
Method Article
実装化学反応ネットワーク用プラスミド由来のDNA鎖置換ゲイツ
要約
This protocol describes a method for deriving DNA strand displacement gates from plasmids and testing them using fluorescence kinetics measurements. Gates can be modularly composed into multi-component systems to approximate the behavior of formal chemical reaction networks (CRN), demonstrating a new use for CRNs as a molecular programming language.
要約
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
概要
ワトソン-クリック塩基対の予測可能性は、動的なDNAのナノテクノロジーは、動的特性1,2と分子デバイスを設計するためのプログラム可能な方法として出現することができました。特に、DNA鎖置換 - プログラム可能な、競合ハイブリダイゼーション反応は - 動的DNAシステムを操作するための強力な機構であることが判明しました。 DNAの鎖置換反応では、入ってくるオリゴヌクレオチドは、相補的結合パートナーからの以前に結合した「出力」鎖を置換します。複数のそのような反応は、個々の反応3ステップの順序やタイミングを制御度の高い多段階反応カスケードに一緒に連鎖させることができます。 DNA鎖変位カスケードは4-7、切り替え可能なナノ構造体8-10、自律分子モーター11-15、および非共有結合触媒アンプ13,16-21デジタルとアナログの分子回路を生成するために使用されています。また、D鎖置換反応を用いてNAデバイスをシミュレートし、コンピュータ支援設計ソフトウェア22-24を使用して 、多様な用途のために設計することができます。
現在、化学的に合成されたDNAは、DNAナノテクノロジーのための主材料として機能します。しかし、DNA合成プロセスにおけるエラー、そして得られた不完全なオリゴヌクレオチドは、誤った副反応を引き起こすことにより、動的DNAデバイスの性能を制限すると考えられています。例えば、「リーク」反応も反応トリガーの非存在下での出力オリゴヌクレオチドの放出をもたらすことができます。このような効果は、最初のリークであっても最小限の量は、最終的にカスケード19,20の完全な活性化につながる自己触媒反応のカスケードに最も明白です。一部のコンポーネントが意図した入力7,25の存在下でさえも誘発しないので逆に、反応はしばしば活性化の期待されるレベルに到達することができません。 DNAベースの性能を作成するにはそれらの生物学的なタンパク質ベースの対応物に匹敵するナノデバイスは、エラーモードが劇的に低減する必要があります。
細菌プラスミドまたは他の生物学的DNAは、ナノテクノロジーのアプリケーションのための高純度のDNAの比較的安価な供給源として働く可能性があります。 DNAの大量の細菌中で複製することにより生成することができ、生物系の固有の校正能力は、得られたDNAの純度を確保します。実際には、いくつかの最近の論文は、ナノテクノロジーのアプリケーション21,26-28のための生物学的DNAの潜在的な有用性を認識しています。しかしながら、プラスミドDNAの完全に二本鎖の性質は、これまで一般的に複数のオリゴヌクレオチドからなり、二本鎖および一本鎖ドメインの両方を含む動的DNAデバイスを製造するための材料としての使用を禁止しています。最近の論文29この問題が対処された、主にニックの入った二本鎖DNA(ndsDNA)で構成されて新たなDNAのゲートアーキテクチャでは紹介しましたD。
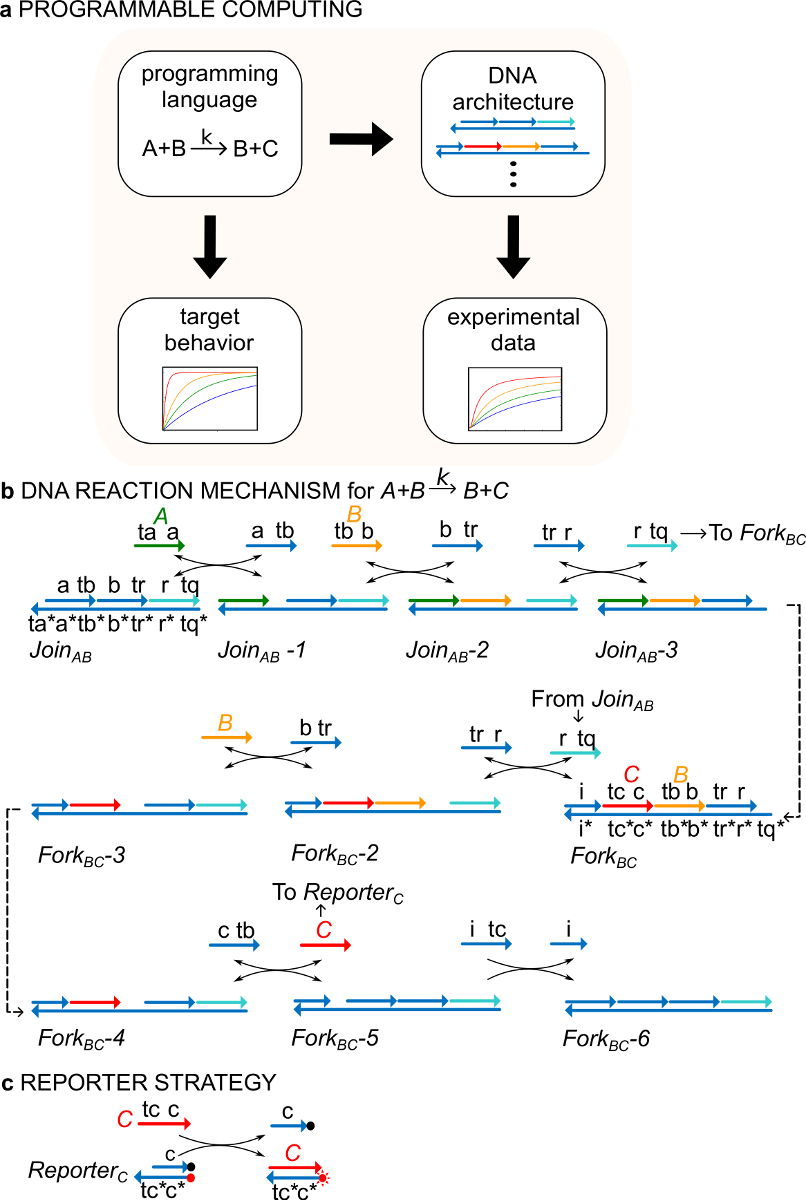
重要なことには、ndsDNAゲートのシステムは、正式な化学反応ネットワーク(CRN)29で指定されたダイナミクスを実現するように設計することができます。 ndsDNAゲートは、このように振動とカオス、双安定性とメモリ、ブール論理またはアルゴリズムの挙動30-38を示 す動的システムを作成するには、原則として、使用することができます。例えば、参考文献29は、「コンセンサス」プロトコル、分散コンピューティングアルゴリズム29,39,40のタイプの分子の実装を提供三反応CRNを示しました。この作品は、最初に急速に機能性分子システムを合成するための「プログラミング言語」( 図1A)などのCRNの形式化のための新規な使用を実証しました。
ここでは、プラスミドDNAからndsDNAゲートを導出するための詳細なプロトコルが提供されます。最初のシーケンス設計プロセスのレビューです。そして、どのように含む合成オリゴヌクレオチドの説明を次のゲート配列は、細菌培養物を介して検証され、増幅されたプラスミドとの配列中にクローニングされます。 ndsDNAゲートは、酵素処理によりプラスミドから誘導することができる方法を次に、それが示されている( 図2参照)。最後に、蛍光動力学アッセイを用いてゲートの動作をテストするための方法を概説します。
反応機構
例として、プロトコルは、触媒化学反応A + B-> B + Cに焦点を当てています。種A、B、およびC(「シグナル」、 図1B)は、すべての異なる一本鎖DNA分子に対応します。これらの分子の配列は完全に独立しており、鎖は互いに直接反応しません。すべての信号のシーケンス、すなわち二つの異なる機能的ドメイン、鎖置換反応において一緒に作用する部分配列を有する:1)短い足掛かりドメイン(ラベル鎖置換rの開始のために使用され、TB、TC)TA信号の同一性を決定するeaction、及び2)長いドメイン(A、B、Cのラベル)。
信号鎖間の相互作用は、ニックの入った二本鎖DNA(ndsDNA)ゲート複合体(ABとフォークBCに参加呼ばれる)と補助一本鎖種(、、と<私のTCによって媒介されます>)。正式な反応A + B-> B + Cは、各反応工程は、その後の反応( 図1B)のための足掛かりを公開鎖置換反応の一連のステップを介して実行されます。信号Cは、フォークゲートに結合している間に、この例では、信号A及びBは、最初に溶液中で遊離しています。反応BとCの終了時に溶液中にあります。より一般的には、ゲートにバインドされた信号は、溶液中で遊離している信号がアクティブな間に、つまり、彼らは鎖置換反応などに参加することができ、非アクティブです入力。反応の時間経過は、蛍光レポーター戦略( 図1C)を使用して続いています。以前の研究29には、この反応機構は、正しい化学量論だけでなく、目的とする反応の速度を実現するだけでなく、ことが示されました。
プロトコル
1.配列設計
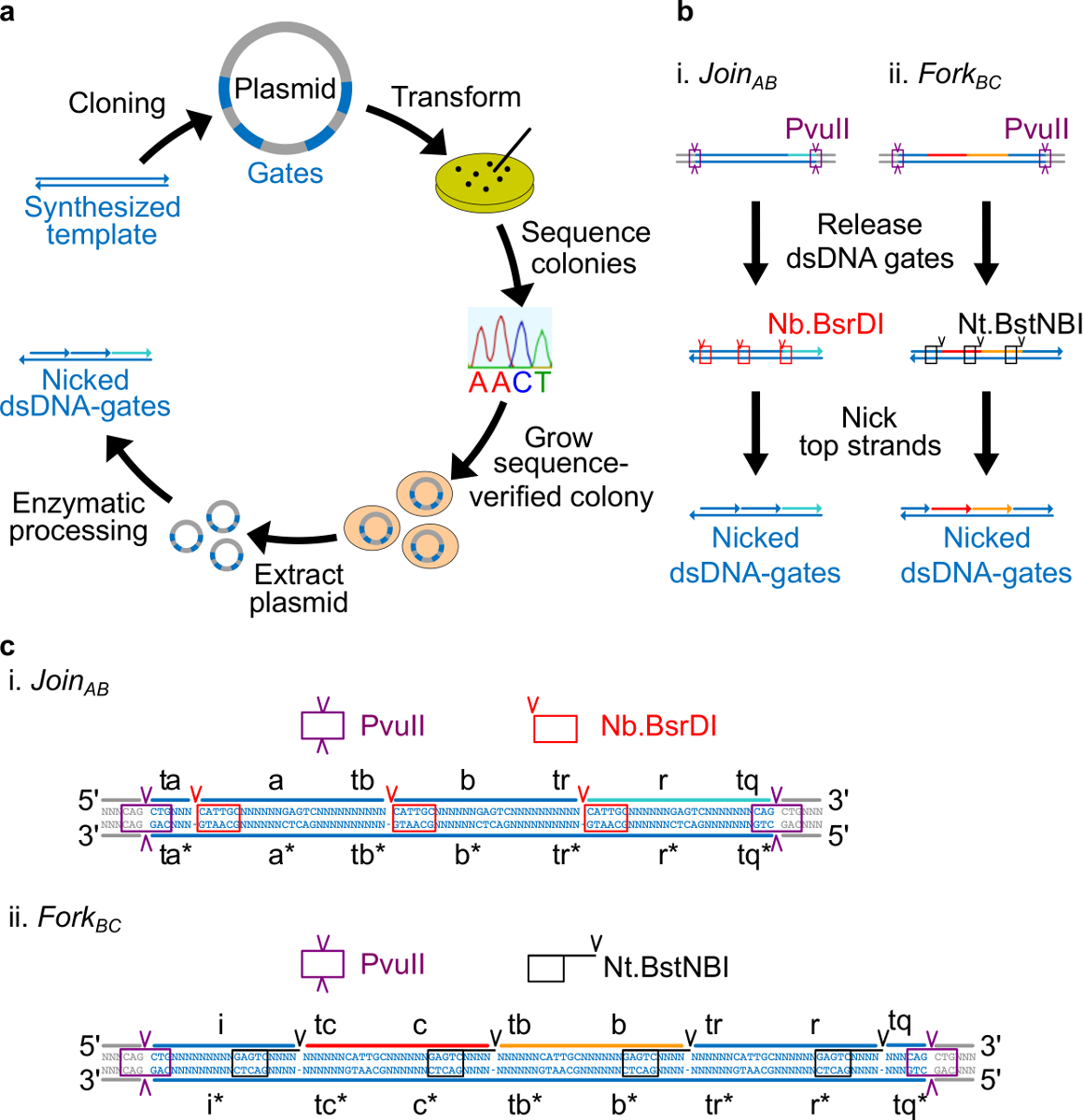
注意:シーケンスの設計の概要を:このセクションでは、プラスミド由来のDNAのゲートを設計するための戦略が記載されています。酵素部位は消化後に完全に二本鎖のゲートのリリースを可能にするために、ゲートの両端に配置されました。ニッキング部位は、その後、酵素は、最終的なndsDNAゲートを作成するために、上の鎖にニックを作成するように配置されています。最後に、残りの配列は、独立したドメインが互いに直交しており、二次構造を示さないように選択されます。
- 4つのヌクレオチド離れて長いドメイン(A、B、C、R、及びi)の3 '末端からNt.BstNBIニッキングサイトを配置します。各長いドメインの5 '末端にNb.BsrDIニッキング部位を配置(A、B、C、およびr。ドメインが、私は任意のNb.BsrDIニッキングサイトを持っていないことに注意してください)。 図2Cは、一連の詳細図を示していますABとフォークBCゲートに参加。
- PvuII消化プラスミドからゲートを解放することができるようにndsDNAゲートの両端にPvuII制限部位を配置する( 図2Cを参照)。
- 2つの原則に従うことによって、他の制約のないシーケンスを設計します(A)鎖は、(DNA構造がNupack 41を用いて予測することができます)二次構造を示してはならない、(b)は、すべてのドメインは、クロストークを最小化するために直交する必要があります。
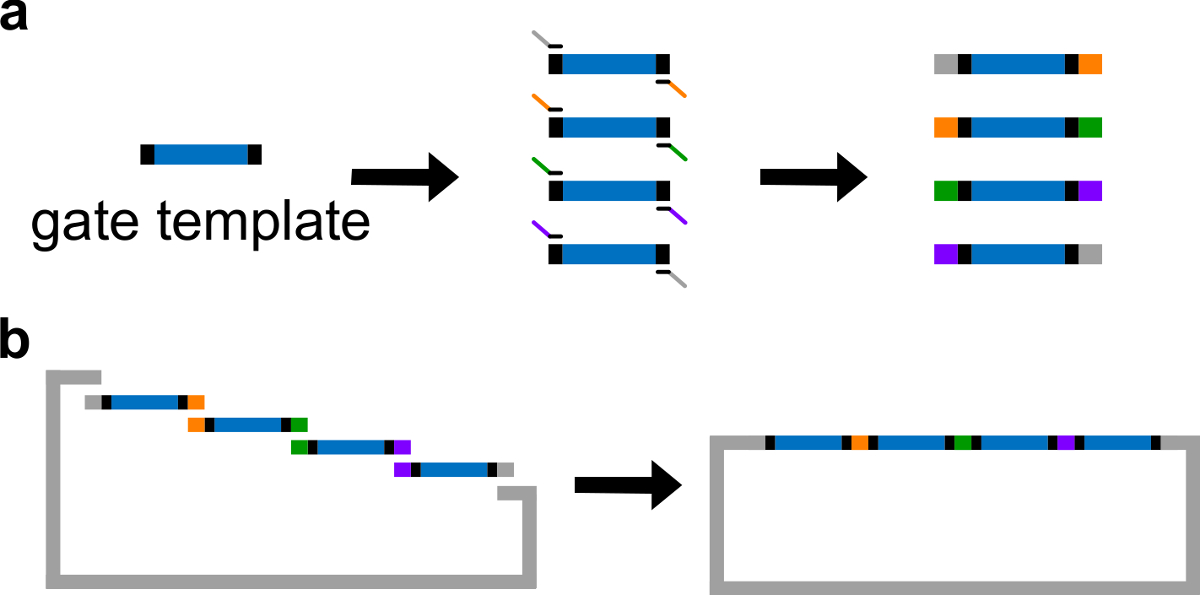
- ゲートテンプレートの中央にndsDNAシーケンスを配置します。ゲートテンプレートの両端に30〜40塩基対のランダムスペーサー配列を配置し、各スペーサは、以下のポリメラーゼ連鎖反応(PCR)のための固有の結合部位として働きます。
プラスミドへのNdsDNAゲイツの2クローニング
注:このセクションでは、プラスミド骨格にゲートの4コピーを挿入するためのギブソンのクローニング方法について説明します。
- (ゲート鋳型配列が示されているDNAのメーカーからの二本鎖ゲノムブロックとして注文ndsDNAゲートテンプレート表1で、ストランドは、 表2に示す ndsDNAゲートで発生します。ドメインレベルの配列)を表3に示します 。
- 順序付けられたDNAを受信した後、すべての乾燥したDNAは、チューブの底にあることを保証するために1分間10,000-14,000×gでのゲノムのブロックを含むチューブをスピン。
- 10 NG /μlの最終濃度を達成するために、DNアーゼを含まない水で乾燥したゲノムのブロックを再懸濁します。
注:または、DNAは1×トリスエチレンジアミン四酢酸(EDTA)バッファー使用して再懸濁させることができる(TE緩衝液を10 mMトリスおよび1mMのEDTA、pH8.0)に。しかし、EDTAは二価の陽イオンのためのキレート剤であり、PCRを阻害し得ます。 - 高忠実度DNAポリメラーゼによる標準的PCR( 図3A参照 )を介して、他の重複領域4ゲート断片を生成します。プライマー配列を表4に詳述されている(これらのプライマーの融解温度は62℃です)。
- 1で2%アガロースゲルを実行しますRTで30分間、40 Vゲルから断片を増幅した(詳細なアガロースゲル電気泳動プロトコルの42を参照)、各PCRに対応するバンドを切り取り。次いで、製造業者の説明書に従ってゲル抽出キット(材料を参照)を用いたゲルスライスを精製します。
- 製造業者のプロトコルに従ってのPvuII-HFと1時間37℃でのPstI-HFと高コピー数のプラスミド骨格(材料を参照)( 表5参照 )ダイジェスト。 PvuII-HFとPstI-HFは劇的に非特異的なカットを削減する、高忠実度の制限酵素です。
- 線形化バックボーンを1.5%アガロースゲルを実行し、カット(通常は室温で30〜40分間、140Vでゲルを実行します)。次いで、製造業者の説明書に従ってゲル抽出キットを用いてゲルスライスからDNAを抽出します。
- 線状化ベクターおよび精製されたPCR断片とギブソンアセンブリ43を実行します ( 表6および図3Bを参照してください )。
- (100μg/ mlの濃度で)アンピシリン抗生物質を含む溶原性ブロス(LB)寒天プレート上の大腸菌 (Escherichia coli)とプレートにステップ2.8からギブソン組立製品を変換します。エレクトロポレーションまたは熱ショック方法44,45を介して変換を実行し、適切なE.を使用大腸菌菌株。例えば、Eを使用し熱ショック形質転換のための大腸菌 JM109株、およびDH5αエレクトロEを使用エレクトロポレーションのための大腸菌細胞。
注意:使用するプラスミド骨格は、アンピシリン耐性カセットを含みます。異なる選択マーカーを使用している場合は、代わりにアンピシリンの適切な抗生物質を使用しています。
3.細菌培養増幅および品質管理
注:このセクションでは、品質管理後のDNAゲートを含むプラスミドの大量生産及び単離を記載しています。
- 単一のコロニーを選択してくださいステップ2.9からのアンピシリン選択プレートから3 mlのアンピシリン抗生物質を含む濃縮培地の文化をインキュベート(100μg/ mlの濃度で)。それは、その後の実験工程に再利用することができるように、コロニーをマークします。激しく振盪(200〜300 rpm)しながら37°のCO / Nで培養物を成長させます。一般的に、16〜24時間インキュベートします。
- 製造者の指示に従ってミニプレップキットを用いて細菌培養物からプラスミドDNAを抽出します。
- 製造業者の指示に従って、分光光度計を使用して精製したプラスミドDNAを測定します。典型的な収率は50〜1000 ngの/μLの範囲です。
- DNAシーケンシング会社にサンプルを送ることによって配列抽出したプラスミドDNAを取得します。配列決定プライマーは、上流および配列決定される領域の下流約100ヌクレオチドを配置する必要があります。 ATTACCGCCTTTGAGTGAGC:プラスミドための配列決定プライマーは、(プラスミドの材料を参照)以下の配列を有します。
ノーTE:シーケンスエラーまたは挿入ndsDNAゲートにおける再結合がある場合は、ステップ2.9からのプレートは異なるコロニーを選択します。フォローは、挿入されたゲートのシーケンスが正しいことを確認するために、3.1から3.4を繰り返します。 - シーケンスが正しいことを確認した後、(ステップ2.9から)アンピシリン選択プレートから対応するコロニーを選択し、(100μg/ mlの濃度で)アンピシリン抗生物質を含む800ミリリットルテリフィックブロス(TB)の文化をインキュベートします。激しく振盪(200〜300 RPM)で16〜24時間、37℃で培養物を成長させます。 TBは、特に高収率プラスミド生産によく適しています。
注:また、LBはまた、プラスミド収量は、問題になる可能性があるが、細菌を成長させるために使用することができます。 - 製造業者の説明書に従ってマキシプレップキットを用いてDNAを精製します。
- シーケンスが正しいかどうかを確認するために、ステップ3.3から3.4に従ってください。いずれかの組換えが発生した場合は、次の注を参照してください。それ以外の場合は、ステップ4に進みます
注:ここでの一つの可能性のある問題は、プラスミド中に挿入ゲートの複数のコピーが、DNA修復のために再結合することです。この問題に対処するために、使用してE.そのようなJM109やDH5αなどのRecAタンパク質(DNA修復に関連するタンパク質)を欠いた大腸菌株は、(任意の配列エラーと再結合することなく、すなわち )は、以前に配列検証したプラスミドを変換します。次に、このプレートから1つのコロニーを選択し、DNA配列決定の会社にサンプルを送信することにより、プラスミド配列を確認します。
4.酵素処理
注:このセクションでは、彼らはカットし、正しい位置にニックが入っおよび動力学実験のために使用する準備ができているように、プラスミドを消化するための方法を説明します。
- 37°C( 表7参照 )で1時間制限酵素のPvuII-HFとステップ3.7から精製されたプラスミドDNAを消化し ます。一般的に、プラスミドの1 1mg当たりのPvuII-HFの4台でプラスミドを消化。ハイFID彼らは劇的に非特異カットを減らすためelity制限酵素を使用することをお勧めします。
- サンプルにエタノール沈殿を行います。
- サンプルに氷冷無水エタノールの2同等のボリュームを追加します。
- 少なくとも1時間(この混合物はまた、O / Nのために-80℃で座ることができる)のために-80℃で混合物をインキュベートします。
- 0℃で30分間10,000-14,000×gで遠心分離。
- 上清を取り除きます。
- サンプルにRTの95%エタノール千μl添加し、10〜15回転倒。
- 10分間4℃で10,000-14,000×gで遠心分離。
- 10〜20分間ベンチに上清と空気乾燥を削除します。
- ヌクレアーゼフリーのH 2 O(通常は100〜200μL)の適切な量でDNAペレットを再懸濁します。以上の200μlのを追加すると、一般的にサンプルがあまりに動態実験で使用するために希釈するようになります。
- メートル以下の分光光度計を用いて再懸濁したDNAを測定anufacturerの指示。
- ダイジェストは、( 表8を参照 )1μgのプラスミドあたりの酵素の4台を使用して1時間65℃で酵素Nb.BsrDIをニッキングとゲートに加わります。 ( 表9を参照)1μgのプラスミドあたりの酵素の8単位を用いて、1時間55℃で酵素Nt.BstNBIをニッキングとフォークゲートを消化。
注意:手順4.2は、酵素消化緩衝液を除去し、反応速度実験のためのゲートを集中することができます。制限酵素PvuIIで-HF及びニッキング酵素の両方が同じ消化緩衝液を共有Nb.BsrDIので、ゲートに参加するためのステップ4.2をスキップすることができます。 EDTAは、二価の陽イオンのためのキレート剤であり、これらのイオンは、機能するために必要な制限酵素を阻害することができるので、ステップ4.2.8において、ヌクレアーゼフリーのH 2 Oの代わりにTEのに使用されます。
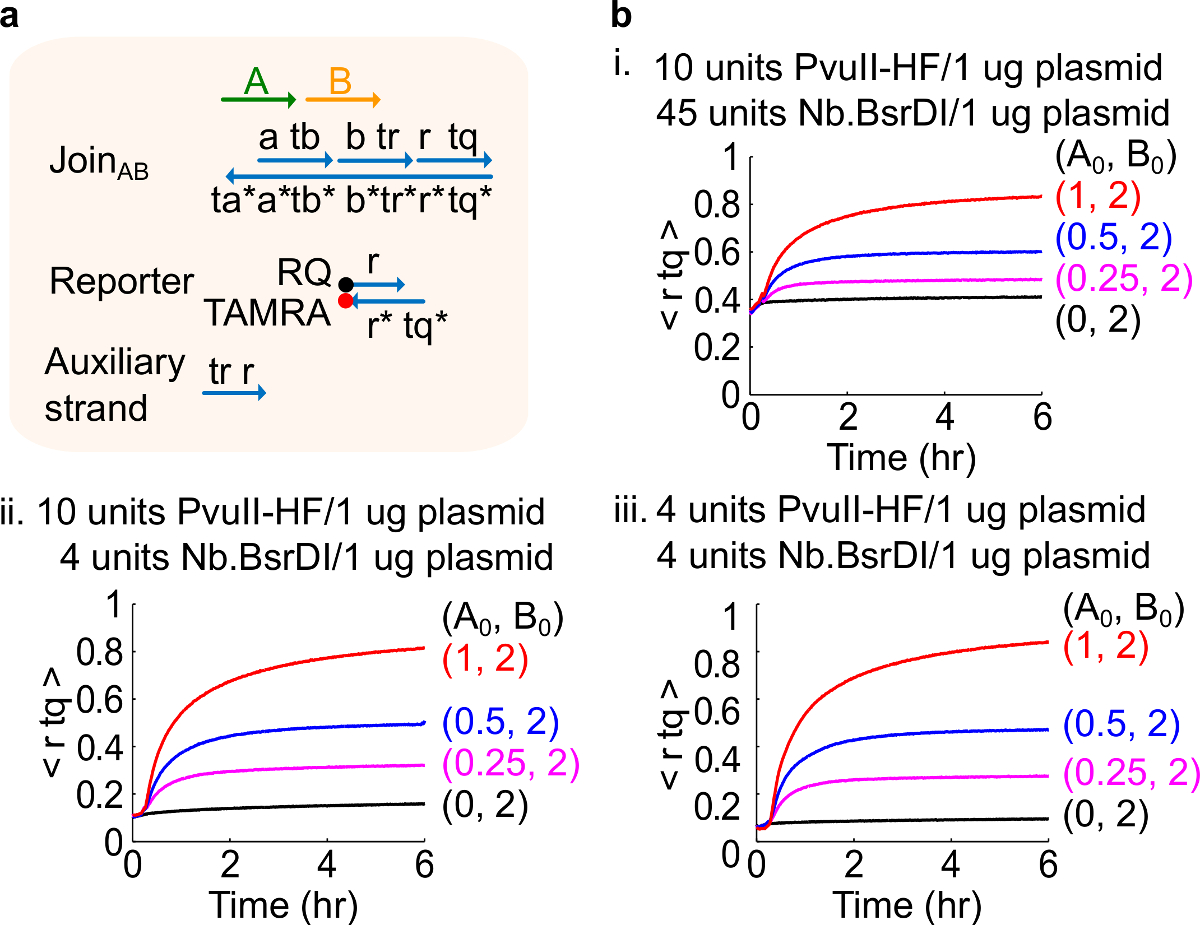
注:酵素の過剰量の添加は、おそらく過剰消化46によって引き起こされる初期の回路漏れ( 図4)の高い量をもたらすことができます。この問題は、CAN( 図4参照)、酵素量を最適化することによって対処されます。酵素の典型的な範囲は1〜10単位/ 1μgのプラスミドからです。
一本鎖オリゴヌクレオチドの5準備
注:このセクションでは、信号のストランドと補助ストランドのために使用される化学的に合成し、一本鎖DNA(ssDNAの)を再懸濁し、定量化するためのプロトコルについて説明します。鎖配列については、表10を参照してください 。以下のプロトコルは、10μMの一本鎖DNAを調製する一例であることに注意してください。一本鎖DNAの他の濃度も同様に調製することができます。
- DNAメーカーからオリゴを受け取った後、すべての乾燥したDNAがチューブの底にあることを保証するために1分間10,000-14,000×gでDNAを含むチューブをスピン。
- 100μMの最終濃度を達成するために:1×トリスエチレンジアミン四酢酸(EDTA)緩衝液(10mMトリスおよび1mMのEDTA、pH8.0のTE緩衝液)を用いてDNAを再懸濁します。ための例えば、TE緩衝液80μlのDNAの8ナノモルを再懸濁します。
- 10μMの最終濃度を達成しなければならないマイクロ遠心チューブ中の分子の水を90μlを、100μMでDNAを10μlを混ぜます。
- 製造者の指示に従って分光光度計を用いてDNA試料の正確な濃度を測定します。以下のプロトコルは、DNA濃度を測定する方法の例を示します。
- 分子の水を2μlの分光光度計のブランク。
- DNAサンプルの260 nmの(260)での吸光度を測定します。ストック濃度を計算するために、以下の式を使用します。
注:試料濃度はM 260 /吸光係数=れます。吸光係数は、DNAの製造者によって仕様データシートに記載されています。
蛍光レポーターの6準備
注:このセクションでは、説明レポーターCを調製するためのプロトコルは、他の蛍光レポーターは同様に組み立てることができます。
- 高速液体クロマトグラフィー(HPLC)で精製したオリゴヌクレオチドを注文ROX- *>(レポーターCの上の鎖)と -rq DNAメーカーから(レポーターCのボトム鎖が)(配列について、表10を参照してください )。
- 合成されたオリゴヌクレオチドを受信した後、再懸濁し、ステップ5で説明したようにサンプルを定量します。
- 12.5 mMのMgをレポータートップと1×トリス-酢酸-EDTA(TAE)の一番下の鎖(すなわち、ROX- と -rq)ミックス2+(詳細なレシピについては、表11を参照してください )。ここでは、30%の過剰クエンチャーラベルストランド -rqがすべてのフルオロフォアで標識された鎖があっても不完全な化学量論でクエンチされることを保証するレポーターを、組み立てるために追加されていることに注意してください。
- 1℃/分の速度で95°Cから20°Cまで冷却し、サーマルサイクラーを用いて、 レポーターC複合体をアニールします。サンプルを4℃で保存することができます。
7.蛍光測定
(実験手順については図5を参照)セクションには、蛍光反応速度測定のための一般的なプロトコルを記述し、このプロトコルは、手順8で使用される、9、および10はまた、このプロトコルは、蛍光分光計を使用するためのものであることに注意してください。注意してください。感度、ウェル間の変動および長期実験における温度制御の欠如が問題になる可能性があるが、代替的に、これらの実験はまた、プレートリーダーで実施することができます。
- 25℃の温度コントローラを設定し、温度が安定するのを待ちます。温度コントローラを使用して、温度変化に起因し得る信号の変動を低減することができます。
- FOを設定し、適切なパラメータ分光蛍光光度計のデータ収集ソフトのR動態測定。次のように詳細な設定例は以下のとおりです。
- 励起と発光モノクロメータの両方に2.73 nmのスリット幅を設定します。
- 60秒ごとの時点のために10秒に積分時間を設定します。 24時間に全測定時間を設定します。
- 実験に用いたフルオロフォアに適合するように、励起/発光波長を設定します。 ROX(588ナノメートル/ 608 nm)であり、TAMRA(559ナノメートル/ 583 nm)を次のように例の波長があります。
- ヌクレアーゼフリーのH 2 Oおよび合成石英セルに125 mMのMg 2+を(10倍TAE /マグネシウム2+)を含む10倍トリス-酢酸-EDTA緩衝液を追加します。例ボリュームが使用するために、表12、図13、図14を参照してください。
- 〜1μM(表12、13、およびボリュームの14)の最終濃度を達成するために、ポリTストランドを追加し、合成をボルテックス10〜15秒のための石英セル。一般的に、ピペットチップは、非特異的にDNAに結合します。ポリTストランドの高濃度を追加すると、この非特異的結合のエラーを低減することができます。
- 記者や補助ストランドを追加します。例ボリュームが使用する表12、図13、図14を参照してください。レポーターキャリブレーションのために、何の補助ストランドが必要とされないことに注意してください。
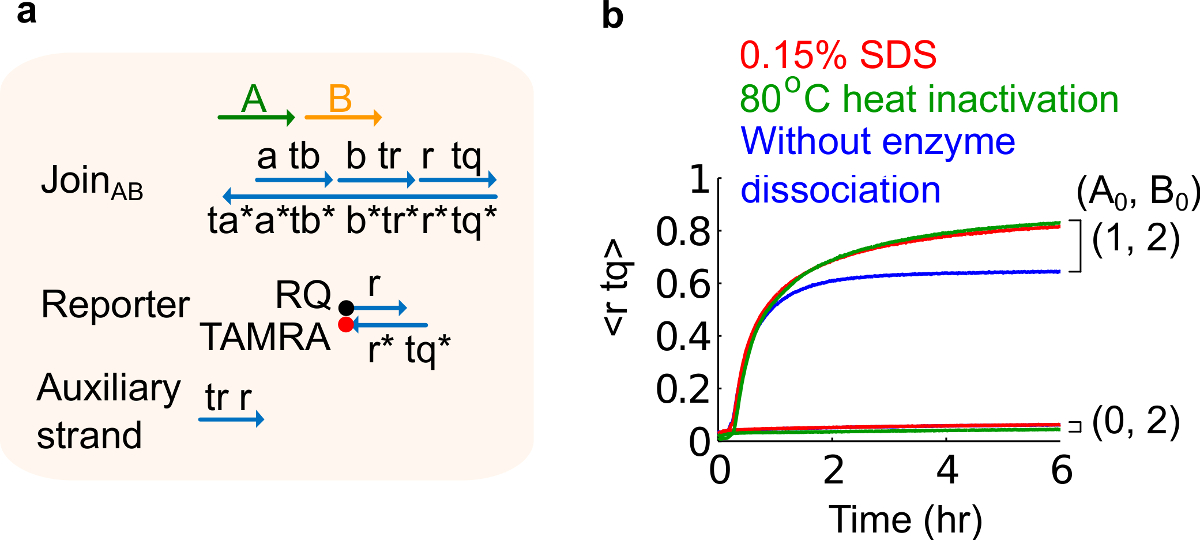
- 0.15%SDSの最終濃度を達成するために、10%のドデシル硫酸ナトリウム(SDS)を加えます。注:SDSは、酵素は、鎖置換反応( 図6参照)に干渉する可能性があるため、プラスミド由来のゲートから酵素を解離させるために使用されます。 SDSは悪影響回路機能に影響を与える可能性がある、ゲート鎖の解離、誤った再結合を回避するために、代わりに酵素の熱変性のここで推奨されています。
- 【レポーターキャリブレーションについて、このステップをスキップします。]
- 参加追加とフォークゲート( 表13、およびボリュームの14を参照してください )合成石英セルへと20倍以上(SDSでボルテックスソリューションは蛍光動態の測定に影響を与える、気泡が生じる可能性があるため、キュベットをボルテックスしないでください)のためにそれをピペットでダウン溶液を混合。
- リーク反応は合成石英セルに参加し、フォークゲートの添加直後に開始するため、また、できるだけ早く次の測定ステップに移動します。
- 分光蛍光計のチャンバー内に合成石英セルを配置します。
- 反応速度の測定を開始します。
- 測定の5分後、合成石英セルに入力ストランドを(ボリューム表12、図13、図14参照)を追加して、少なくとも20倍のためにピペッティングによって、およびダウン反応混合。サンプルは気泡を避けるために、穏やかに混合されるべきであることに注意してください。データ取得プログラムは、外部によってトリガ信号を測定避けるために一時停止している間に、この手順を実行します光。
- それは定常状態に達するまで反応速度を記録します。反応速度は、コンピュータ上に表示されます。
8.キャリブレーション蛍光レポーター
注:このセクションでは、蛍光レポーターの検量線を作成するためのプロトコルについて説明します。較正曲線は、モル濃度信号に任意の蛍光単位を変換するために使用されます。
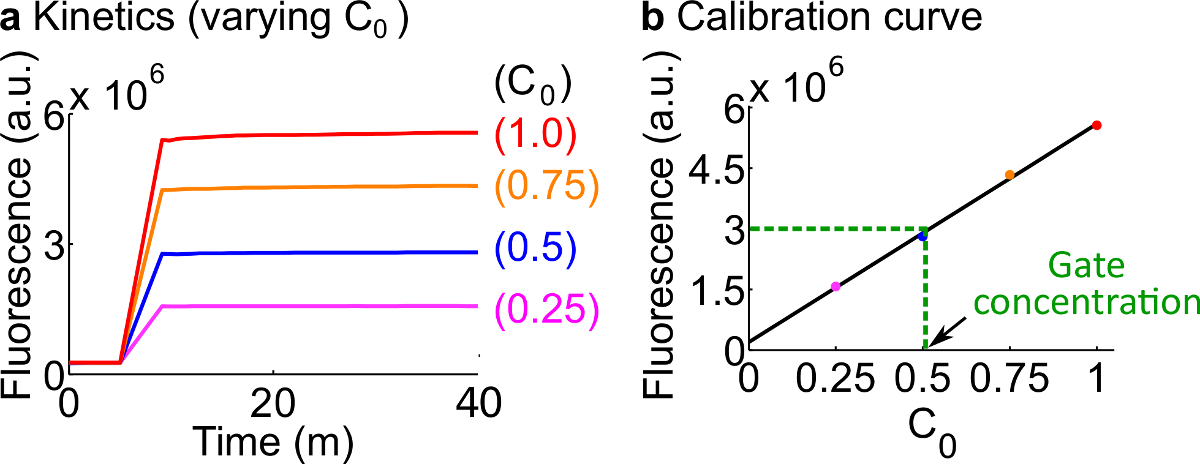
- ステップ7に記載のプロトコルに従って、蛍光レポーターのキャリブレーション表12にまとめたように、反応物およびバッファのボリュームを使用します。この例のための標準的な濃度は、50 nMの(1×)です。記者は3倍です。入力は1倍です。入力は0.25×、0.5倍、0.75xである場合には、600μlのように、各反応の最終容量を維持するためにそれに応じて2 OヌクレアーゼフリーのHの音量を調整します。例えばデータは、 図7Aに示されています。
- の検量線を作成します信号Cの初期濃度に対する最終的な蛍光値の線形近似によるレポーターC(例えば、較正曲線は、 図7Bに示されています)。この検量線は、対応する信号の濃度になるように、任意の蛍光単位を変換するために使用することができます。
9.プラスミド由来ndsDNAゲイツの濃度を定量化
機能ゲートの異なる歩留まりのプラスミド由来のndsDNAゲート結果のそれぞれ独立してバッチ処理を、このセクションでは、プラスミド由来ndsDNAゲートの濃度を定量化するためのプロトコルについて説明します。注意してください。
- 表13にまとめたようにステップ7.試薬の体積に記載のプロトコル以下のプラスミド由来のndsDNAゲートの濃度を定量注: 表13は、 フォーク、BCの定量化のための一例のレシピを説明しましょう 。 ABと他のゲートは同様に実施するが、異なる入力ストランド、補助ストランドと記者を使用することができます。
- ステップ8.2からの検量線を用いて信号Cの濃度がこの実験で測定された最終的な蛍光値を変換します。その後ndsDNAゲート濃度を逆算。例えば、 図7(b)の検量線に基づいて、25 nMの信号C(0.5倍)に対応するゲートの定量実験の最終的な蛍光値。 フォークBCの在庫は、この反応で40倍に希釈されているので、 フォークBCゲートのストック濃度は1μMです。
反応 A + B-> B + C 10.動力学的な測定
注:このセクションでは、蛍光動態の測定値を使用して、正式な化学反応のDNAの実現をテストするためのプロトコルについて説明します。
- パー表14にまとめたように、試薬および緩衝液のステップ7.ボリュームに記載のプロトコルに従うことにより、フォーム動態測定。
結果
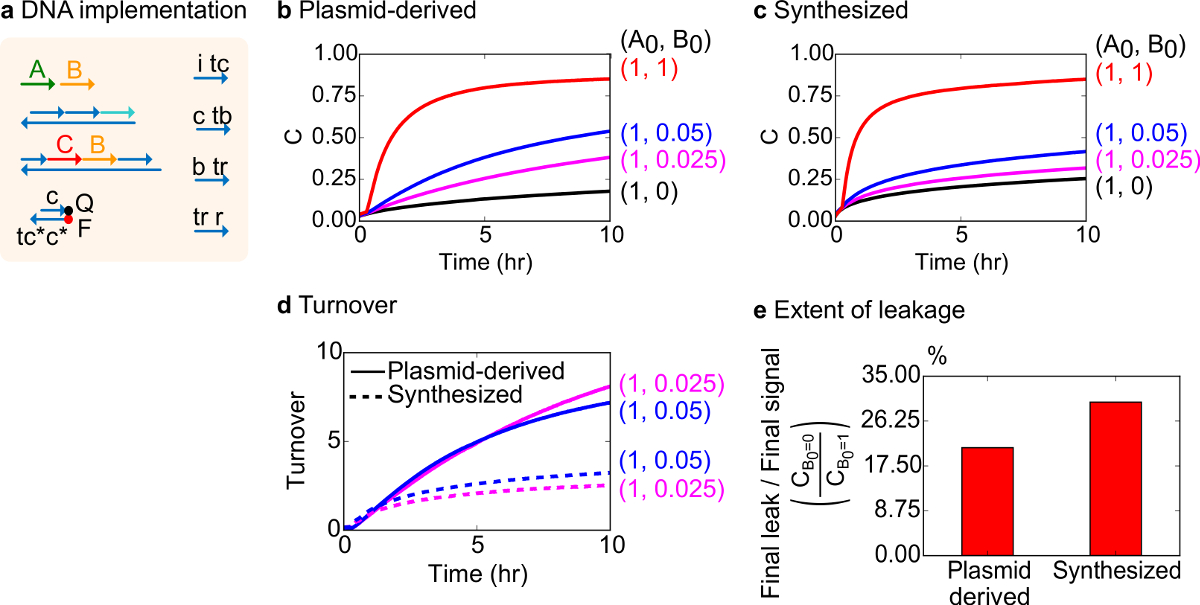
機能テストのために、二分子触媒反応( すなわち、A + B-> B + C)のDNAの実装が作成されました。プラスミド由来のゲートの性能は、合成DNAから組み立てゲートと比較しました。障害のあるゲートは不可逆的にトラップ生成物の量に不釣り合いな影響を引き起こす触媒は、18,19を生成できるため、触媒反応は、ゲート純度のための良いテストです。これと同時に、触媒の信号のuntriggered放出をもたらす小さなリーク反応が直線的に不均衡なエラー信号につながる、増幅されます。プラスミド由来の合成ゲートの実験データを、それぞれ、 図8B及び図8Cに示されています。触媒の信号Bの量を変化させながら実験では、信号の鎖Aの濃度が固定されています。信号Cは、触媒を中断することなく、反応の進行を読み出すために使用されサイクル。反応もAの量よりもはるかに小さい触媒Bの量の完了に近づくので、触媒は、データで観察することができます。 SDSが合成されたシステムで行われた実験に追加されなかったので、反応速度が(それは、SDSの添加によって影響を受ける可能性がある)と比較されていないと分析の焦点は、(次のように詳述)の代わりに触媒回転率です。
この反応の触媒代謝回転の更なる分析を行いました。代謝回転は、所与の時間に各触媒Bの生成量信号Cとして定義されます。具体的には、売上高は、Bを添加し 、触媒の初期量によってリーク減算信号Cを分割することによって我々の実験データから計算しました。理想的な触媒系では、この代謝回転数は直線的に時間の経過とともに増大するはずであると限り基板が制限されていないような触媒の量に依存しないように。実際のシステムでは、障害のあるゲートは猫を無効にすることができます alysts、および代謝回転は、すべての利用可能な基質が生成物に変換されても最大値に達します。最大ターンオーバー値は、触媒(信号B)は不活性化になる前に変換することができるどのように多くの基質(信号A)を示しています。ここでは、合成されたシステムは、プラスミド由来のシステムよりもはるかに早く売上高の理想的な直線的な増加から外れることが観察される望ましくない副反応( 図8D)を介し触媒の隔離を示し、実行します。売上高の比較はためだけの触媒の高濃度で低濃度のために示されている、すべてのゲートがトリガされ、 信号 C を解放します。回路の漏れも比較され、それはプラスミド由来のゲートを使用して漏れ信号の比は、反応( 図8E)の10時間後に合成されたゲートを使用してより約8%以下であることが観察されます。
ジル/ ftp_upload / 53087 / 53087fig1.jpg "/>
図1(A)CRNsは、規範的なプログラミング言語としての役割を果たす。 DNA反応ネットワークは、正式なCRNの動力学を近似するように操作することができる、例えば化学命令の(B)DNAの実装:A + B-> B + C。 DNA鎖は、3 '末端に矢印の付いたラインとして描かれており、*は、相補性を示しています。すべての信号は(緑>)、B(<結核B>、オレンジ)、 および C(、赤)は(TA、TB、およびTCと表記)1足掛かりドメインから構成されているストランドと(A、B、およびCとラベル付け)は、1つの同一ドメイン。二分子反応A + B-> B + Cは、二つの多重鎖複合体はABとフォークBCに参加が必要であり、4つの補助ストランド、、、および B。各ステップの開始鎖置換、の7つのステップを経て反応が進行します足掛かり結合。(C)レポーター戦略との。反応は、下の鎖は、フルオロフォア(レッドドット)で標識され、上の鎖は、クエンチャー(黒丸)に結合しているレポーターを使用して続いています。なぜなら、フルオロフォアおよびクエンチャーの共局在化を、レポーター蛍光は、無傷のレポーターで急冷されます。 信号 C は、蛍光の増加につながる、レポーターの上の鎖を置き換えることができます。 (この図は、参考文献29から変更されている。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

細菌プラスミドDNAから作られた図2(A)NdsDNAゲート。二本鎖ndsDNAゲートテンプレートのいくつかのコピーがプラスミドにクローニングします。クローン化されたプラスミドでありますnは、Eに変換コリ細胞とプレート上のコロニーを配列を確認しています。配列が確認されたら、プラスミドDNAを増幅し、抽出します。最後に、二本鎖プラスミドを、酵素処理により、所望のndsDNAゲートに加工される。ndsDNAゲートの(B)酵素処理。制限酵素PvuIIでは、プラスミドからゲートを解放するために使用されます。解放ゲートはさらにニッキング酵素を用いて処理される。Nb.BsrDIが参加AB(パネルI)でニックを生成するために使用されます。 Nt.BstNBIはフォークBC(パネルII)のためにニックを生成するために使用されます。制限及びニッキング部位は、色分けされたボックスとして示されている。 参加AB(パネルI)およびフォークBC(パネルII)のゲートテンプレートの(C)シーケンス図である。(紫色のボックスで強調表示)PvuII制限部位が両端にありndsDNAゲートの。 Nb.BsrDIおよびNT.BstNBIニッキング部位は、それぞれ、赤と黒のボックスで強調されています。カットの位置は矢印の頭が付いています。シーケンスNは任意のヌクレオチドです。 (この図は、参考文献29から許可を得て変更されています。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

DNAゲートテンプレートの図3(A)PCR。 DNAゲートテンプレートは、両端の中央にndsDNAゲートシーケンス(青色領域)、及びスペーサー配列(;これら二つの末端配列が直交する黒領域)が含まれています。プライマーは(重複配列を図に色分けされている)は、ゲートテンプレートのスペーサー配列に結合し、PCRを通じて4オーバーラップDNAフラグメントを生成することができます。(B)ギブソンアセンブリを。 4増幅したDNA fragmenTSは、その後、ギブソンの組み立て方法43を介して線状プラスミドバックボーンに組み立てられます。 (この図は、参考文献29から許可を得て変更されています。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

異なる酵素量図4.回路性能。プラスミド由来の参加ABと(A)ゲートを簡単に示し、レポーター、補助ストランド、および対応する実験に使用される信号ストランド。(B)キネティクス実験は異なる酵素量で処理。私。 PvuII-HFと1μgのプラスミドあたりNb.BsrDIの45単位、10単位; II。 PvuII-HFと1μgのあたりNb.BsrDIの4台の10台プラスミド; III。 PvuII-HFと1μgのプラスミドあたりNb.BsrDIの4台の4台。すべての補助鎖は2倍(1倍= 10nMの)でした。ゲート複合体は1.5倍であり、実験は、1×TAE / Mg 2+を35°Cで行いました。 (この図は、参考文献29から許可を得て変更されています。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図5動態実験のチャートフローブルー:マテリアルキュベットに追加する(0.875ミリリットル合成石英セル)。 > B + C -特定のボリュームのリファレンス表14は、A + Bの運動実験のために追加します。緑:(SPEXと表記)分光光度計の指示。赤:指示をミキシング。53087fig5large.jpg「ターゲット= "_空白">この図の拡大版をご覧になるにはこちらをクリックしてください。

図6.酵素解離し、回路の動作。(A)単純化されたゲートの表現、レポーター、補助ストランド、および対応する実験に使用される信号ストランド。(B)プラスミド由来の速度論実験は、80℃の熱を利用して、ABに参加不活化(緑のトレース)、0.15%ドデシル硫酸ナトリウム(SDS)(赤)、および熱不活性化またはSDS(青)を添加しないコントロール。標準濃度は1×= 10 nmであった、とすべての補助ストランドと入力Bは2倍でした。ゲート複合体は1.5倍であり、実験は、12.5 mMのMg 2+を(1×TAE /のMg 2+を含む1×トリス-酢酸-EDTA緩衝液中で35℃で行いましたアップ。 (この図は、参考文献29から変更されている。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図7. Reporterの校正。(A) レポーターC 動態。測定終了点(40分)でのレポーター濃度信号Cの(1×= 50 nM)を3回であって、信号Cの初期濃度は、図に示されている。(B)の蛍光レベルを有する線形の関係を示しています信号Cの初期濃度。 フォークBCゲート(緑の破線)の定量化の例では、 フォークBCの蛍光値は測定しましたD検量線に基づいて、25 nMの(0.5倍)に相当する3×10 6(AU)、として。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図8。二分子触媒反応速度論(A + B-> B + C)(A)Aは、対応する実験に用いられるゲート、レポーター、補助鎖、およびシグナル鎖の表現を簡略化します。実験は、12.5 mMのMg 2+を(1×TAE /のMg 2+)を含む1×トリス-酢酸-EDTA緩衝液中で行いました。すべてのゲート複合体は、75 nMの濃度(1.5倍)であった、と補助鎖は(2×)、100nMの濃度でした。合成されたゲートのプラスミド由来のゲートおよびデータのための反応速度データは、(B)及び(C)に示されています強いです。信号は、50 nMの(1×)でした。の信号(触媒)の様々な量がシステムに導入し、反応は35℃で試験した。(D)プラスミド由来のゲートは、入力の少量を添加した合成DNAゲートより高い売上高を示した。(E)程度漏れ。棒グラフは、エンドポイント(10時間)での最終信号(C B0 = 0 / C B0 = 1)への最終的な漏れの割合を示しています。 (この図は、参考文献29から変更されている。) この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
| 門テンプレート | シーケンス | 長さ(NT) |
| JOINAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
表1。ndsDNAゲートテンプレートの配列。
| ゲート | ストランド | ボトム鎖の長さ(NT) |
| JOINAB | JOINABボトムは、、、 | 87 |
| ForkBC | ForkBCボトム、、、<結核B>、 | |
| 108 |
表2。ABとフォークBCに参加備えるストランド。(この表には、参考文献29から変更されています。)
| ドメイン | シーケンス | 長さ(NT) |
| TA | CTGCTA | 6 |
| TB | TTCCAC | 6 |
| TC | TACCCA | 6 |
| TR | TCCTAC | 6 |
| TQ | AACCAG | 6 |
| A | CATTGCTTCTACGAGTCATCC | 21 |
| B | CATTGCACCTTAGAGTCCGAA | 21 |
| C言語 | CATTGCCACATCGAGTCCCTT | 21 |
| R | CATTGCTTAACCGAGTCTCAC | 21 |
| 私 | CTGCCATCATAAGAGTCACCA | 21 |
| プライマーの鎖 | シーケンス | 長さ(NT) |
| フォワードプライマー-1 | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| リバースプライマー1 | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| フォワードプライマー2 | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| リバースプライマー2 | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| フォワードプライマー3 | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| リバースプライマー3 | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| フォワードプライマー-4 | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| リバースプライマー4 | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
表4。ndsDNAゲートテンプレートのPCR用プライマー配列。
| 試薬 | 1X反応のための容量(μL) |
| 高コピープラスミド骨格(〜300 ngの/μL) | 10 |
| PvuII-HF(2万台/ ml)を | 2 |
| PstI-HF(2万台/ ml)を | 2 |
| 10倍のカットスマートバッファ | 2 |
| H 2 O | 4 |
| 全容積 | 20 D> |
表5。プラスミド骨格ダイジェストのためのプロトコル。
| 試薬 | 1X反応のための容量(μL) |
| DNAベクター(〜50 ngの/μL) | 1 |
| PCRは断片-1(〜50 ngの/μl)を増幅しました | 1 |
| PCRは、フラグメント2(〜50 ngの/μl)を増幅しました | 1 |
| PCRは断片-3(〜50 ngの/μl)を増幅しました | 1 |
| PCRは断片-4(〜50 ngの/μl)を増幅しました | 1 |
| 2倍ギブソン・アセンブリのマスターミックス | 5 |
| 全容積 | 10 |
| 試薬 | 1X反応のための容量(μL) |
| プラスミドDNA(〜を1μg/μlの濃度) | 千 |
| PvuII-HF(2万台/ ml)を | 200 |
| 10倍のカットスマートバッファ | 133.3 |
| 全容積 | 1333.3 |
ndsDNAゲートについては、表7。プロトコルは、プラスミドを制限酵素PvuIIで-HFで消化挿入しました。
| 試薬 | 容量(μL) |
| 参加ゲート(〜5μgの/μlの濃度) | 150 |
| Nb.BsrDI(10,000単位/ ml) | 300 |
| 10倍のカットスマートバッファ | 50 |
| 全容積 | 500 |
表8。ゲートは、酵素Nb.BsrDIをニッキングでダイジェストに参加するためのプロトコル。
| 試薬 | 容量(μL) |
| フォークゲート(〜5μgの/μlの濃度) | 150 |
| Nt.BstNBI(10,000単位/ ml) | 600 |
| 10倍NEBバッファー3.1 | 83.3 |
| 全容積 | 833.3 |
表9議定フォークゲートの酵素Nt.BstNBIをニッキングで消化。
| ストランド | ドメイン | シーケンス | 長さ(NT) |
| JOINABボトム | TQの*のR * TA * TR * b *のTB * * | CTGGTT GTGAGACTCGGTTAAGCAATG GTAGGA TTCGGACTCTAAGGTGCAATG GTGGAA GGATGACTCGTAGAAGCAATG TAGCAG | 87 |
| FORKBCボトム | TQ *のR *のtr * b *の結核*のC *のTCの*私は* | CTGGTT GTGAGACTCGGTTAAGCAATG GTAGGA TTCGGACTCTAAGGTGCAATG GTGGAA AAGGGACTCGATGTGGCAATG TGGGTA TGGTGACTCTTATGATGGCAG | 108 |
| <タ> | CTGCTA CATTGCTTCTACGAGTCATCC | 27 | |
| <結核B> | A TA | TTCCAC CATTGCACCTTAGAGTCCGAA | 27 |
| 結核B | TACCCA CATTGCCACATCGAGTCCCTT | 27 | |
| TCのC | CATTGCTTCTACGAGTCATCC TTCCAC | 27 | |
| TB | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 | |
| B TRを | CATTGCTTAACCGAGTCTCAC AACCAG | 27 | |
| RのTQ | CTGCCATCATAAGAGTCACCA | 21 | |
| 私 | TCCTAC CATTGCTTAACCGAGTCTCAC | 27 | |
| <私はTC> | TRのR | CTGCCATCATAAGAGTCACCA TACCCA | 27 |
| 私は、TC | CATTGCCACATCGAGTCCCTT TCCTAC | 27 | |
| C TRを | CATTGCCACATCGAGTCCCTT TTCCAC | 27 | |
| CのTB | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 | |
| <私はTB> | B TRを | CTGCCATCATAAGAGTCACCA TTCCAC | 27 |
| 私はTB | CATTGCACCTTAGAGTCCGAA TTCCAC | 27 | |
| BのTB | CATTGCACCTTAGAGTCCGAA TACCCA | 27 | |
| BのTC | CATTGCCACATCGAGTCCCTT TCCTAC | 27 | |
| C TRを | CATTGCACCTTAGAGTCCGAA TCCTAC | 27 | |
| ROX- | B TRを | / 56-ROXN / AAGGGACTCGATGTGGCAATG TGGGTA | 27 |
| -rq | C * TC * | CATTGCCACATCGAGTCCCTT / 3IAbRQSp / | 21 |
| - TAMRA | C | CTGGTT GTGAGACTCGGTTAAGCAATG / 36-TAMTSp / | 27 |
| RQ- | TQの* rを* | / 5IAbRQ / CATTGCTTAACCGAGTCTCAC | 21 |
| R |
表10鎖配列を化学反応Aを実現するための+ B - > B + C(。この表は、参考文献29から変更されています)
| 試薬 | 容量(μL) | 最終濃度 |
| 100μMでROX- | 10 | 10μM(1×) |
| 100μで -rq; M | 13 | 13μM(1.3倍) |
| 125 mMのMg 2+を持つ10倍TAE | 10 | 12.5 mMのMg 2+を持つ1×TAE |
| H 2 O | 67 | - |
| 全容積 | 100 | 10μM(1×) |
レポーターCを組み立てるための表11のプロトコル。
| 試薬 | 容量(μL) | 最終濃度 |
| H 2 O | 514 | - |
| 125 mMのMg 2+を持つ10倍TAE | 60 | 12.5 mMのMg 2+を持つ1×TAE |
| 300μでポリT; M | 2 | 1μM |
| 10μMでレポーターC | 9 | 150nMの(3×) |
| 10%SDS | 9 | 0.15% |
| 5μMで | 6 | 50 nMの(1×) |
| 全容積 | 600 | - |
レポーターCのキャリブレーションのための表12。プロトコル。ここで提供されるボリュームは600μL(0.875 mlの合成石英セルを使用することに相当する)の全反応容積のためであるが、異なるサイズのセルで動作するように調整することができます。
| 試薬 | 容量(μL) | 最終的なコンセンタリング |
| H 2 O | 493 | - |
| 125 mMのMg 2+を持つ10倍TAE | 60 | 12.5 mMのMg 2+を持つ1×TAE |
| 300μMでポリT | 2 | 1μM |
| 10μMでレポーターC | 9 | 150nMの(3×) |
| 100μMで<私はTC> | 3 | 10倍 |
| 100μMで | 3 | 10倍 |
| 100μMで | 3 | 10倍 |
| 10%SDS | 9 | 0.15% |
| 〜1μMでフォークBC(濃度不明) | 15 | 〜0.5倍 |
| 100μMで | 3 | 10倍 |
| 全容積 | 600 | - |
表13。 フォークBCのキャリブレーションのためのプロトコル。ここで提供されるボリュームは、600μlの総反応容量のためであるが、異なるサイズのセルで動作するように調整することができます。
| 試薬 | 容量(μL) | 最終濃度 | |
| H 2 O | 407.2 | - | |
| 125 mMのMg 2+を持つ10倍TAE | 52.8 | 12.5 mMのMg 2+を | |
| 300μMでポリT | 2 | 1μM | |
| 10μMでレポーターC | 9 | 150nMの(3×) | |
| 10μMで<私はTC> | 6 | 100nMの(2×) | |
| 10μMで | 6 | 100nMの(2×) | |
| 10μMで | 6 | 100nMの(2×) | |
| 6 | 100nMの(2×) | ||
| 10%SDS | 9 | 0.15% | |
| 1μMでABに参加 | 45 | 75 nMの(1.5倍) | |
| 1μMでフォークBC | 45 | 75 nMの(1.5倍) | |
| 10μMで<タ> | 3 | 50 nMの(1×) | |
| 10μMで<結核B> | 3 | 50 nMの(1×) | |
| 全容積 | 600 | - | |
化学反応A + B-> B + C表14.プロトコル。ここで提供されるボリュームは、600μlの総反応容量のためであるが、異なるサイズのセルで動作するように調整することができます。
| 合成ゲート | プラスミド由来のゲート | ||||
| 説明 | コスト | ゲートに参加 | フォークゲート | ||
| PAGE精製した長い鎖(100 NTは、ゲートのボトム鎖を務めました) | 〜$ 75 | 説明</強いです> | コスト | 説明 | コスト |
| PAGE精製された短鎖(〜30ヌクレオチド、ゲートの上部の鎖を務めました) | 〜$ 185 | ゲートテンプレート | 〜$ 100 | ゲートテンプレート | 〜$ 100 |
| トータル | 〜$ 260 | プラスミド抽出キット | 〜$ 26 | プラスミド抽出キット | 〜$ 26 |
| 制限酵素(のPvuII-HF) | 〜$ 11 | 制限酵素(のPvuII-HF) | 〜$ 11 | ||
| ニッキング酵素(Nt.BsrDI、ゲートに参加) | 〜$ 29 | ニッキング酵素(Nt.BstNBI、フォークゲート) | 〜$ 62 | ||
| トータル | 〜$ 166 | トータル | 〜$ 199 |
表15プラスミド由来のゲートと合成ゲート間の比較をコスト (この表は、参考文献29から変更されています。)。
| 合成ゲート | プラスミド由来のゲート | ||
| 処理 | 処理時間 | 処理 | 処理時間 |
| アニーリング | 1時間 | クローニング | 5時間 |
| PAGE精製 | 2時間 | プラスミド抽出 | 2時間 |
| トータル | 3時間 | 酵素消化の二段階 | 0.5時間 |
| エタノール沈殿 | 1時間 | ||
| トータル | 8.5時間 | ||
表16。プラスミド由来のゲートとゲート間の合成処理時間の比較。(この表には、参考文献29から変更されています。)
ディスカッション
本論文では、高純度のプラスミドDNAからndsDNAゲートを導出するための方法を説明します。また、プロトコルは、蛍光動力学アッセイを用いて、ゲート性能を特徴付けるために提示されています。実験データは、プラスミド由来の系、合成系は、ポリアクリルアミドゲル電気泳動(PAGE)を用いて精製したストランドから組み立てられていても、その合成対応物よりも優れていることを示しています。多分、プラスミド由来のゲートの性能の改善は、主に生物学的DNAの非常に高純度に起因するものです。合成DNAは、さまざまなエラーが含まれ、特に、長さN-1のオリゴヌクレオチド、およびそのような副生成物を生じる欠失は、典型的には、完全PAGEまたは高速液体クロマトグラフィー(HPLC)精製手順で除去されません。ここで報告されたものと同様の改善はまた、生物学的供給源21に由来するDNAを使用した触媒ヘアピン増幅器の以前の研究で観察されました。
しかし、プラスミド由来のゲートを用いても、完全に少なくとも2つの理由がありますするためのゲート性能のエラー、排除することはできません。あまりにも多くてゲートにつながることができ、最初の過消化またはカット精度の欠如を間違った位置にニックまたはニック。いずれの場合においても、ゲートが望ましくない反応に関与する可能性が高いです。このような問題は、( 図4を参照)を使用する酵素の量を最適化することにより緩和することができます。第二に、これらの実験では、ほとんどの入力および補助鎖合成DNAであり、したがって、欠失および変異を含みました。原則として、すべての一本鎖入力と補助鎖はまた、事前符号化M13ウイルスゲノム26のニッキング酵素消化を介してファージミドDNAから得ることができます。おそらく回路性能は、さらに、細菌ゲノムに由来する一本鎖DNAを用いて改善することができます。
プラスミド由来のゲートの使用は、回路性能を改善することが見出されたが、分析コストや処理時間のプラスミド由来のゲートの製造がやや安い( 表15)であるが、それは商業的に合成されたオリゴ( 表16)からゲートのアセンブリおよび精製に比べて2~3倍長い処理時間を要することが明らかになりました。プラスミド由来のゲートの主要コストは、遺伝子合成、制限酵素の使用です。 (30 nmで15の反応のために十分な)ゲートの300ピコモルのために、参加ゲートの推定コストはフォークゲート、違いによるニッキング酵素の使用にいるコストの差は約$ 170と$ 200です。これとは対照的に、PAGE精製料込み$ 260の周りに同じゲート費用鎖の化学合成。プラスミド由来のゲートのための主要な時間コストは、単にDNA合成と同様に、遺伝子合成会社に委託することができ、クローニング手順、です。しかし、一旦組み立て、プラスミド由来のゲートは、宿主プラスミドを容易に複製することができるという利点を有しますND細菌のグリセロールストックの形で保存することができます。これは、上にゲートを何度も再利用することができます。
楽しみにして、プラスミド由来のゲートの改良された性能を実験的にDNA CRNsで、これまでに実証されているよりも、力学挙動のはるかに大きな範囲を可能にすることができます。例えば、最近の理論的研究47,48は、マクロスケールでの自己組織化の空間パターンは、反応拡散機構を介してDNA CRNsで実現することができることを示唆しました。ここで紹介する方法は、自己パターニングDNA材料の基礎となる分子成分を構築するための実行可能なパスを提供します。挑戦が、プログラム可能な方法で、マクロスケールの形態を開発するバイオマテリアルの研究から再生医療に至るまでの分野で重要な意味を持っているでしょう。
開示事項
The authors declare no competing financial interests.
謝辞
図1、2、3、4、6、8及び表2、3、10、15、16は、参考文献29から変更されています。この作品は、国立科学財団(NSF-GSにCCF 1117143およびNSF-CCF 1162141を付与する)によってサポートされていました。 Y.-JCは、台湾政府のフェローシップによってサポートされていました。 SDRは、国立科学財団大学院研究フェローシッププログラム(GRFP)によってサポートされていました。
資料
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S | |
| PvuII-HF | NEB | R3151L | |
| PstI-HF | NEB | R3140S | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Terrific Broth, Modified | SIGMA-ALDRICH | T0918-250G | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAGEN Hispeed Maxi-prep Kit | QIAGEN | 12662 | |
| Nb.BsrDI | NEB | R0648L | |
| Nt.BstNBI | NEB | R0607L | |
| NanoDrop 2000c | Thermo Scientific | ||
| Double-stranded Genomic Blocks | IDT | ||
| Horiba Jobin-Yvon Spex Fluorolog-3 Fluorimeter | Horiba/Jobin Yvon | ||
| Synthetic Quartz Cells | Starna | 23-5.45-S0G-5 | |
| QIAGEN Gel Extraction Kit | QIAGEN | 28706 | |
| Plasmid Backbones | BioBrick | E0240-pSB1A2 | High copy number plasmid with Ampicillin resistance. Sequence can be found from http://parts.igem.org |
参考文献
- Zhang, D. Y., Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103-113 (2011).
- Krishnan, Y., Simmel, F. C. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 50, 3124-3156 (2011).
- Zhang, D. Y., Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303-17314 (2009).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475, 368-372 (2011).
- Qian, L., Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 332, 1196-1201 (2011).
- Zadegan, R. M., Jepsen, M. D., Hildebrandt, L. L., Birkedal, V., Kjems, J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 11, 1811-1817 (2015).
- Seelig, G., Soloveichik, D., Zhang, D. Y., Winfree, E. Enzyme-free nucleic acid logic circuits. Science. 314, 1585-1588 (2006).
- Zadegan, R. M., et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 6, 10050-10053 (2012).
- Andersen, E. S., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 459, 73-76 (2009).
- Zhang, D. Y., Hariadi, R. F., Choi, H. M., Winfree, E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 4, (1965).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406, 605-608 (2000).
- Green, S. J., Lubrich, D., Turberfield, A. J. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 91, 2966-2975 (2006).
- Venkataraman, S., Dirks, R. M., Rothemund, P. W., Winfree, E., Pierce, N. A. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2, 490-494 (2007).
- Green, S. J., Bath, J., Turberfield, A. J. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 101, 238101 (2008).
- Omabegho, T., Sha, R., Seeman, N. C. A bipedal DNA Brownian motor with coordinated legs. Science. 324, 67-71 (2009).
- Turberfield, A. J., et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 90, 118102 (2003).
- Dirks, R. M., Pierce, N. A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 101, 15275-15278 (2004).
- Seelig, G., Yurke, B., Winfree, E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 128, 12211-12220 (2006).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318, 1121-1125 (2007).
- Yin, P., Choi, H. M., Calvert, C. R., Pierce, N. A. Programming biomolecular self-assembly pathways. Nature. 451, 318-322 (2008).
- Chen, X., Briggs, N., McLain, J. R., Ellington, A. D. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 110, 5386-5391 (2013).
- Phillips, A., Cardelli, L. A programming language for composable DNA circuits. J. R. Soc. Interface. 6, S419-S436 (2009).
- Lakin, M. R., Youssef, S., Polo, F., Emmott, S., Phillips, A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 27, 3211-3213 (2011).
- Lakin, M. R., Youssef, S., Cardelli, L., Phillips, A. Abstractions for DNA circuit design. J. R. Soc. Interface. 9, 470-486 (2012).
- Zhang, D. Y., Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182-4197 (2010).
- Ducani, C., Kaul, C., Moche, M., Shih, W. M., Hogberg, B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 10, 647-652 (2013).
- Lin, C., et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 105, 17626-17631 (2008).
- Bhatia, D., et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 48, 4134-4137 (2009).
- Chen, Y. J., et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 8, 755-762 (2013).
- Arkin, A., Ross, J. Computational functions in biochemical reaction networks. Biophys. J. 67, 560-578 (1994).
- Érdi, P., Tóth, J. . Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. , (1989).
- Magnasco, M. O. Chemical kinetics is Turing universal. Phys. Rev. Lett. 78, 1190 (1997).
- Oishi, K., Klavins, E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 5, 252-260 (2011).
- Senum, P., Riedel, M. Rate-independent constructs for chemical computation. PLoS One. 6, (2011).
- Soloveichik, D., Cook, M., Winfree, E., Bruck, J. Computation with finite stochastic chemical reaction networks. Natural Computing. 7, 615-633 (2008).
- Soloveichik, D., Seelig, G., Winfree, E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 107, 5393-5398 (2010).
- Tyson, J. J., Chen, K. C., Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 15, 221-231 (2003).
- Cardelli, L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 23, 247-271 (2013).
- Angluin, D., Aspnes, J., Eisenstat, D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 21, 87-102 (2008).
- Cardelli, L., Csikasz-Nagy, A. The cell cycle switch computes approximate majority. Sci. Rep. 2, 656 (2012).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170-173 (2011).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. , (2012).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. , e253 (2007).
- Lessard, J. C. Transformation of E. coli via electroporation. Methods Enzymol. 529, 321-327 (2013).
- Nasri, M., Thomas, D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 15, 7677-7687 (1987).
- Dalchau, N., Seelig, G., Phillips, A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. , 84-99 (2014).
- Scalise, D., Schulman, R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2, 55-66 (2014).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved