、細胞と足場との間の接触を改善し、最終的に細胞の付着を高めることができる。細胞播種のプロセスは、細胞の損傷および/ または細胞死16が形成されることがあるので、将来の研究はまた、細胞生存率を検討すべきである。そのような生/死アッセイは、例えば、生存能力のレベルを強調と同じように細胞DNAアッセイは、生存と非生存細胞を区別しません。
この調査は、既知量の播種にもかかわらず、足場に付着した細胞の実際の数に意識を発生させます。細胞の開始番号に依存していた研究のためには、研究者が実際に関心のある基板に接着しない正確にどのように多くのその数字の知っていることが非常に重要です。
開示事項
The authors declare that they have no competing financial interests.
謝辞
著者は、この研究に資金を提供するための医学研究審議会に感謝し、感謝したい - MRC-DPFSグラントコードG1000788-98812。
資料
| Name | Company | Catalog Number | Comments |
| Distilled water | in-house supply | n/a | |
| Ethanol | Merck | 1117271000 | |
| Phosphate Buffered Saline solution | Life Technologies | 70013016 | |
| Human mesenchymal stem cells | PromoCell GmbH | C-12974 | |
| MSC culture media | PromoCell GmbH | C-28010B | Warmed to 37 oC before use |
| Supplement mix | PromoCell GmbH | C-39810 | Add to culture media |
| Antibiotic/antimyotic mix | Sigma-Aldrich | A5955 | Add to culture media |
| Trypsin (0.05%) EDTA (0.02%) | Sigma-Aldrich | 59417C | Warmed to 37 °C before use |
| Cell culture flasks (T75) | Becton Dickinson Ltd | 353110 | |
| Low binding 6-well plates | Costar Corning | 3471 | |
| 6-well CellCrowns | Scaffdex | C00003S | |
| Petri-dish 50 ml deep | Sterilin | 124 | |
| PTFE troughs | in-house production | n/a | |
| Disposable RCCS vessels 10 ml | Synthecon | D-410 | |
| 4 Vessel Rotary Cell Culture System bioreactor | Synthecon | RCCS-4DQ | |
| Shaker plate | Stuart | SSM1 | Mini Orbital Shaker |
| Haemocytometer | Digital Bio | DHC-F01 | Disposable C-Chip |
| Centrifuge tube | Deltalab | 352096, 429901 | 15 ml and 50 ml |
| Centrifuge | Hettich | Rotafix 32 A | |
| Syringe 3 ml | Shield Medicare Ltd | 3039820 | |
| Pipettes | Sterilin | 40305, 47310, 40125 | 5, 10 and 25 ml |
| Gilson pipettes | SLS | F144801, F144802, F123600, F123601, F123602 | P2 - P1000 |
| Pipette tips | SLS | PIP7852, PIP7834, PIP7840 | |
| Micro test tube 1.5 ml | Eppendorf | 30125.15 | |
| Triton X-100 | Sigma | 9002-93-1 | |
| PicoGreen Assay | Invitrogen | P7589 | Assay set-up in the dark |
| Black 96-well plate | Greiner Bio One | 655086 | |
| Fluorescent plate-reader | BGM Labtech | FLUOstar Optima | |
| Glutaraldehyde 25% | TAAB Laboratories | G002 | Made to a concentration of 1.5% v/v in PBS |
| Hexamethyldisilazane (HMDS) | Sigma | 999-97-3 | |
| Aluminium stubs (SEM) | Agar Scientific | G301 | |
| Carbon tabs (SEM) | Agar Scientific | G3347N | |
| Gold sputter coater | Edwards | S150B | |
| Scanning Electron Microscope (SEM) | Phenom World | Phenom Pro |
参考文献
- Jauregui, H. O. Cell adhesion to biomaterials. The role of several extracellular matrix components in the attachment of non-transformed fibroblasts and parenchymal cells. ASAIO transactions/American Society for Artificial Internal Organs. 33 (2), 66-74 (1986).
- Puca, A., Russo, G., Giordano, A. Properties of Mechano-Transduction via Simulated Microgravity and its Effects on Intracellular Trafficking of VEGFR's. Oncotarget. 3 (4), 426 (2012).
- Vincent, L., Avancena, P., Cheng, J., Rafii, S., Rabbany, S. Y. Simulated microgravity impairs leukemic cell survival through altering VEGFR-2/VEGF-A signaling pathway. Annals of biomedical engineering. 33 (10), 1405-1410 (2005).
- Rungarunlert, S., Klincumhom, N., Tharasanit, T., Techakumphu, M., Pirity, M. K., Dinnyes, A. Slow Turning Lateral Vessel Bioreactor Improves Embryoid Body Formation and Cardiogenic Differentiation of Mouse Embryonic Stem Cells. Cellular Reprogramming. 15 (5), 443-458 (2013).
- Wu, X., Li, S. H., Lou, L. M., Chen, Z. R. The Effect of the Microgravity Rotating Culture System on the Chondrogenic Differentiation of Bone Marrow Mesenchymal Stem Cells. Molecular biotechnology. 54 (2), 331-336 (2013).
- Wang, Y., et al. Rotating Microgravity-Bioreactor Cultivation Enhances the Hepatic Differentiation of Mouse Embryonic Stem Cells on Biodegradable Polymer Scaffolds. Tissue Engineering Part A. 18 (21-22), 2376-2385 (2012).
- Lv, Q., Deng, M., Ulery, B. D., Nair, L. S., Laurencin, C. T. Nano-ceramic Composite Scaffolds for Bioreactor-based Bone Engineering. Clinical Orthopaedics and Related Research. 471 (8), 2422-2433 (2013).
- Hammond, T. G., Hammond, J. M. Optimized suspension culture: the rotating-wall vessel. American Journal of Physiology-Renal Physiology. 281 (1), F12-F25 (2001).
- Bosworth, L. A., Alam, N., Wong, J. K., Downes, S. Investigation of 2D and 3D electrospun scaffolds intended for tendon repair. Journal of Materials Science: Materials in Medicine. 24 (6), 1605-1614 (2011).
- Dormer, N. H., Qiu, Y., Lydick, A. M., Allen, N. D., Mohan, N., Berkland, C. J., Detamore, M. S. Osteogenic differentiation of human bone marrow stromal cells in hydroxyapatite-loaded microsphere-based scaffolds.. Tissue Engineering Part A. 18 (7-8), 757-767 (2011).
- Rayatpisheh, S., Heath, D. E., Shakouri, A., Rujitanaroj, P. O., Chew, S. Y., Chan-Park, M. B. Combining cell sheet technology and electrospun scaffolding for engineered tubular, aligned, and contractile blood vessels. Biomaterials. 35 (9), 2713-2719 (2014).
- Wismer, N., Grad, S., Fortunato, G., Ferguson, S. J., Alini, M., Eglin, D. Biodegradable electrospun scaffolds for annulus fibrosus tissue engineering: effect of scaffold structure and composition on annulus fibrosus cells in vitro. Tissue Engineering Part A. (3-4), 672-682 (2014).
- Yavin, E., Yavin, Z. Attachment and culture of dissociated cells from rat embryo cerebral hemispheres on polylysine-coated surface. The Journal of cell biology. 62 (2), 540-546 (1974).
- Chen, H. Guan, of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proceedings of the National Academy of Sciences. 91 (21), 10148-10152 (1994).
- Grant, D. S., Tashiro, K. -. I., Segui-Real, B., Yamada, Y., Martin, G. R., Kleinman, H. K. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell. 58 (5), 933-943 (1989).
- Carrier, R. L., Papadaki, M., Rupnick, M., Schoen, F. J., Bursac, N., Langer, R., Freed, L. E., Vunkaj-Novakovic, G. Cardiac tissue engineering: cell seeding, cultivation parameters, and tissue construct characterization. Biotechnology and bioengineering. 64 (5), 580-589 (1999).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved