
Method Article
Plasmidi derivati da DNA Strand Displacement Cancelli per reti di reazione implementazione chimici
In questo articolo
Riepilogo
This protocol describes a method for deriving DNA strand displacement gates from plasmids and testing them using fluorescence kinetics measurements. Gates can be modularly composed into multi-component systems to approximate the behavior of formal chemical reaction networks (CRN), demonstrating a new use for CRNs as a molecular programming language.
Abstract
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
Introduzione
La prevedibilità di Watson-Crick base di abbinamento è permesso nanotecnologia del DNA dinamico di emergere come un modo programmabile per la progettazione di dispositivi molecolari con proprietà dinamiche 1,2. In particolare, lo spostamento filamento di DNA - una reazione di ibridazione competitiva programmabile - ha dimostrato di essere un potente meccanismo di ingegneria dei sistemi dinamici di DNA. In una reazione spostamento filamento di DNA, un oligonucleotide arrivo sposta una ciocca precedentemente legato "uscita" da un partner di legame complementare. Molteplici tali reazioni possono essere concatenati in cascate di reazione multi-step con un alto grado di controllo sull'ordine e la tempistica della reazione individuale passi 3. DNA cascate spostamento filone sono stati utilizzati per creare circuiti digitali e analogici molecolari 4-7, 8-10 nanostrutture commutabili, motori molecolari autonome 11-15, e non covalenti amplificatori catalitiche 13,16-21. Inoltre, DDispositivi NA utilizzando reazioni di spostamento dei filamenti possono essere simulati e progettati per diverse applicazioni utilizzando il software di progettazione assistita da computer 22-24.
Attualmente, il DNA sintetizzato chimicamente serve come il materiale principale per le nanotecnologie del DNA. Tuttavia, gli errori nel processo di sintesi del DNA, e le conseguenti oligonucleotidi imperfetti, si ritiene che limitare le prestazioni dei dispositivi di Dynamic DNA, provocando reazioni collaterali errati. Ad esempio, reazioni "fuga" possono causare il rilascio di un oligonucleotide uscita anche in assenza di un trigger reazione. Tali effetti sono più evidenti in cascate di reazione autocatalitiche dove anche una minima quantità di perdita iniziale eventualmente sfociare nella piena attivazione della cascata 19,20. Viceversa, reazioni spesso non riescono a raggiungere il livello atteso di attivazione perché alcuni componenti non attivano anche in presenza dell'ingresso previsto 7,25. Per rendere le prestazioni di DNA-basednanodispositivi paragonabili alle loro controparti a base di proteine biologiche, tali modalità di errore devono essere drasticamente ridotto.
Plasmidi batterici o altro DNA biologico potrebbe servire come fonte relativamente a buon mercato di DNA altamente puro per applicazioni nanotecnologiche. Grandi quantità di DNA possono essere generati dalla replica nei batteri e le capacità di correzione di bozze intrinseche dei sistemi viventi garantiscono la purezza del DNA risultante. In effetti, diversi lavori recenti hanno riconosciuto la potenziale utilità del DNA biologico per applicazioni nanotecnologiche 21,26-28. Tuttavia, la natura completamente a doppio filamento di DNA plasmide ha finora vietato l'uso come materiale per la realizzazione di dispositivi di DNA dinamici, normalmente rappresentato più oligonucleotidi e contengono entrambi i domini doppio filamento e singolo filamento. In un recente lavoro 29 questo problema è stato affrontato e una nuova architettura porta DNA che consiste principalmente di intaccate DNA a doppia elica (ndsDNA) era introdurred.
È importante sottolineare che i sistemi di porte ndsDNA possono essere progettati che realizzano le dinamiche specificato da una rete formale reazione chimica (CRN) 29. cancelli ndsDNA potrebbero quindi essere utilizzati, in linea di principio, di creare sistemi dinamici che presentano oscillazioni e il caos, bistabilità e la memoria, la logica booleana o comportamenti algoritmici 30-38. Ad esempio, Rif. 29 hanno dimostrato una CRN tre reazione che ha fornito un'implementazione molecolare di un protocollo "consenso", un tipo di algoritmo di calcolo distribuito 29,39,40. Questo primo lavoro ha dimostrato un nuovo uso per il formalismo CRN come un "linguaggio di programmazione" per la rapida sintesi di sistemi molecolari funzionali (Figura 1A).
Qui, è previsto un protocollo dettagliato per ricavare porte ndsDNA dal plasmide DNA. Il primo è una revisione del processo di progettazione sequenza. Segue una spiegazione di oligonucleotidi sintetici contenenti comele sequenze del cancello vengono clonati in plasmidi e la sequenza verificate e amplificati tramite coltura batterica. Successivamente, è mostrato come ndsDNA porte possono essere derivati da plasmidi di trattamento enzimatico (vedere Figura 2). Infine, un metodo per testare il comportamento cancello utilizzando saggi cinetici fluorescenza è delineato.
Meccanismo di reazione
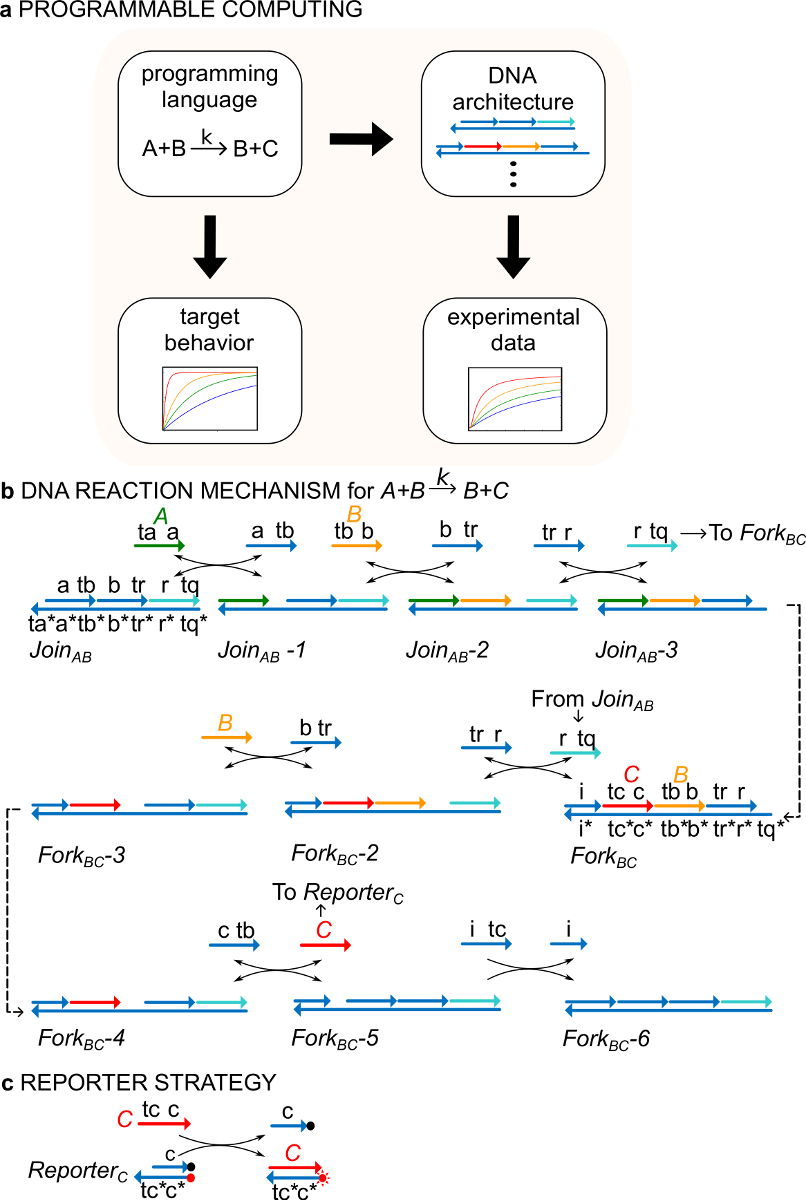
Come esempio, il protocollo si concentra sulla reazione chimica catalitica A + B-> B + C. Le specie A, B, e C ("segnali", Figura 1B) tutti corrispondono ad una diversa molecola di DNA a singolo filamento. Le sequenze di queste molecole sono completamente indipendenti ed i fili non reagiscono uno con l'altro direttamente. Le sequenze di tutti i segnali hanno due differenti domini funzionali, cioè sottosequenze che agiscono insieme a reazioni di spostamento strand: 1) un punto d'appoggio a dominio breve (etichette ta, tb, tc) che viene utilizzato per l'avvio di filo spostamento reaction, e 2) una lunga dominio (etichette a, b, c), che determina l'identità del segnale.
Le interazioni tra fili del segnale sono mediati da intaccate DNA (ndsDNA) complessi a doppia elica del cancello (chiamati Join AB e forcella aC) e ausiliari di specie a singolo filamento (, , e ). La reazione A + B- formale> B + C viene eseguito attraverso una serie di stadi di reazione di spostamento del filo, in cui ogni stadio di reazione espone un punto d'appoggio per una successiva reazione (Figura 1B). In questo esempio, i segnali A e B sono inizialmente libere in soluzione mentre il segnale C è legato al cancello forcella. Al termine della reazione B e C sono in soluzione. Più in generale, i segnali che sono associati a un cancello sono inattive mentre i segnali che sono liberi in soluzione sono attivi, cioè, essi possono partecipare a una reazione di spostamento del filo comeun ingresso. Il decorso della reazione viene seguito mediante una strategia reporter fluorescente (Figura 1C). Nel precedente lavoro 29, è stato dimostrato che questo meccanismo di reazione realizza non solo la stechiometria corretta ma anche la cinetica della reazione di destinazione.
Protocollo
1. Sequenza di design
Nota: Sequenza Panoramica disegno: In questa sezione, la strategia per la progettazione di plasmidi derivati da cancelli di DNA viene descritta. Siti enzimatici posti su entrambe le estremità delle porte per consentire il rilascio di cancelli completamente doppio filamento dopo la digestione. Siti intaccare vengono poi poste in modo tale che gli enzimi creano nick sul filo superiore per creare le porte ndsDNA finali. Infine, le sequenze rimanenti vengono scelti in modo tale che i domini indipendenti sono ortogonali tra loro e non presentano struttura secondaria.
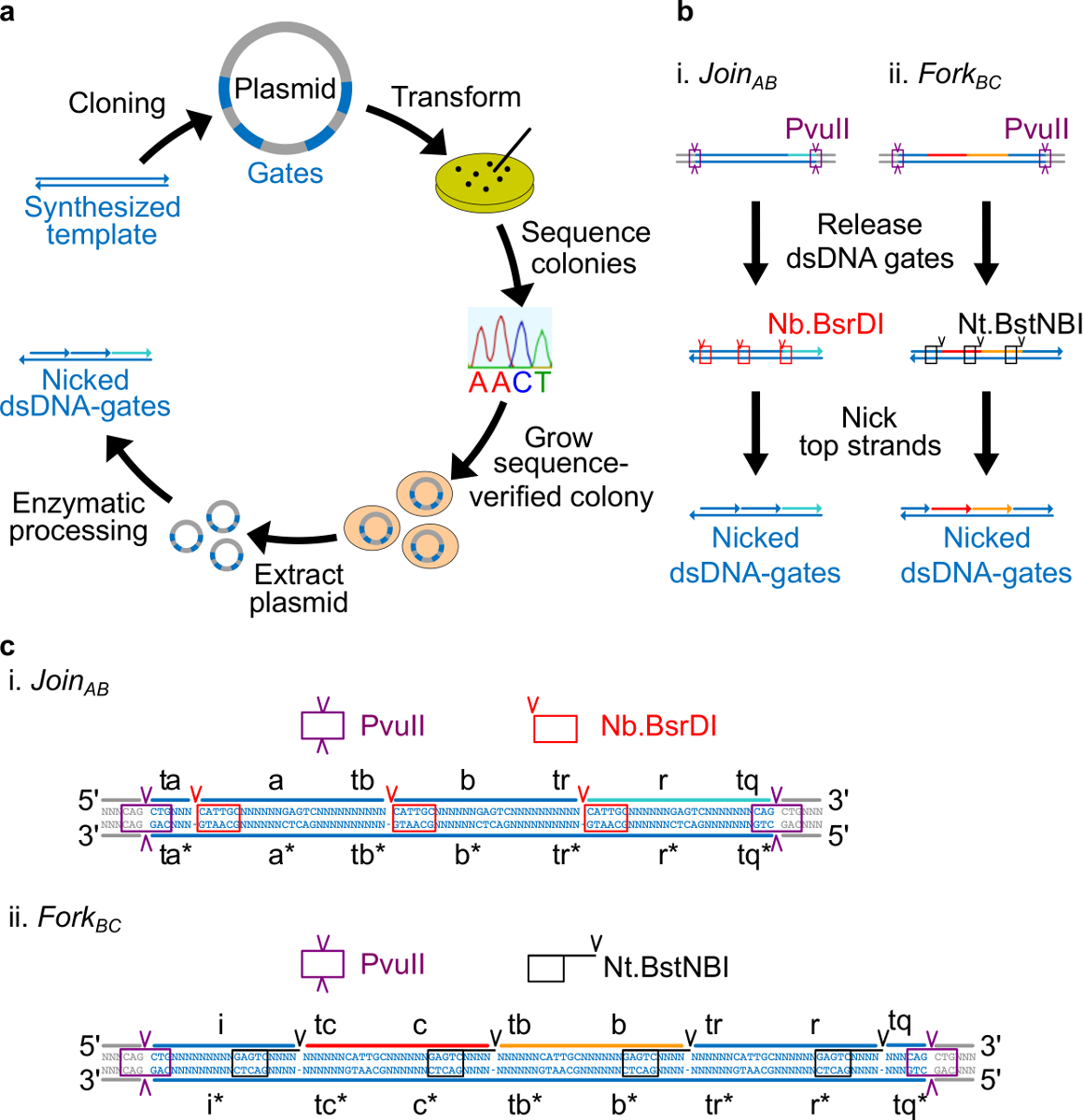
- Posizionare il sito intaccare Nt.BstNBI quattro nucleotidi dal terminale 3 'di ciascun dominio lungo (a, b, c, r, e i). Posizionare il sito intaccare Nb.BsrDI sull'estremità 5 'di ogni dominio lungo (a, b, c, e r. Si noti che il dominio che non ha alcun sito intaccare Nb.BsrDI). Figura 2C mostra la vista dettagliata sequenza di Join AB e forcella BC cancelli.
- Posizionare il sito di restrizione PvuII su entrambe le estremità in modo che cancelli ndsDNA PvuII digestione può rilasciare le porte da plasmidi (vedere la Figura 2C).
- Progettare altre sequenze non vincolati, seguendo due principi: (a) non devono presentare filoni strutture secondarie (strutture di DNA possono essere previsti con Nupack 41), e (b) tutti i domini dovrebbero essere ortogonali per ridurre al minimo le interferenze.
- Posizionare sequenze ndsDNA nel centro di un modello di gate. Posizionare 30-40 sequenze distanziatori bp casuali su entrambe le estremità del modello porta, ciascun distanziatore serve come un sito di legame unico consultato Polymerase Chain Reaction (PCR).
2. Clonazione di NdsDNA Gates in Plasmidi
Nota: Questa sezione descrive il metodo di clonazione Gibson per l'inserimento di 4 copie del cancello in una spina dorsale plasmide.
- Modelli cancello ordine ndsDNA come blocchi genomiche a doppia elica del DNA di un produttore (sequenze modello cancello sono mostratiin Tabella 1; filoni si verificano in porte ndsDNA sono riportati nella tabella 2; sequenze livello di dominio sono mostrati nella Tabella 3).
- Dopo aver ricevuto il DNA ordinata, girare le provette contenenti blocchi genomiche a 10,000-14,000 xg per 1 min per assicurare che tutto il DNA essiccato è al fondo della provetta.
- Risospendere i blocchi genomici secchi in acqua priva di DNasi per ottenere una concentrazione finale di 10 ng / ml.
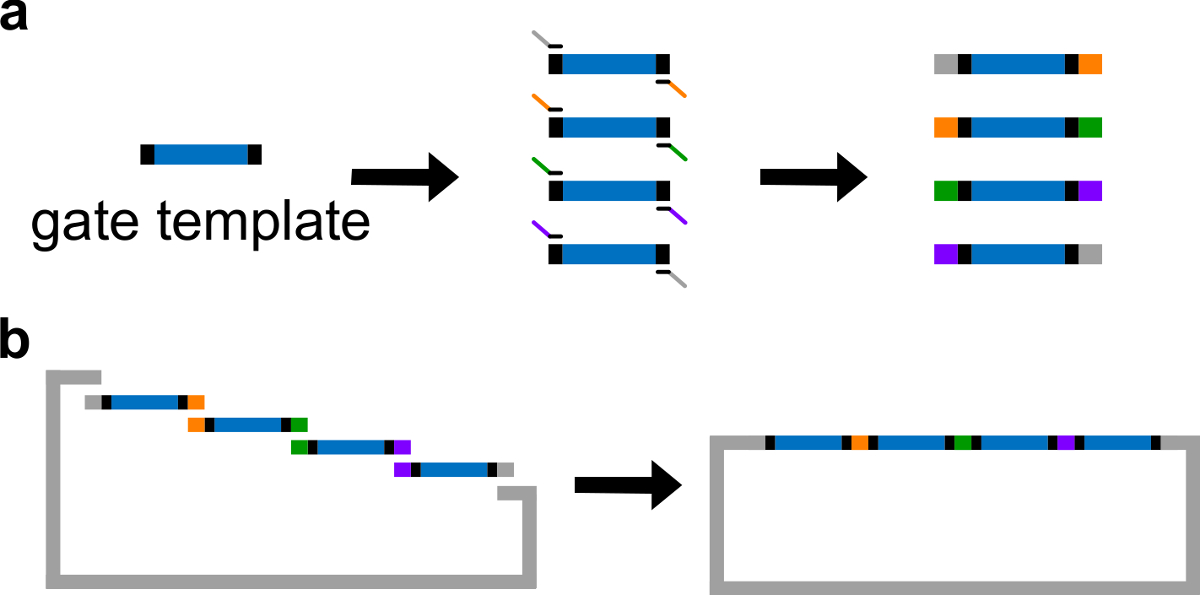
Nota: In alternativa, il DNA può essere risospeso con 1x Tris acido etilendiamminotetraacetico (EDTA) tampone (tampone TE: 10 mM Tris e EDTA 1 mM, pH 8,0). Tuttavia, EDTA è un agente chelante per cationi bivalenti e potrebbe inibire PCR. - Genera 4 frammenti cancello con diverse regioni di sovrapposizione attraverso una PCR standard con un polimerasi ad alta fedeltà del DNA (vedi Figura 3A). Le sequenze dei primer sono dettagliate nella Tabella 4 (temperatura di fusione di questi primer è 62 ° C).
- Eseguire un gel di agarosio 2% a 140 V per 30 minuti a RT (per un protocollo elettroforesi su gel di agarosio dettagliata vedi 42) e tagliare le bande corrispondenti a ciascun frammento amplificato PCR su gel. Poi purificare le fette di gel con un kit di estrazione di gel (vedi Materiali) seguendo le istruzioni del produttore.
- Digerire una dorsale numero plasmide alta copia (vedi materiali) con PvuII-HF e PstI-HF a 37 ° C per 1 ora (vedi Tabella 5) secondo il protocollo del produttore. PvuII-HF e PstI-HF sono enzimi di restrizione ad alta fedeltà, che riducono drasticamente i tagli non specifici.
- Eseguire un gel di agarosio 1,5% e tagliare il backbone linearizzato (tipicamente eseguire il gel a 140 V per 30-40 minuti a temperatura ambiente). Quindi estrarre il DNA dalla fetta gel utilizzando il kit di estrazione gel seguendo le istruzioni del produttore.
- Eseguire Gibson montaggio 43 con il vettore linearizzato e frammenti di PCR purificati (vedi Tabella 6 e la Figura 3B ).
- Trasformare il prodotto assembly Gibson dal punto 2.8 in Escherichia coli (E. coli) e piatto su una piastra di agar lisogenia Broth (LB) contenenti antibiotici ampicillina (ad una concentrazione di 100 ug / ml). Eseguire trasformazione attraverso elettroporazione o un metodo di shock termico 44,45, e utilizzare l'apposito E. coli. Ad esempio, utilizzare E. coli ceppo JM109 per la trasformazione shock termico, e utilizzare DH5α electrocompetent E. coli per elettroporazione.
Nota: La spina dorsale plasmide usato contiene una cassetta di resistenza ampicillina. Se si utilizza un marcatore di selezione diverso, utilizzare gli antibiotici appropriati, invece di ampicillina.
3. batterica Cultura amplificazione e Controllo Qualità
Nota: Questa sezione descrive la produzione di massa e l'isolamento dei plasmidi contenenti le porte del DNA dopo il controllo di qualità.
- Scegli una singola coloniadalla piastra selettiva Ampicillina dal punto 2.9 e incubare una cultura di 3 ml di mezzo arricchito contenente antibiotici ampicillina (ad una concentrazione di 100 ug / ml). Segnare la colonia tale che possa essere utilizzato di nuovo in successive fasi sperimentali. Far crescere la coltura a 37 ° CO / N con vigoroso scuotimento (200-300 giri al minuto). Tipicamente, incubare per 16-24 ore.
- Estrarre il DNA plasmide dalla coltura batterica utilizzando un kit mini-preparazione seguendo le istruzioni del produttore.
- Misurare il DNA plasmidico purificato utilizzando uno spettrofotometro seguendo le istruzioni del produttore. Fasce di resa tipici 50-1,000 ng / ml.
- Prendi il DNA plasmidico estratto sequenziato mediante l'invio del campione ad una società di sequenziamento del DNA. Primer di sequenziamento devono trovarsi circa 100 nucleotidi a monte ea valle della regione da sequenziare; il primer sequenziamento del plasmide (vedi materiali per plasmide) ha la seguente sequenza: ATTACCGCCTTTGAGTGAGC.
Note: se ci è errore di sequenza o la ricombinazione genetica in inserita ndsDNA porte, selezionare una colonia diverso dalla piastra dal punto 2.9. Seguire i passaggi 3,1-3,4 per verificare che le sequenze delle porte inseriti sono corretti. - Dopo aver verificato che le sequenze sono corrette, scegliere il corrispondente colonia dalla piastra selettiva Ampicillina (dal punto 2.9), e incubare una cultura di 800 ml Terrific Broth (TB) contenenti antibiotici ampicillina (ad una concentrazione di 100 ug / ml). Crescere la coltura a 37 ° C per 16-24 ore con agitazione vigorosa (200-300 rpm). TB particolarmente è adatto per la produzione ad alto rendimento plasmide.
Nota: In alternativa, LB potrebbe anche essere utilizzato per far crescere batteri anche se la resa plasmide può essere un problema. - Purificare il DNA utilizzando un kit Maxi-preparazione seguendo le istruzioni del produttore.
- Seguire passo 3,3-3,4 per verificare se le sequenze sono corrette. Se si è verificato alcun ricombinazione, vedere la seguente nota. In caso contrario, passare al punto 4.
Nota: Un possibile problema è che più copie di porte inseriti nella plasmide può ricombinarsi a causa di riparazione del DNA. Per risolvere questo problema, utilizzare un E. coli ceppo senza proteina recA (una proteina correlata alla riparazione del DNA), come JM109 o DH5α trasformare un plasmide precedentemente sequenza verificato (cioè, senza errori di sequenza e ricombinazione). Poi scegliere una colonia da questo piatto e verificare la sequenza plasmide inviando il campione ad una società di sequenziamento del DNA.
4. enzimatica Processing
Nota: Questa sezione descrive il processo per digerire i plasmidi tale che essi sono tagliati e intaccate nelle posizioni corrette e pronti per essere utilizzati per esperimenti cinetici.
- Digerire il plasmide DNA purificato dal punto 3.7 con enzima di restrizione PvuII-HF per 1 ora a 37 ° C (vedi Tabella 7). Tipicamente digerire il plasmide con 4 unità di PvuII-HF per 1 mg di plasmide. Alta fidenzimi di restrizione elity sono raccomandati per l'uso in quanto riducono drasticamente i tagli non specifici.
- Eseguire precipitazione con etanolo sul campione.
- Aggiungere 2 volumi equivalenti di ghiacciata etanolo assoluto al campione.
- Incubare la miscela a -80 ° C per almeno 1 ora (questa miscela può anche sedersi a -80 ° C per O / N).
- Centrifugare a 10,000-14,000 xg a 0 ° C per 30 min.
- Rimuovere il surnatante.
- Aggiungere 1,000 ml di RT 95% di etanolo al campione, e capovolgere 10-15 volte.
- Centrifugare a 10,000-14,000 xga 4 ° C per 10 min.
- Rimuovere il surnatante e aria secca sulla panchina per 10-20 minuti.
- Risospendere il pellet di DNA in un volume adeguato di nucleasi libera H 2 O (in genere 100-200 ml). L'aggiunta di più di 200 ml in genere fare il campione troppo diluito per l'uso in cinetiche esperimenti.
- Misurare il DNA risospeso utilizzando uno spettrofotometro a seguito della mle istruzioni del fabbricante.
- Digest unirsi cancelli intaccare enzima Nb.BsrDI a 65 ° C per 1 ora con 4 unità di enzima per 1 pg di plasmide (vedi tabella 8); digerire forcella cancelli intaccare enzima Nt.BstNBI a 55 ° C per 1 ora usando 8 unità di enzima per 1 pg di plasmide (vedi Tabella 9).
Nota: Passo 4.2 rimuove tampone digestione enzimatica e aiuta a concentrarsi le porte per cinetiche esperimenti. Passo 4.2 può essere saltato per unirsi cancelli perché entrambi enzima di restrizione PvuII-HF e intaccare enzima Nb.BsrDI condividono lo stesso buffer di digestione. Al punto 4.2.8, nucleasi gratuito H 2 O è usato al posto di TE perché EDTA è un agente chelante per cationi bivalenti e può inibire gli enzimi di restrizione che hanno bisogno questi ioni di funzionare.
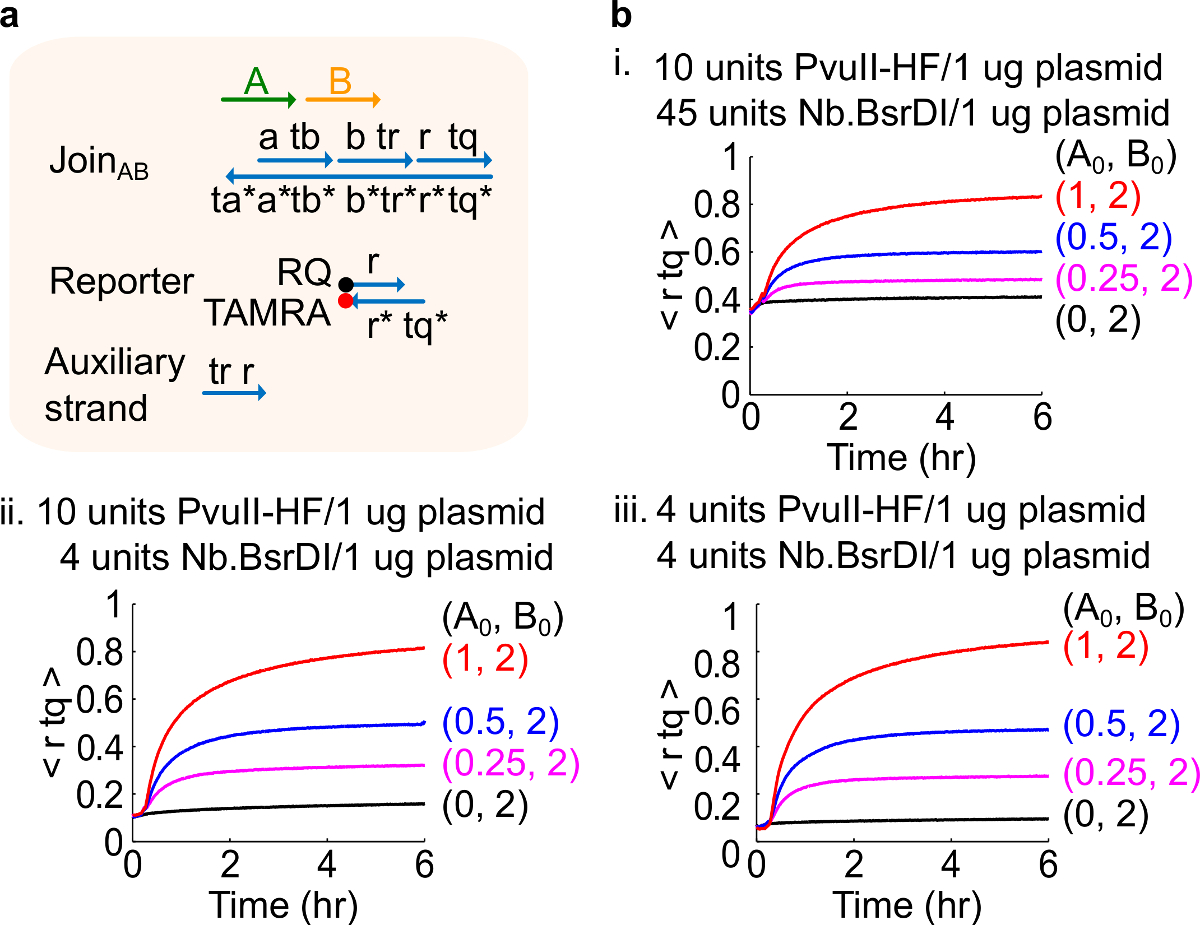
Nota: L'aggiunta di quantità in eccesso di enzimi può portare ad elevate quantità di perdite circuito iniziale (figura 4), che è probabilmente causato da un eccesso di digestione 46. Questo problema can essere indirizzata da ottimizzare i quantitativi enzimatici (vedere Figura 4). Tipico gamma di enzimi è da 1-10 unità / 1 mg plasmidi.
5. Preparazione di oligonucleotidi a singolo filamento
Nota: Questa sezione descrive il protocollo per la risospensione e la quantificazione sintetizzato chimicamente singolo filamento di DNA (ssDNA), che verrà utilizzato per fili di segnale e fili ausiliari. Per le sequenze strand vedi Tabella 10. Si noti che il seguente protocollo è un esempio di preparazione di 10 micron ssDNA. Altre concentrazioni di ssDNA possono essere preparati in modo analogo.
- Dopo aver ricevuto dal produttore oligo DNA, girare le provette contenenti DNA a 10,000-14,000 xg per 1 min per assicurare che tutto il DNA essiccato è al fondo della provetta.
- Risospendere il DNA usando 1x Tris acido etilendiamminotetraacetico (EDTA) tampone (tampone TE: 10 mM Tris e 1mM EDTA, pH 8,0) per ottenere una concentrazione finale di 100 mM. Peresempio, risospendere 8 nmol del DNA in 80 ml di tampone TE.
- Mescolare 10 ml di DNA a 100 mM con 90 ml di acqua molecolare in una provetta da microcentrifuga, che dovrebbe raggiungere una concentrazione finale di 10 pM.
- Misurare la concentrazione esatta del campione di DNA utilizzando uno spettrofotometro seguendo le istruzioni del produttore. Il protocollo seguente fornisce un esempio di come la concentrazione di DNA può essere misurata.
- Vuoto lo spettrofotometro con 2 ml di acqua molecolare.
- Misurare l'assorbanza a 260 nm (A 260) del campione di DNA. Utilizzare la seguente equazione per calcolare la concentrazione magazzino.
Nota: La concentrazione del campione è M = un coefficiente 260 / estinzione. Il coefficiente di estinzione può essere trovato sulla scheda specifiche del costruttore del DNA.
6. Preparazione di Reporters fluorescenti
Nota: Questa sezione descrive laprotocollo per la preparazione di Reporter C, Altri giornalisti fluorescenti possono essere assemblati in modo simile.
- Ordina cromatografia liquida ad alte prestazioni (HPLC) oligonucleotidi purificati ROX- (il filo superiore del Reporter C) e -RQ (il filo inferiore del Reporter C) dal produttore del DNA (vedi Tabella 10 per le sequenze ).
- Dopo aver ricevuto il oligonucleotidi sintetizzati, risospendere e quantitate campioni come spiegato al punto 5.
- Mescolare il top giornalista e fili inferiori (ad esempio, ROX- e -RQ) a 1x Tris-acetato-EDTA (TAE) con 12,5 mg mM 2+ (si veda la tabella 11 per la ricetta dettagliata ). Si noti che qui il 30% in eccesso filo quencher etichettato -RQ viene aggiunto per assemblare il giornalista, che assicura che tutti i fili fluoroforo marcati sono state spente anche con stechiometria imperfetta.
- Ricuocere complesso Reporter C utilizzando un ciclatore termico, raffreddamento da 95 ° C a 20 ° C ad una velocità di 1 ° C / min. I campioni possono essere conservati a 4 ° C.
7. misure di fluorescenza
Nota: La sezione descrive un protocollo generale per cinetiche fluorescenza misurazioni (vedere la Figura 5 per la procedura sperimentale), e questo protocollo verrà utilizzato nei passaggi 8, 9 e 10. Si noti inoltre che questo protocollo è per l'uso di uno spettrofluorimetro. In alternativa, questi esperimenti potranno essere effettuati anche in un lettore di piastre, anche se la sensibilità, ben-to-bene le variazioni e la mancanza di controllo della temperatura in esperimenti a lungo termine possono essere un problema.
- Impostare il regolatore di temperatura a 25 ° C, e attendere che la temperatura si stabilizzi. Utilizzando un regolatore di temperatura può ridurre la variabilità del segnale che può derivare da variazioni di temperatura.
- Impostare i parametri corretti focinetiche r misure sul software di acquisizione dati del spettrofluorimetro. Impostazioni di esempio dettagliate sono le seguenti:
- Impostare la larghezza della fessura di 2.73 nm sia per monocromatori eccitazione e di emissione.
- Impostare il tempo di integrazione per 10 sec per ogni 60 sec time-point. Impostare il tempo di misurazione totale di 24 ore.
- Impostare le lunghezze d'onda di eccitazione / emissione per abbinare i fluorofori utilizzati nell'esperimento. Esempio lunghezze d'onda sono i seguenti: ROX (588 nm / 608 nm), e TAMRA (559 nm / 583 nm).
- Aggiungere nucleasi gratuito H 2 O e 10x tampone Tris-acetato-EDTA contenente 125 mg mM 2+ (10x TAE / Mg 2+) a una cella di quarzo sintetico. Vedere Tabelle 12, 13 e 14 ad esempio volumi da utilizzare.
- Aggiungi fili polyT per ottenere una concentrazione finale di ~ 1 micron (vedi tabella 12, 13, e 14 per i volumi), e poi il vortice sinteticocelle al quarzo per 10-15 secondi. In generale, i puntali delle pipette saranno non specificamente legare il DNA. Aggiunta di alte concentrazioni di trefoli polyT può ridurre l'errore di legame non specifico.
- Aggiungere giornalisti e fili ausiliari. Ad esempio i volumi da utilizzare Vedere Tabella 12, 13, e 14. Si noti che per la calibrazione giornalista, non sono necessari fili ausiliari.
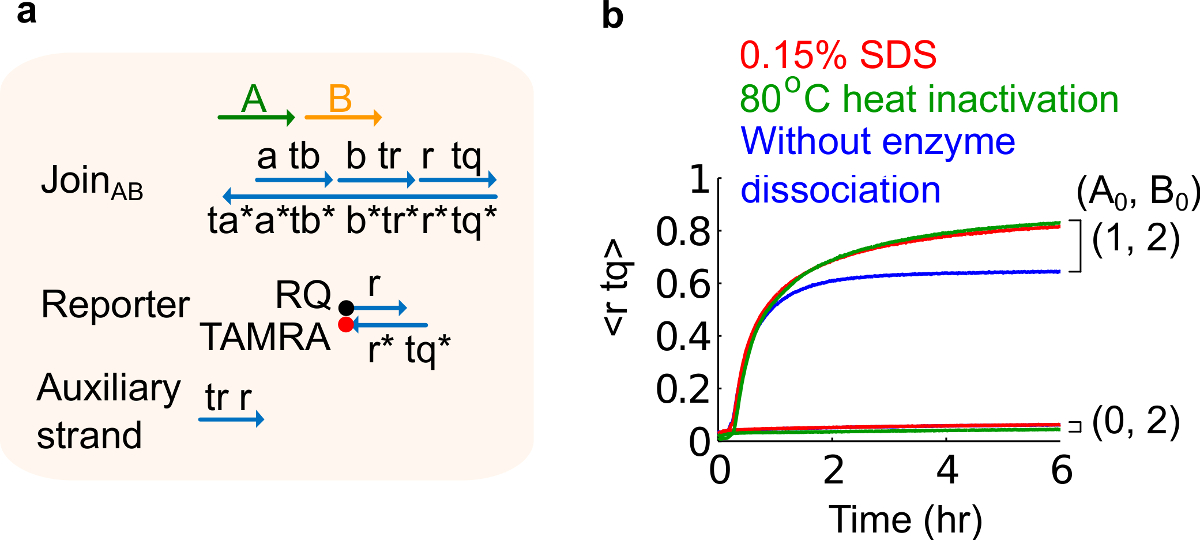
- Aggiungere 10% di solfato di sodio dodecil (SDS) per ottenere una concentrazione finale di 0,15% SDS. Nota: SDS è utilizzato per dissociare enzimi dalle porte plasmide derivato perché enzimi possono interferire con la reazione di spostamento del filo (vedi figura 6). SDS è consigliato qui invece di denaturazione termica di enzimi per evitare la dissociazione e ricombinazione non corretta dei fili cancello, che possono influenzare negativamente la funzione del circuito.
- [Ignorare questo passaggio per la calibrazione giornalista.]
- Aggiungere unirsi e cancelli forcella (vedere Tabella 13, e 14 per i volumi)alla cella di quarzo sintetico e miscelare la soluzione pipettando su e giù per almeno 20 volte (non agitare la cuvetta perché soluzioni Vortexare con SDS possono causare bolle che influenzeranno cinetiche fluorescenza misurazioni).
- Inoltre, spostare la seguente procedura di misurazione non appena possibile perché la reazione perdita avvia immediatamente dopo l'aggiunta di unirsi e forcella porte alla cella di quarzo sintetico.
- Mettere le celle al quarzo sintetico nella camera di un spettrofluorimetro.
- Avviare la misurazione cinetica.
- Dopo 5 min di misura, aggiungere fili di ingresso (vedere Tabella 12, 13, e 14 per i volumi) alla cella di quarzo sintetico e mescolare la reazione pipettando su e giù per almeno 20 volte. Si noti che il campione deve essere miscelato con delicatezza per evitare le bolle. Eseguire questo passaggio, mentre il programma di acquisizione dei dati è in pausa per evitare segnali di misura innescato da esternoleggero.
- Registra la cinetica di reazione fino a raggiungere lo stato stazionario. La cinetica di reazione sono visualizzati sul computer.
8. Calibrazione fluorescenti Reporter
Nota: Questa sezione descrive il protocollo per fare curve di calibrazione di giornalisti fluorescenti. Le curve di calibrazione saranno utilizzati per convertire le unità di fluorescenza arbitrarie alla concentrazione molare del segnale.
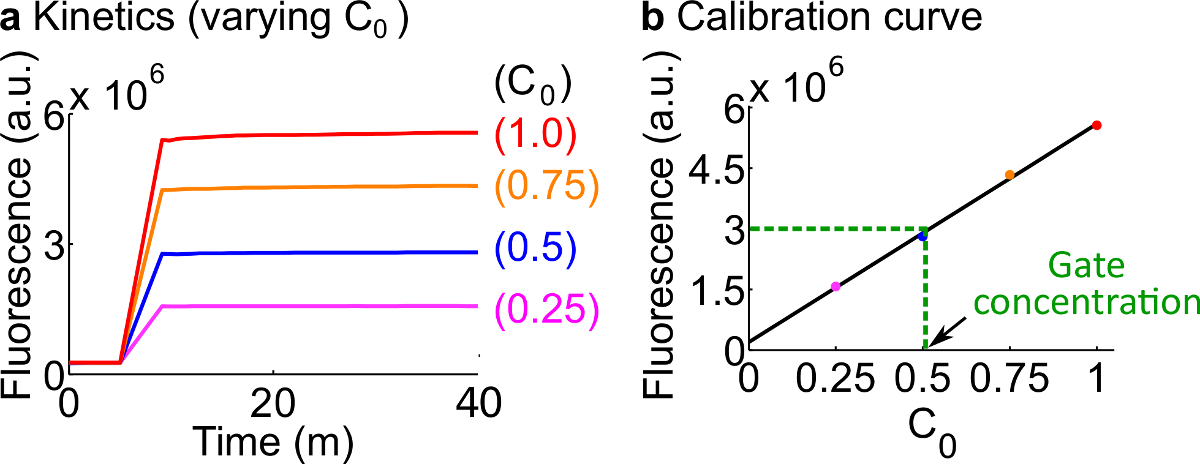
- Calibrare i giornalisti fluorescenti seguendo il protocollo descritto al punto 7. Utilizzare i volumi di reagenti e tamponi come riassunto nella tabella 12 La concentrazione standard per questo esempio è 50 Nm (1x).; i giornalisti sono a 3x; ingresso è 1x. Per i casi in cui l'input sono a 0,25x, 0,5x, 0,75x, regolare il volume di nucleasi libera H 2 O corrispondentemente di tenere il volume finale di ogni reazione ad essere 600 microlitri. Un dato di esempio sono mostrati in Figura 7A.
- Effettuare una curva di calibrazione delReporter C mediante un accoppiamento lineare dei valori finali fluorescenza contro la concentrazione iniziale del segnale C (Una curva di calibrazione esempio è mostrato nella Figura 7B). Questa curva di calibrazione può essere utilizzata per convertire le unità arbitrarie fluorescenza alla concentrazione corrispondente segnale.
9. Quantificare la concentrazione di plasmidi derivati da ndsDNA Gates
Nota: ogni partita trasformata indipendentemente Portone ndsDNA plasmidi derivati da risultati in una resa diversa di porte funzionali, e questa sezione descrive un protocollo per quantificare la concentrazione di porte ndsDNA plasmide-derivati.
- Quantificare la concentrazione di plasmide di derivazione cancelli ndsDNA seguito il protocollo descritto al punto 7. Utilizzare i volumi di reagenti come riassunto nella tabella 13. Nota: La tabella 13 descrive un esempio ricetta per Forcella aC quantificazione Registrati. AB e altre porte possono essere eseguite in modo simile, ma utilizzando diversi filoni di ingresso, fili ausiliari e giornalisti.
- Convertire il valore di fluorescenza finale misurato in questo esperimento per una concentrazione di segnale C utilizzando la curva di calibrazione dal punto 8.2. Poi back-calcolare la concentrazione porta ndsDNA. Ad esempio, un valore di fluorescenza finale per l'esperimento di quantificazione porta corrisponde a 25 nM segnale C (0,5x) sulla base della curva di calibrazione in Figura 7B. Dal momento che lo stock di Forcella aC è diluito 40 volte in questa reazione, la concentrazione della della porta Fork BC è 1 micron.
10. Cinetica Misure per la reazione A + B-> B + C
Nota: Questa sezione descrive un protocollo per testare la realizzazione DNA di una reazione chimica convenzionale usando cinetiche fluorescenza misurazioni.
- Performa cinetica misura seguendo il protocollo descritto al punto 7. Utilizzare volumi di reagenti e tamponi come riassunto nella tabella 14.
Risultati
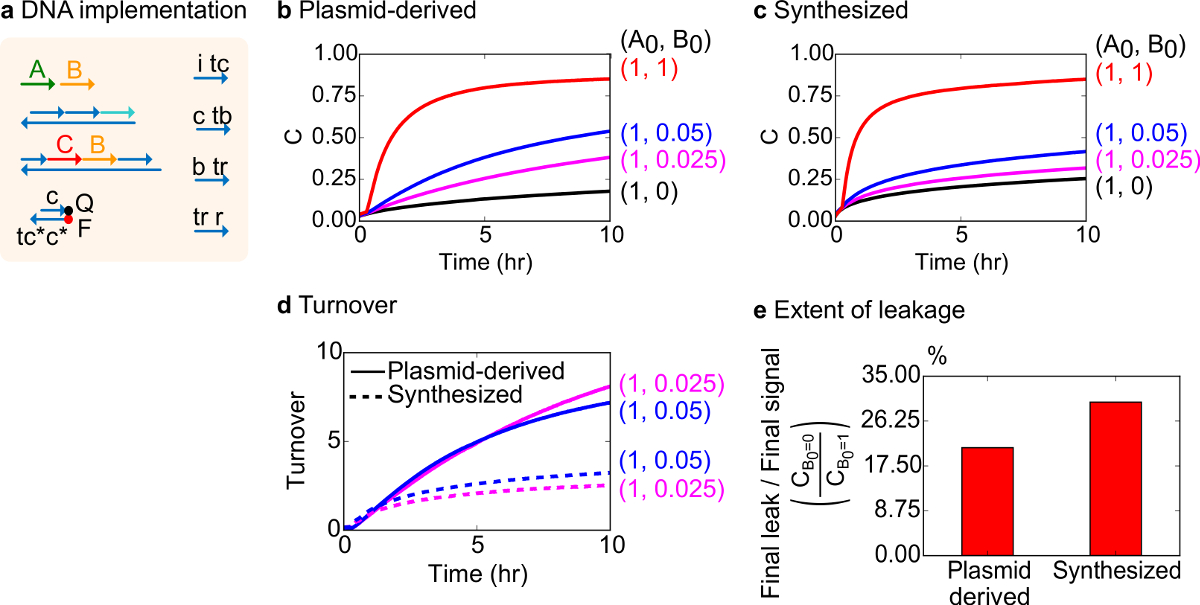
Per un test funzionale, un'implementazione DNA della reazione catalitica bimolecolare (cioè, A + B-> B + C) è stato creato. Le prestazioni di cancelli plasmide derivato è stato confrontato con porte assemblati da DNA sintetico. Reazioni catalitiche sono un buon test per cancello purezza perché un cancello difettoso può irreversibilmente intrappolare un catalizzatore, provocando un effetto sproporzionato sulla quantità di prodotto fabbricato 18,19. Allo stesso tempo, una piccola reazione perdita conseguente rilascio non sollevato del segnale catalitico sarà linearmente amplificato, portando ad un segnale di errore sproporzionata. Dati sperimentali per cancelli plasmide derivato e sintetizzati sono mostrati in Figura 8B e 8C, rispettivamente. Negli esperimenti, la concentrazione del segnale di filo A è fissata mentre la quantità di segnale catalitico B è variata. Segnale C viene utilizzato per leggere l'andamento della reazione senza interrompere cataliticociclo. Catalysis si osserva nei dati dal completamento reazioni avvicinano anche con quantità di catalizzatore B molto più piccola della quantità di A. Poiché SDS non è stato aggiunto a esperimenti fatti con il sistema di sintesi, la velocità di reazione (che potrebbe essere influenzata da l'aggiunta di SDS) non viene confrontata e l'attenzione analitica è invece sul fatturato catalitica (così dettagliati).
Ulteriori analisi del fatturato catalitica di questa reazione è stata condotta. Fatturato è definita come la quantità di segnale C prodotti per ciascun catalizzatore B in un determinato momento. In particolare, il fatturato è stato calcolato dai nostri dati sperimentali dividendo la perdita del segnale C sottratto dal valore iniziale di catalizzatore B aggiunto. Per un sistema catalitico ideale, questo numero fatturato deve aumentare linearmente con il tempo e di essere indipendente dalla quantità di catalizzatore finché il substrato non è limitante. In un sistema reale, cancelli difettosi possono disabilitare gatto alysts, e il fatturato raggiungerà un valore massimo, anche se non tutti disponibili substrato viene convertito al prodotto. Il valore massimo fatturato indica quanti substrati (segnale A) un catalizzatore (segnale B) in grado di convertire prima di diventare inattivato. Qui, si osserva che il sistema sintetizzato discosta dalla incremento lineare ideale di turnover molto prima che il sistema plasmide derivato fa, indicando il sequestro del catalizzatore attraverso una reazione collaterale indesiderato (Figura 8D). Il confronto fatturato viene visualizzato solo per le basse concentrazioni, perché ad alte concentrazioni di catalizzatori, tutti i cancelli saranno attivati e rilasciano il segnale C. La perdita circuito è inoltre confrontato, e si osserva che il rapporto del segnale di perdita impiegano porte plasmide derivato è di circa 8% inferiore a quella che impiegano porte sintetizzati dopo 10 h di reazione (figura 8E).
iles / ftp_upload / 53087 / 53087fig1.jpg "/>
Figura 1. (A) CRNS servire come un linguaggio di programmazione prescrittiva. Reti di reazione del DNA possono essere progettati per approssimare la dinamica di un CRN formale di attuazione (B) del DNA di una istruzione chimica esempio. A + B-> B + C. Filamenti di DNA sono disegnati come linee con frecce a 'estremità 3 e * indica complementarità. Tutti i segnali delle sezioni A (, verde), B (, arancio) e C (, rosso) sono costituiti da un dominio appiglio (etichettato come ta, tb, e tc) e un dominio di identità (etichettato come a, b, e c). La reazione bimolecolare A + B-> B + C richiede due complessi multi-stranded Unisciti AB e forcella aC, e quattro fili ausiliari , , , e . La reazione procede attraverso sette fasi di spostamento Strand, dove ogni inizio passos con appiglio vincolante. (C) strategia Reporter. La reazione viene seguita mediante un reporter in cui il filo di fondo è etichettato con un fluoroforo (punto rosso) e il filamento superiore è collegata a un quencher (punto nero). A causa della co-localizzazione del fluoroforo e il quencher, giornalista fluorescenza viene raffreddato in reporter intatta. Il segnale C può sostituire il filo superiore del reporter, portando ad un aumento della fluorescenza. (Questa cifra è stata modificata da Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2. cancelli (A) NdsDNA realizzati batterica DNA plasmidico. Diverse copie del filamento modello doppia porta ndsDNA vengono clonati in un plasmide. I plasmidi clonati sono iln trasformato in E. coli e le colonie sulla piastra sono sequenza di verifica. Una volta che la sequenza viene confermata, il plasmide DNA viene amplificato ed estratta. Infine, il doppio filamento plasmide è trasformato nelle porte ndsDNA desiderati attraverso lavorazione enzimatica. (B) il trattamento enzimatico dei cancelli ndsDNA. L'enzima di restrizione PvuII viene utilizzata per sbloccare il cancello dal plasmide. I cancelli rilasciati sono ulteriormente trattati con enzimi intaccare: Nb.BsrDI viene utilizzato per generare nick per Join AB (Panel i); Nt.BstNBI è usato per generare nicks per Fork BC (Pannello ii). Siti di restrizione e intaccare vengono indicate come caselle con codice colore. (C) vista Sequenza del modello porta di Join AB (Panel i) e Forcella BC (Pannello ii). Il sito di restrizione PvuII (evidenziato nella casella viola) è su entrambe le estremità dei cancelli ndsDNA. Il Nb.BsrDI e NtSiti intaccare .BstNBI sono evidenziati in scatole rosse e nere, rispettivamente. Le posizioni di taglio sono contrassegnati con frecce. Sequenza N è un qualsiasi nucleotide. (Questa cifra è stata modificata con il permesso di Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 3. (A) PCR di un modello di gate DNA. Un modello di cancello di DNA contiene le sequenze ndsDNA cancello del centro (una regione blu), e sequenze distanziali su entrambe le estremità (regioni nere, queste due sequenze finali sono ortogonali). Primer possono legarsi alle sequenze distanziatori del modello porta, e generare quattro frammenti di DNA sovrapposti attraverso PCR (sequenze sovrapposti sono a colori e in figura). Di montaggio (B) Gibson. I quattro frammentazione del DNA amplificatits vengono poi assemblati in una dorsale plasmide linearizzato attraverso Gibson metodo di assemblaggio 43. (Questa cifra è stata modificata con il permesso di Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 4. Circuito prestazioni con diverse quantità di enzimi. (A) Una rappresentazione semplificata del cancello, reporter, fili ausiliari, e segnali trefoli utilizzato per gli esperimenti corrispondenti. (B) Cinetica esperimenti con plasmide derivato Unisciti AB trattati con diverse quantità di enzima . io. 10 unità di PvuII-HF e 45 unità di Nb.BsrDI per 1 mg di plasmide; ii. 10 unità di PvuII-HF e 4 unità di Nb.BsrDI per 1 mg di plasmide; iii. 4 unità di PvuII-HF e 4 unità di Nb.BsrDI per 1 mg di plasmide. Tutti i fili ausiliari erano a 2x (1x = 10nm). Il complesso di gate è 1,5x, e gli esperimenti sono stati eseguiti a 35 ° C in TAE 1x / Mg 2+. (Questa cifra è stata modificata con il permesso di Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

. Figura 5 Schema di flusso cinetici esperimenti Blu:. Materiali da aggiungere alla cuvetta (cella di quarzo 0,875 ml sintetico). Tabella di riferimento 14 per volumi specifici da aggiungere per l'esperimento cinetica di A + B -> B + C. Verde: le istruzioni di un spettrofluorimetro (etichettato come SPEX). Rosso: miscelazione istruzioni.53087fig5large.jpg "target =" _ blank "> Clicca qui per vedere una versione più grande di questa figura.

Figura 6. Enzima di dissociazione e il circuito di comportamento. (A) Una rappresentazione semplificata del cancello, giornalista, fili ausiliari, e di segnale fili utilizzati per gli esperimenti corrispondenti. (B) Cinetica esperimenti del 80 ° C di calore plasmide-derivato Join AB utilizzando inattivazione (tracce verdi), 0,15% di solfato di sodio dodecil (SDS) (rossi), e un controllo senza inattivazione al calore o l'aggiunta di SDS (blu). La concentrazione standard era 1x = 10 nm, e tutti i fili ausiliari e l'ingresso B erano a 2x. Il complesso di gate è 1,5x, e gli esperimenti sono stati eseguiti a 35 ° C in tampone Tris-acetato 1x-EDTA contenente 12,5 mM Mg 2+ (1x TAE / Mg 2+ ). (Questa cifra è stata modificata da Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 7. calibrazione Reporter. (A) Reporter C cinetica. La concentrazione giornalista era a 3x (1x = 50 nM), e la concentrazione iniziale di segnale C è indicato in figura. (B) I livelli di fluorescenza di segnale C al punto finale di misurazione (40 min) mostra una relazione lineare con la concentrazione iniziale di segnali C. In un esempio di quantificazione Fork BC gate (verde linea tratteggiata), il valore di fluorescenza di Fork BC era misurad come 3 x 10 6 (au), che corrisponde a 25 Nm (0.5x) in base alla curva di calibrazione. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 8. Bimolecolari cinetica di reazione catalitica (A + B-> B + C). (A) Una rappresentazione semplificata del cancello, reporter, fili ausiliari, e fili di segnale utilizzato per gli esperimenti corrispondenti. Gli esperimenti sono stati eseguiti in 1x tampone Tris-acetato-EDTA contenente 12,5 mg mM 2+ (1x TAE / Mg 2+). Tutti i complessi del cancello erano a concentrazione 75 Nm (1,5x), e fili ausiliari erano in concentrazione 100 nM (2x). Cinetica dati per cancelli e dati plasmide derivato per cancelli sintetizzati sono mostrati in (B) e (C) , rispettivamente. Segnale era a 50 nm (1x). Diverse quantità di segnale (catalizzatore) sono stati introdotti nel sistema, e la reazione è stato testato a 35 ° C. Porte (D) Plasmid-derivati esposti fatturato superiore sintetizzati porte DNA quando venivano aggiunte piccole quantità di input. (E) Portata perdite. Il grafico a barre mostra la proporzione della finale di tenuta il segnale finale (C B0 = 0 / C B0 = 1) alle estremità (10 hr). (Questa cifra è stata modificata da Rif. 29) Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
| Modelli Cancello | Sequenze | Lunghezza (nt) |
| JoinAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
"always"> Tabella 1. Sequenze di modelli cancello ndsDNA.
| cancello | Filo | Lunghezza del fondo filamento (nt) |
| JoinAB | JoinAB-Bottom, , , | 87 |
| ForkBC | ForkBC-basso, , , , | |
| 108 |
Tabella 2. Fili comprendenti registrazione AB e Forcella aC. (La tabella è stata modificata da Rif. 29)
| Dominio | Sequenza | Lunghezza (nt) |
| ta | CTGCTA | 6 |
| tb | TTCCAC | 6 |
| tc | TACCCA | 6 |
| tr | TCCTAC | 6 |
| tq | AACCAG | 6 |
| un | CATTGCTTCTACGAGTCATCC | 21 |
| b | CATTGCACCTTAGAGTCCGAA | 21 |
| c | CATTGCCACATCGAGTCCCTT | 21 |
| r | CATTGCTTAACCGAGTCTCAC | 21 |
| io | CTGCCATCATAAGAGTCACCA | 21 |
| Strand Primer | Sequenze | Lunghezza (nt) |
| Forward Primer-1 | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| Reverse Primer-1 | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward Primer-2 | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse Primer-2 | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward Primer-3 | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse Primer-3 | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward Primer-4 | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse Primer-4 | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
Tabella 4. Sequenze primer per la PCR di ndsDNA modelli cancello.
| Reagente | Volume per reazione 1x (ml) |
| -Alta copia plasmide spina dorsale (~ 300 ng / ml) | 10 |
| PvuII-HF (20.000 unità / ml) | 2 |
| Pstl-HF (20.000 unità / ml) | 2 |
| Tampone intelligente 10x Cut | 2 |
| H 2 O | 4 |
| Volume totale | 20 d> |
Tabella 5. Protocollo per la spina dorsale plasmide digest.
| Reagente | Volume per reazione 1x (ml) |
| Vettore DNA (~ 50 ng / ml) | 1 |
| PCR amplificato frammento 1 (~ 50 ng / ml) | 1 |
| PCR amplificato frammento 2 (~ 50 ng / ml) | 1 |
| PCR amplificato frammento-3 (~ 50 ng / ml) | 1 |
| PCR amplificato frammento-4 (~ 50 ng / ml) | 1 |
| 2x Gibson Assemblea Master Mix | 5 |
| Volume totale | 10 |
| Reagente | Volume per reazione 1x (ml) |
| Plasmidi DNA (~ 1 mg / ml di concentrazione) | 1.000 |
| PvuII-HF (20.000 unità / ml) | 200 |
| Tampone intelligente 10x Cut | 133,3 |
| Volume totale | 1.333,3 |
Tabella 7. Protocollo per cancelli ndsDNA inseriti plasmide digerire con restrizione enzimatica PvuII-HF.
| Reagente | Volume (ml) |
| Join cancelli (~ 5 mg / ml di concentrazione) | 150 |
| Nb.BsrDI (10.000 unità / ml) | 300 |
| Tampone intelligente 10x Cut | 50 |
| Volume totale | 500 |
Tabella 8. Protocollo per unirsi cancelli digeriscono intaccare enzima Nb.BsrDI.
| Reagente | Volume (ml) |
| Forcella cancelli (~ 5 mg concentrazione / ml) | 150 |
| Nt.BstNBI (10.000 unità / ml) | 600 |
| Buffer 10x NEB 3.1 | 83.3 |
| Volume totale | 833,3 |
Tabella 9 Protocollo per la forcella cancelli digerire intaccare enzima Nt.BstNBI.
. Tabella 10 sequenze Strand per l'attuazione della reazione chimica A + B -.> B + C (. La tabella è stata modificata da Rif 29)
| Reagente | Volume (ml) | Concentrazione finale |
| ROX- a 100 micron | 10 | 10 micron (1x) |
| -RQ a 100 μ; M | 13 | 13 micron (1.3x) |
| 10x TAE con 125 Mg mM 2 + | 10 | 1x TAE con 12,5 mg mM 2 + |
| H 2 O | 67 | - |
| Volume totale | 100 | 10 micron (1x) |
Tabella 11. Protocollo per l'assemblaggio di Reporter C.
| Reagente | Volume (ml) | Concentrazione finale |
| H 2 O | 514 | - |
| 10x TAE con 125 Mg mM 2 + | 60 | 1x TAE con 12,5 mg mM 2 + |
| PolyT a 300 μ; M | 2 | 1 pM |
| Reporter C a 10 mM | 9 | 150 Nm (3x) |
| 10% SDS | 9 | 0,15% |
| a 5 micron | 6 | 50 Nm (1x) |
| Volume totale | 600 | - |
Tabella 12. Protocollo per la calibrazione di Reporter C. I volumi forniti qui sono per un volume totale di 600 microlitri di reazione (corrispondente all'utilizzo di una cella di quarzo sintetico ml 0,875), ma può essere regolato per funzionare con celle di dimensioni diverse.
| Reagente | Volume (ml) | Con finalecentrazione |
| H 2 O | 493 | - |
| 10x TAE con 125 Mg mM 2 + | 60 | 1x TAE con 12,5 mg mM 2 + |
| polyT a 300 micron | 2 | 1 pM |
| Reporter C a 10 mM | 9 | 150 Nm (3x) |
| a 100 micron | 3 | 10x |
| a 100 micron | 3 | 10x |
| a 100 micron | 3 | 10x |
| 10% SDS | 9 | 0,15% |
| Forcella aC ~ 1 micron (concentrazione sconosciuto) | 15 | ~ 0.5x |
| a 100 micron | 3 | 10x |
| Volume totale | 600 | - |
Tabella 13. Protocollo per la calibrazione di Fork BC. I volumi forniti qui sono per un volume totale di 600 microlitri di reazione, ma può essere regolato per funzionare con celle di dimensioni diverse.
| Reagente | Volume (ml) | Concentrazione finale | |
| H 2 O | 407,2 | - | |
| 10x TAE con 125 Mg mM 2 + | 52.8 | 12.5 Mg mM 2 + | |
| polyT a 300 micron | 2 | 1 pM | |
| Reporter C a 10 mM | 9 | 150 Nm (3x) | |
| a 10 micron | 6 | 100 nM (2x) | |
| a 10 micron | 6 | 100 nM (2x) | |
| a 10 micron | 6 | 100 nM (2x) | |
| 6 | 100 nM (2x) | ||
| 10% SDS | 9 | 0,15% | |
| Unitevi a 1 micron AB | 45 | 75 Nm (1.5x) | |
| Forcella aC a 1 micron | 45 | 75 Nm (1.5x) | |
| a 10 micron | 3 | 50 Nm (1x) | |
| a 10 micron | 3 | 50 Nm (1x) | |
| Volume totale | 600 | - | |
Tabella 14. Protocollo per una reazione chimica A + B-> B + C. I volumi forniti qui sono per un volume totale di 600 microlitri di reazione, ma può essere regolato per funzionare con celle di dimensioni diverse.
| Cancelli sintetizzati | Cancelli plasmidi di derivazione | ||||
| Descrizione | Costo | Join cancelli | Cancelli Fork | ||
| PAGE filamento lungo purificato (100 nt, servito come i fili inferiori di un cancello) | ~ 75 $ | Descrizione </ strong> | Costo | Descrizione | Costo |
| PAGINA filo breve purificato (~ 30 nt, servito come top fili di una porta) | ~ $ 185 | Modello di cancello | ~ $ 100 | Modello di cancello | ~ $ 100 |
| Totale | ~ $ 260 | Kit di estrazione plasmide | ~ 26 $ | Kit di estrazione plasmide | ~ 26 $ |
| Enzima di restrizione (PvuII-HF) | ~ 11 $ | Enzima di restrizione (PvuII-HF) | ~ 11 $ | ||
| Intaccare enzima (Nt.BsrDI, Join cancelli) | ~ 29 $ | Intaccare enzima (Nt.BstNBI, cancelli Fork) | ~ 62 $ | ||
| Totale | ~ $ 166 | Totale | ~ $ 199 |
fo:.. keep-con-previous.within-page = "always"> Tabella 15 Confronto dei costi tra i gate plasmidi di derivazione e cancelli sintetizzati (. La tabella è stata modificata da Rif 29)
| Cancelli sintetizzati | Cancelli plasmidi di derivazione | ||
| Elaborazione | Tempo di elaborazione | Elaborazione | Tempo di elaborazione |
| Ricottura | 1 ora | Clonazione | 5 ore |
| Purificazione PAGINA | 2 ore | Estrazione plasmide | 2 ore |
| Totale | 3 ore | Due passi di digestione enzimatica | 0,5 ore |
| Etanolo precipitazioni | 1 ora | ||
| Totale | 8.5 hr | ||
Confronto Tabella 16. Tempo di elaborazione tra i gate plasmidi di derivazione e cancelli sintetici. (La tabella è stata modificata da Rif. 29)
Discussione
Questo documento descrive un metodo per derivare cancelli ndsDNA da DNA plasmidico altamente puro. Inoltre, un protocollo è presentato per caratterizzare prestazioni cancello usando un test cinetica di fluorescenza. Dati sperimentali mostrano che il sistema plasmide derivato supera la sua controparte sintetica anche se il sistema di sintesi viene assemblato con trefoli purificato usando SDS-PAGE (PAGINA). Probabilmente, il miglioramento delle prestazioni di cancelli plasmide derivato è dovuta principalmente alla elevatissima purezza del DNA biologica. DNA sintetico contiene una varietà di errori, in particolare delezioni che provocano oligonucleotidi di lunghezza tali prodotti collaterali n-1, e sono tipicamente non completamente rimosso in PAGE o cromatografia liquida ad alta prestazione (HPLC) procedure di purificazione. Sono stati inoltre osservati miglioramenti simili a quelli riportati qui in un precedente studio di un amplificatore tornante catalizzata che usati DNA di origine biologica 21.
Tuttavia, anche l'utilizzo di porte plasmide derivato non può eliminare completamente gli errori nell'esecuzione porta, per cui ci sono almeno due motivi: il primo over-digestione o la mancanza di precisione di taglio può portare a cancelli con troppe scheggiature o nick nelle posizioni sbagliate. In entrambi i casi, i gate sono più probabilità di partecipare a reazioni indesiderate. Tali problemi possono essere alleviati ottimizzando la quantità di enzima utilizzato (vedere Figura 4). In secondo luogo, in questi esperimenti, la maggior parte degli ingressi e trefoli ausiliari erano DNA sintetico e quindi contenevano delezioni e mutazioni. In linea di principio, tutti gli input a singolo filamento e fili supplementari potrebbero essere ottenuti anche dal DNA fagemide attraverso un enzima digestione intaccare del genoma virale M13 pre-codificato 26. Forse le prestazioni del circuito può essere ulteriormente migliorata utilizzando ssDNA derivata dal genoma batterico.
Mentre è stato trovato l'uso di porte plasmidi derivati per migliorare le prestazioni del circuito, un'analisidei costi e tempi di lavorazione rivelato che mentre la produzione di porte plasmide derivato è leggermente più economico (Tabella 15), richiede 2-3 volte più lungo tempo di lavorazione rispetto al montaggio e purificazione di porte da oligonucleotidi sintetizzati in commercio (Tabella 16). I costi primari di porte plasmide derivato sono sintesi genica e l'impiego di enzimi di restrizione. Per 300 pmoli di cancelli (sufficienti per 15 reazioni 30 nm), il costo stimato per Iscriviti al cancello è di circa $ 170 e $ 200 per Forcella cancelli, la differenza di costo è dovuta all'utilizzo di diversi enzimi intaccare. Al contrario, la sintesi chimica dei trefoli per gli stessi costi di gate intorno $ 260 compresa una commissione purificazione PAGE. Il costo orario principale per cancelli plasmide derivato è nella procedura di clonazione, che, come la sintesi del DNA, può essere affidata a una società di sintesi gene. Tuttavia, una volta assemblati, cancelli plasmide derivato hanno il vantaggio che i plasmidi ospitanti possono facilmente essere replicate unnd possono essere memorizzati in forma di scorte glicerolo batteriche. Ciò consente di riutilizzare le porte più volte.
Guardando al futuro, il miglioramento delle prestazioni dei cancelli plasmide-derivato potrebbe consentire una gamma molto più ampia di dinamica dei comportamenti che sono stati sperimentalmente dimostrato finora con CRNS DNA. Ad esempio, di recente 47,48 lavoro teorico ha suggerito che i modelli spaziali autorganizzati alla macro-scala possono essere realizzate con DNA CRNS attraverso un meccanismo di diffusione di reazione. Il metodo qui presentato fornisce un percorso praticabile per costruire i componenti molecolari sottostanti per tali materiali DNA auto-patterning. Anche se impegnativo, lo sviluppo di morfologie macro-scala in modo programmabile avrebbe implicazioni significative in settori che vanno dalla ricerca biomateriali alla medicina rigenerativa.
Divulgazioni
The authors declare no competing financial interests.
Riconoscimenti
Figure 1, 2, 3, 4, 6, 8 e tabelle 2, 3, 10, 15, 16 vengono modificati dal Ref 29. Questo lavoro è stato sostenuto dal National Science Foundation (NSF concedere-CCF 1.117.143 e NSF-CCF 1.162.141 a GS). Y.-JC è stata sostenuta da borse di studio del governo di Taiwan. SDR è stato sostenuto dalla Science Foundation Graduate Research Fellowship Program Nazionale (GRFP).
Materiali
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S | |
| PvuII-HF | NEB | R3151L | |
| PstI-HF | NEB | R3140S | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Terrific Broth, Modified | SIGMA-ALDRICH | T0918-250G | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAGEN Hispeed Maxi-prep Kit | QIAGEN | 12662 | |
| Nb.BsrDI | NEB | R0648L | |
| Nt.BstNBI | NEB | R0607L | |
| NanoDrop 2000c | Thermo Scientific | ||
| Double-stranded Genomic Blocks | IDT | ||
| Horiba Jobin-Yvon Spex Fluorolog-3 Fluorimeter | Horiba/Jobin Yvon | ||
| Synthetic Quartz Cells | Starna | 23-5.45-S0G-5 | |
| QIAGEN Gel Extraction Kit | QIAGEN | 28706 | |
| Plasmid Backbones | BioBrick | E0240-pSB1A2 | High copy number plasmid with Ampicillin resistance. Sequence can be found from http://parts.igem.org |
Riferimenti
- Zhang, D. Y., Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103-113 (2011).
- Krishnan, Y., Simmel, F. C. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 50, 3124-3156 (2011).
- Zhang, D. Y., Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303-17314 (2009).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475, 368-372 (2011).
- Qian, L., Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 332, 1196-1201 (2011).
- Zadegan, R. M., Jepsen, M. D., Hildebrandt, L. L., Birkedal, V., Kjems, J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 11, 1811-1817 (2015).
- Seelig, G., Soloveichik, D., Zhang, D. Y., Winfree, E. Enzyme-free nucleic acid logic circuits. Science. 314, 1585-1588 (2006).
- Zadegan, R. M., et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 6, 10050-10053 (2012).
- Andersen, E. S., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 459, 73-76 (2009).
- Zhang, D. Y., Hariadi, R. F., Choi, H. M., Winfree, E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 4, (1965).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406, 605-608 (2000).
- Green, S. J., Lubrich, D., Turberfield, A. J. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 91, 2966-2975 (2006).
- Venkataraman, S., Dirks, R. M., Rothemund, P. W., Winfree, E., Pierce, N. A. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2, 490-494 (2007).
- Green, S. J., Bath, J., Turberfield, A. J. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 101, 238101 (2008).
- Omabegho, T., Sha, R., Seeman, N. C. A bipedal DNA Brownian motor with coordinated legs. Science. 324, 67-71 (2009).
- Turberfield, A. J., et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 90, 118102 (2003).
- Dirks, R. M., Pierce, N. A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 101, 15275-15278 (2004).
- Seelig, G., Yurke, B., Winfree, E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 128, 12211-12220 (2006).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318, 1121-1125 (2007).
- Yin, P., Choi, H. M., Calvert, C. R., Pierce, N. A. Programming biomolecular self-assembly pathways. Nature. 451, 318-322 (2008).
- Chen, X., Briggs, N., McLain, J. R., Ellington, A. D. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 110, 5386-5391 (2013).
- Phillips, A., Cardelli, L. A programming language for composable DNA circuits. J. R. Soc. Interface. 6, S419-S436 (2009).
- Lakin, M. R., Youssef, S., Polo, F., Emmott, S., Phillips, A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 27, 3211-3213 (2011).
- Lakin, M. R., Youssef, S., Cardelli, L., Phillips, A. Abstractions for DNA circuit design. J. R. Soc. Interface. 9, 470-486 (2012).
- Zhang, D. Y., Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182-4197 (2010).
- Ducani, C., Kaul, C., Moche, M., Shih, W. M., Hogberg, B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 10, 647-652 (2013).
- Lin, C., et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 105, 17626-17631 (2008).
- Bhatia, D., et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 48, 4134-4137 (2009).
- Chen, Y. J., et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 8, 755-762 (2013).
- Arkin, A., Ross, J. Computational functions in biochemical reaction networks. Biophys. J. 67, 560-578 (1994).
- Érdi, P., Tóth, J. . Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. , (1989).
- Magnasco, M. O. Chemical kinetics is Turing universal. Phys. Rev. Lett. 78, 1190 (1997).
- Oishi, K., Klavins, E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 5, 252-260 (2011).
- Senum, P., Riedel, M. Rate-independent constructs for chemical computation. PLoS One. 6, (2011).
- Soloveichik, D., Cook, M., Winfree, E., Bruck, J. Computation with finite stochastic chemical reaction networks. Natural Computing. 7, 615-633 (2008).
- Soloveichik, D., Seelig, G., Winfree, E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 107, 5393-5398 (2010).
- Tyson, J. J., Chen, K. C., Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 15, 221-231 (2003).
- Cardelli, L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 23, 247-271 (2013).
- Angluin, D., Aspnes, J., Eisenstat, D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 21, 87-102 (2008).
- Cardelli, L., Csikasz-Nagy, A. The cell cycle switch computes approximate majority. Sci. Rep. 2, 656 (2012).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170-173 (2011).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. , (2012).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. , e253 (2007).
- Lessard, J. C. Transformation of E. coli via electroporation. Methods Enzymol. 529, 321-327 (2013).
- Nasri, M., Thomas, D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 15, 7677-7687 (1987).
- Dalchau, N., Seelig, G., Phillips, A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. , 84-99 (2014).
- Scalise, D., Schulman, R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2, 55-66 (2014).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati