
Method Article
Plasmid stammende DNA-Strand Displacement Gatter zum Implementieren Chemical Reaction Networks
In diesem Artikel
Zusammenfassung
This protocol describes a method for deriving DNA strand displacement gates from plasmids and testing them using fluorescence kinetics measurements. Gates can be modularly composed into multi-component systems to approximate the behavior of formal chemical reaction networks (CRN), demonstrating a new use for CRNs as a molecular programming language.
Zusammenfassung
DNA nanotechnology requires large amounts of highly pure DNA as an engineering material. Plasmid DNA could meet this need since it is replicated with high fidelity, is readily amplified through bacterial culture and can be stored indefinitely in the form of bacterial glycerol stocks. However, the double-stranded nature of plasmid DNA has so far hindered its efficient use for construction of DNA nanostructures or devices that typically contain single-stranded or branched domains. In recent work, it was found that nicked double stranded DNA (ndsDNA) strand displacement gates could be sourced from plasmid DNA. The following is a protocol that details how these ndsDNA gates can be efficiently encoded in plasmids and can be derived from the plasmids through a small number of enzymatic processing steps. Also given is a protocol for testing ndsDNA gates using fluorescence kinetics measurements. NdsDNA gates can be used to implement arbitrary chemical reaction networks (CRNs) and thus provide a pathway towards the use of the CRN formalism as a prescriptive molecular programming language. To demonstrate this technology, a multi-step reaction cascade with catalytic kinetics is constructed. Further it is shown that plasmid-derived components perform better than identical components assembled from synthetic DNA.
Einleitung
Die Vorhersehbarkeit der Watson-Crick-Basenpaarung konnten dynamischen DNA-Nanotechnologie als eine programmierbare Weg zu molekularen Geräte mit dynamischen Eigenschaften 1,2 entwerfen entstehen. Insbesondere DNA Strangverdrängung - eine programmierbare, wettbewerbsfähige Hybridisierungsreaktion - hat sich als leistungsfähigen Mechanismus für das Engineering dynamischer DNA Systeme sein. In einem DNA-Strang Verdrängungsreaktion, verdrängt ein eingehender Oligonukleotid eine zuvor gebunden "output" Strang aus einem komplementären Bindungspartner. Mehrere solche Reaktionen können zusammen in mehrstufigen Reaktorkaskaden mit einem hohen Grad an Kontrolle über die Reihenfolge und das Timing der einzelnen Reaktionsschritte 3 verkettet werden. DNA-Strangverdrängungs Kaskaden verwendet worden, um digitale und analoge Schaltungen Molekular 4-7, umschaltbar Nanostrukturen 8-10 autonome molekulare Motoren 11-15, und nicht-kovalente katalytischer Verstärker 13,16-21 erstellen. Außerdem DNA Geräte mit Strangverdrängungsreaktionen können simuliert und für vielfältige Anwendungen unter Verwendung von computergestützten Design-Software 22-24 ausgelegt sein.
Derzeit ist chemisch synthetisierte DNA als Hauptmaterial für die DNA-Nanotechnologie. Jedoch Fehler in der DNA-Syntheseverfahren und die so erhaltenen unvollkommen Oligonukleotiden wird angenommen, dass die Leistung von dynamischen DNA von Geräten durch fehlerhafte Nebenreaktionen zu begrenzen. Zum Beispiel "Leck" Reaktionen können in der Freisetzung eines Ausgangs Oligonucleotid selbst in der Abwesenheit eines Reaktionstrigger führen. Solche Effekte werden in autokatalytischen Reaktionskaskaden, wo auch nur eine minimale Menge von anfänglichen Leck wird schließlich in die vollständige Aktivierung des Kaskaden 19,20 führen offensichtlichsten. Umgekehrt Reaktionen oft nicht zu dem erwarteten Ausmaß der Aktivierung zu erreichen, da einige Komponenten nicht einmal in Anwesenheit von die beabsichtigte Eingabe 7,25 auszulösen. Um die Leistung zu machen DNA basierendenNanovorrichtungen vergleichbar zu biologischen Proteinbasis benötigen solche Fehlerarten, drastisch reduziert werden.
Bakterielle Plasmide oder andere biologische DNA konnte als relativ billige Quelle für hochreine DNA für Nanotechnologie-Anwendungen dienen. Große Mengen von DNA können durch Replikation in Bakterien erzeugt werden, und die Grenzkorrekturlesen Fähigkeiten lebender Systeme sorgen für die Reinheit des erhaltenen DNA. In der Tat haben einige neue Papiere den potenziellen Nutzen der biologischen DNA für Nanotechnologie-Anwendungen 21,26-28 anerkannt. Jedoch die vollständig doppelsträngige Art der Plasmid-DNA hat bisher die Verwendung als Material zur Herstellung von dynamischen DNA-Geräte, die typischerweise aus mehreren Oligonukleotiden und enthalten sowohl doppelsträngige und einzelsträngige Bereiche gestattet. In einem kürzlich erschienenen Papier 29 wurde diese Frage angesprochen und eine neue DNA-Gate-Architektur, die in erster Linie aus geklaut doppelsträngige DNA (ndsDNA) aus war einzuführend.
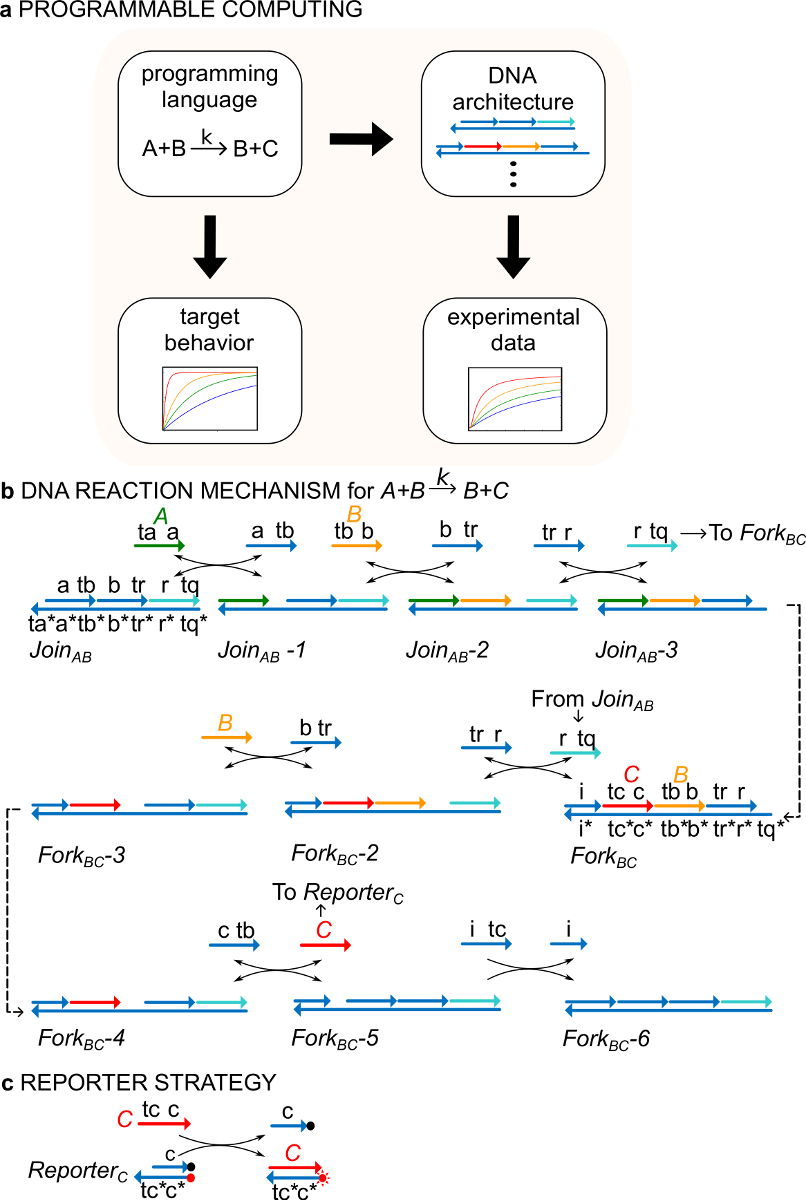
Wichtig ist, dass Systeme der ndsDNA Tore ausgelegt sein, dass die durch jede formale chemische Reaktion Netzwerk (CRN) 29 angegeben Dynamik zu realisieren. ndsDNA Gates könnte also verwendet werden, grundsätzlich auf dynamische Systeme, die Schwingungen und Chaos, Bistabilität und Gedächtnis, Boolesche Logik oder algorithmische Verhaltensweisen 30-38 aufweisen erstellen. Zum Beispiel 29 Ref. Gezeigt, eine dreiReaktions CRN, die ein Molekular Implementierung eines "Konsensus" Protokoll an, das der verteilten Rechenalgorithmus 29,39,40 vorgesehen. Diese Arbeit erstmals gezeigt werden eine neue Verwendung für die CRN-Formalismus als eine "Programmiersprache" zur schnellen Synthese von funktionalen molekularen Systemen (Abbildung 1A).
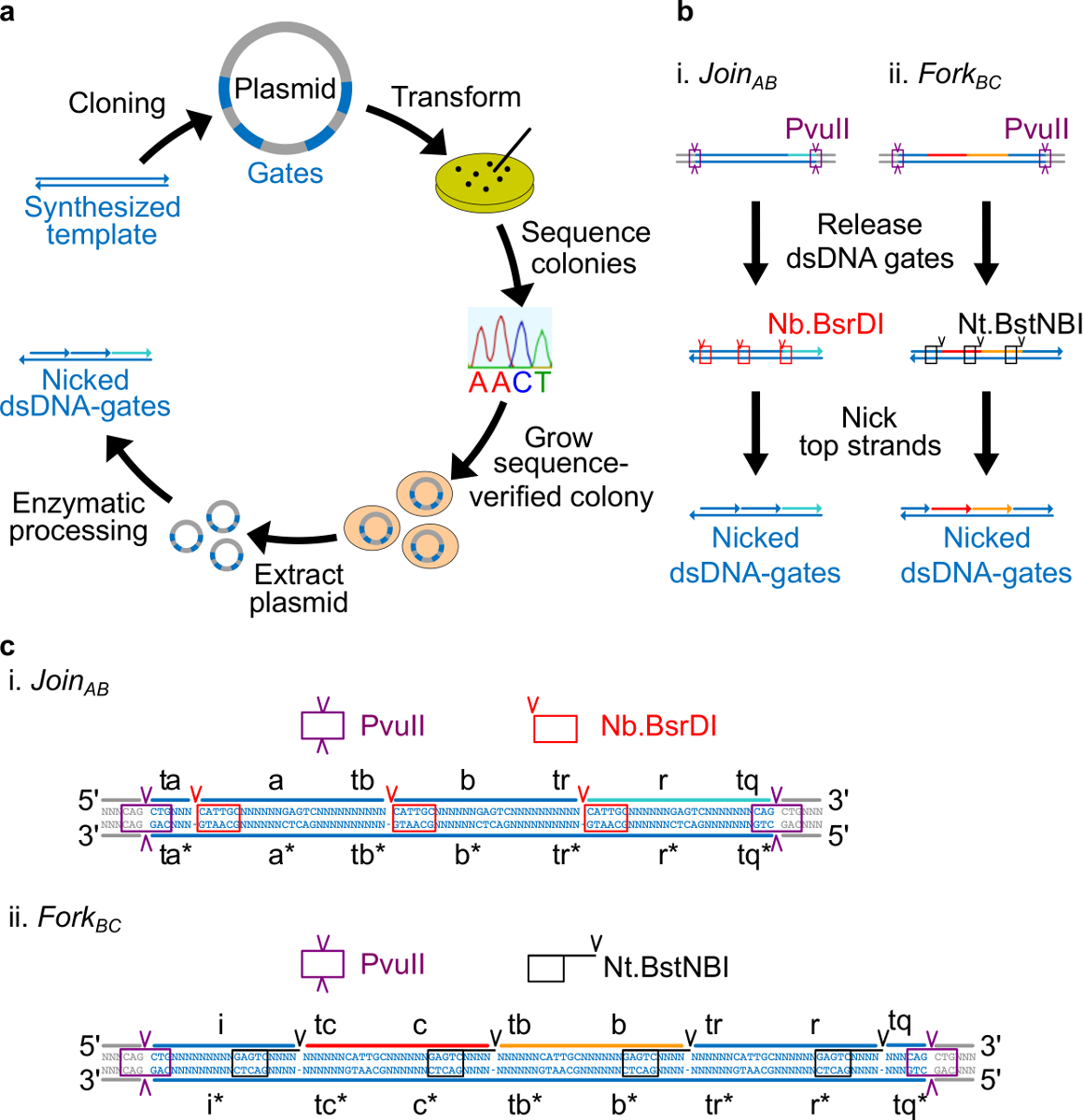
Hier ist ein detailliertes Protokoll zum Ableiten ndsDNA Gates von Plasmid-DNA ist. Zunächst ist eine Überprüfung der Sequenz Design-Prozess. Es folgt eine Erläuterung, wie synthetische Oligonukleotide enthaltendie Gate-Sequenzen in Plasmide kloniert und die Sequenz verifiziert und durch Bakterienkultur amplifiziert. Als nächstes wird gezeigt, wie ndsDNA Tore können aus den Plasmiden durch enzymatische Verarbeitung abgeleitet werden (siehe Abbildung 2). Schließlich wird ein Verfahren zum Testen von Gate Verhaltens mit Fluoreszenzkinetiken Assays beschrieben.
Reaktionsmechanismus
Als ein Beispiel setzt das Protokoll auf die katalytische chemische Reaktion A + B-> B + C. Die Arten A, B und C ("Signale", 1B) entsprechen alle einem anderen Einzelstrang-DNA-Moleküls. Die Sequenzen dieser Moleküle sind völlig unabhängig und die Stränge nicht direkt miteinander reagieren. Die Sequenzen aller Signale weisen zwei verschiedene funktionelle Domänen, das heißt, Teilsequenzen, die in Strangverdrängungsreaktionen zusammenwirken: 1) eine kurze toehold Domäne (labels ta, tb, tc), die für die Initiation der Strangverdrängung r verwendet wirdeAction, und 2) eine lange Domain (Etiketten a, b, c), die das Signal Identität bestimmt.
Wechselwirkungen zwischen Signalstränge durch geklaut doppelsträngige DNA (ndsDNA) Gate-Komplexe vermittelte (genannt anmelden AB und Gabel BC) und Hilfseinzelstrang-Arten (, , und ). Die formale Reaktions A + B-> B + C wird durch eine Reihe von Strangverdrängungsreaktionsschritten, wobei jeder Reaktionsschritt setzt einen Ansatzpunkt für eine anschließende Reaktion (1B) ausgeführt. In diesem Beispiel werden Signale A und B sind anfänglich frei in Lösung, während das Signal C dem Gabel Gate gebunden. Am Ende der Reaktion B und C sind in Lösung. Allgemeiner Signale, die an ein Gate gebunden sind inaktiv, während Signale, die frei in Lösung aktiv sind, das heißt, sie können in einer Strangverdrängungsreaktion als teilnehmenein Eingang. Der zeitliche Verlauf der Reaktion wird unter Verwendung eines fluoreszierenden Reporterstrategie (Abbildung 1C), gefolgt. In früheren Arbeiten 29 wurde gezeigt, dass diese Reaktion Mechanismus realisiert sowohl die korrekte Stöchiometrie als auch die Kinetik der Zielreaktion.
Protokoll
1. Reihenfolgen-Entwurf
Hinweis: Sequence Design Übersicht: In diesem Abschnitt wird die Strategie für die Gestaltung von Plasmid stammende DNA-Gattern beschrieben. Enzymstellen an beiden Enden der Tore angeordnet, um für die Freisetzung von vollständig doppelsträngig Gatter nach Verdau zu ermöglichen. Nicking Websites werden dann solche in Verkehr gebracht, dass Enzyme zu erstellen Kerben auf dem oberen Strang, um die endgültigen ndsDNA Tore erstellen. Schließlich werden die verbleibenden Sequenzen gewählt ist, dass unabhängige Domänen sind orthogonal zueinander und haben die Sekundärstruktur nicht aufweisen.
- Legen Sie die Nt.BstNBI Strangbruchstelle vier Nukleotide vom 3'-Ende jedes langen Domain (a, b, c, r und i). Legen Sie die Nb.BsrDI Strangbruchstelle am 5'-Ende jedes lange Domain (a, b, c und r. Beachten Sie, dass die Domain hat i keinen Nb.BsrDI Strangbruchstelle). 2C zeigt die detaillierte Abfolge Ansicht Registriert AB und BC Fork Tore.
- Legen Sie die PvuII-Restriktionsstelle an beiden Enden des ndsDNA Toren, so dass PvuII der Verdauung können die Tore von Plasmiden zugeben (siehe Abbildung 2C).
- Gestalten Sie anderen unbeschränkte Sequenzen durch folgende zwei Prinzipien: (a) Stränge nicht ausstellen Sekundärstrukturen (DNA-Strukturen können mit Nupack 41 vorhergesagt werden kann), und (b) alle Domänen sollten orthogonal zu Übersprechen zu minimieren.
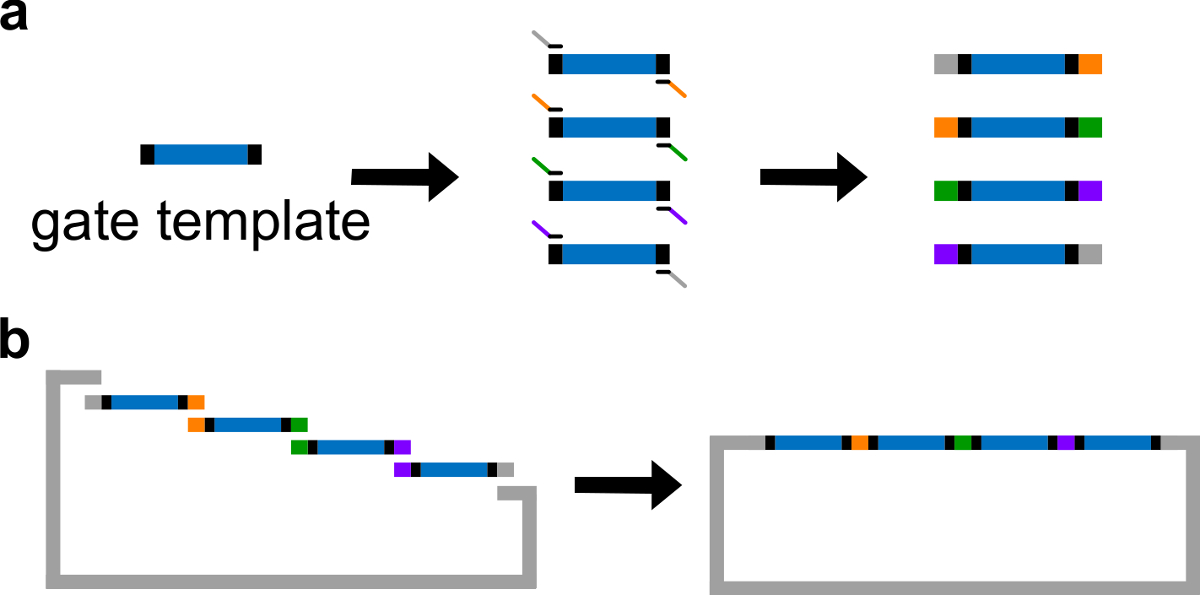
- Platzieren ndsDNA Sequenzen in der Mitte einer Gate-Vorlage. Platzieren 30-40 bp Zufalls Spacer-Sequenzen an beiden Enden des Gate-Template dient jeder Abstandhalter als eine einzigartige Bindungsstelle für die folgende Polymerasekettenreaktion (PCR).
2. Klonierung von NdsDNA Tore in Plasmide
Hinweis: Dieser Abschnitt beschreibt die Gibson Klonen Verfahren zum Einsetzen 4 Kopien des Tores in ein Plasmid-Rückgrat.
- Sortieren ndsDNA Gate-Vorlagen als Doppelstrang-Genom-Bausteine aus einer DNA-Hersteller (Gate Template-Sequenzen werden gezeigtin Tabelle 1; Stränge treten ndsDNA Gatter werden in Tabelle 2 gezeigt; Domain-Ebene-Sequenzen sind in Tabelle 3).
- Nach dem Empfang des geordneten DNA spin die Rohre, die genomische Blöcke an 10.000-14.000 × g für 1 min, um sicherzustellen, daß alle getrockneten DNA ist an der Unterseite des Rohres.
- Resuspendieren der getrockneten genomischen Blöcke in DNase-freiem Wasser auf eine Endkonzentration von 10 ng / & mgr; l zu erreichen.
Anmerkung: Alternativ kann DNA mit 1x Tris Ethylendiamintetraessigsäure (EDTA) resuspendiert werden (TE-Puffer: 10 mM Tris und 1 mM EDTA, pH 8,0). Jedoch ist EDTA einen Chelatbildner für zweiwertige Kationen und konnte PCR hemmen. - Generieren 4 Gate-Fragmente mit unterschiedlichen Überlappungsbereichen durch eine Standard-PCR mit einer High-Fidelity DNA-Polymerase (siehe 3A). Die Primersequenzen sind in Tabelle 4 detailliert (Schmelztemperatur dieser Primer ist 62 ° C).
- Ausführen eines 2% igen Agarosegel in 140 V für 30 min bei RT (für eine detaillierte Agarosegelelektrophorese Protokoll siehe 42), und schneiden die Bänder entsprechend jedem PCR amplifizierte Fragment aus dem Gel. Dann reinigen die Gelscheiben mit einem Gelextraktionskit (beachten Sie bitte die Materialien beziehen) nach den Anweisungen des Herstellers.
- Verdauen eine hohe Kopienzahl Plasmidrückgrat (siehe Materialien) mit PvuII und PstI-HF-HF bei 37 ° C für 1 h (siehe Tabelle 5) gemß dem Protokoll des Herstellers. PvuII-HF und PstI-HF sind High-Fidelity-Restriktionsenzymen, die drastisch reduzieren unspezifische Schnitte.
- Führen Sie einen 1,5% Agarosegel und schneiden Sie die linearisierte Backbone (in der Regel führen Sie das Gel bei 140 V für 30 bis 40 min bei RT). Sodann wird die DNA aus dem Gelstück mit Gelextraktionskit folgenden Anweisungen des Herstellers.
- Zuführen Gibson Anordnung 43 mit den linearisierten Vektor und gereinigten PCR-Fragmente (siehe Tabelle 6 und 3B ) bei 50 ° C für 1 Stunde.
- Wandeln Sie die Gibson Montageprodukt aus Schritt 2.8 in Escherichia coli (E. coli) und Teller auf einem LB-Medium (LB) Agarplatte Ampicillin Antibiotika (bei einer Konzentration von 100 ug / ml). Führen Transformation durch Elektroporation oder Hitzeschock-Methode 44,45, und verwenden Sie die entsprechende E. coli-Stamm. Verwenden Sie beispielsweise E. coli-Stamm JM109 für Hitzeschock-Transformation, und verwenden Sie DH5a elektrokompetente E. coli-Zellen für die Elektroporation.
Hinweis: Das Plasmidrückgrat verwendet ein Ampicillin-Resistenz-Kassette enthält. Wenn mit einem anderen Selektionsmarker, verwenden Sie die entsprechenden Antibiotika anstelle von Ampicillin.
3. Bakterienkultur Amplification und Qualitätskontrolle
Hinweis: Dieser Abschnitt beschreibt die Massenproduktion und die Isolierung von Plasmiden, die die DNA-Gates nach der Qualitätskontrolle.
- Wählen Sie eine Einzelkolonievom Ampicillin selektive Platte aus Schritt 2.9 und Inkubation einer Kultur von 3 ml angereichertem Medium, das Ampicillin-Antibiotika (bei einer Konzentration von 100 ug / ml). Markieren die Kolonie, so daß er wieder in den nachfolgenden Versuchsschritte genutzt werden. Die Kultur bei 37 ° CO / N unter kräftigem Schütteln (200-300 UpM). Typischerweise Inkubation für 16-24 Stunden.
- Entpacken Sie die Plasmid-DNA aus der Bakterienkultur mit Hilfe eines Mini-Prep-Kit nach den Anweisungen des Herstellers.
- Messen die gereinigte Plasmid-DNA unter Verwendung eines Spektrophotometers nach Herstelleranweisungen. Typische Ausbeute liegt im Bereich von 50-1000 ng / ul.
- Holen Sie sich das durch das Senden der Probe auf eine DNA-Sequenzierung Unternehmen sequenziert extrahierte Plasmid-DNA. Sequenzierprimer sollte ungefähr 100 Nukleotiden stromaufwärts und stromabwärts von der Region sequenziert werden; Die Sequenzierungsprimer für das Plasmid (siehe Materialien für Plasmid) hat die folgende Sequenz: ATTACCGCCTTTGAGTGAGC.
Neinte: Wenn es Folgefehler oder Rekombination im eingesetzten ndsDNA Tore, wählen Sie eine andere Kolonie von der Platte aus Schritt 2.9. Führen Sie die Schritte 3,1 bis 3,4, um zu überprüfen, dass die Sequenzen der eingesetzten Gates korrekt sind. - Nach der Überprüfung, dass die Sequenzen korrekt sind, wählen Sie die entsprechende Kolonie aus dem Ampicillin selektive Platte (aus Schritt 2.9), und Inkubation einer Kultur der 800 ml Terrific Broth (TB), das Ampicillin-Antibiotika (bei einer Konzentration von 100 ug / ml). Die Kultur bei 37 ° C für 16-24 h unter starkem Schütteln (200-300 UpM). TB besonders für High-Yield-Plasmid-Produktion geeignet.
Hinweis: Alternativ LB könnte auch verwendet werden, um Bakterien wachsen, obwohl die Plasmid Ausbeute kann ein Problem sein. - Reinigen die DNA mit einem Maxi-Prep-Kit nach den Anweisungen des Herstellers.
- Folgen Sie Schritt 3,3 bis 3,4, um zu überprüfen, ob die Sequenzen korrekt sind. Wenn eine Rekombination aufgetreten ist, finden Sie im folgenden Hinweis. Andernfalls fahren Sie mit Schritt 4.
Hinweis: ist ein mögliches Problem hier, dass mehrere Kopien der einge Toren im Plasmid kann aufgrund der DNA-Reparatur zu rekombinieren. Um dieses Problem zu lösen, verwenden Sie ein E. coli Stamm ohne das RecA-Protein (ein Protein, um die DNA-Reparatur Bezug), wie JM109 oder DH5 eine zuvor sequenz verifiziert Plasmid Transformation (dh ohne Folgefehler und Rekombination). Dann wählen Sie eine Kolonie von dieser Platte und überprüfen Sie die Plasmid-Sequenz durch das Senden der Probe auf eine DNA-Sequenzierung Unternehmen.
4. Enzymatische Verarbeitungs
Hinweis: Dieser Abschnitt beschreibt das Verfahren für die Verdauung der Plasmide, so dass sie ausgeschnitten und an den richtigen Stellen eingekerbt und bereit, für Kinetik Experimente verwendet werden.
- Verdauen die gereinigte Plasmid-DNA aus Stufe 3.7 mit dem Restriktionsenzym PvuII-HF für 1 h bei 37 ° C (siehe Tabelle 7). Typischerweise Digest des Plasmids mit 4 Einheiten PvuII-HF pro 1 mg des Plasmids. Hohe fidelity Restriktionsenzyme sind für den Einsatz empfohlen, da sie drastisch reduzieren unspezifische Schnitte.
- Führen Ethanol-Fällung auf die Probe.
- In 2 äquivalente Volumina eiskaltem absolutem Ethanol auf die Probe.
- Inkubieren der Mischung bei -80 ° C für mindestens 1 Stunde (Diese Mischung kann auch bei -80 ° C für O / N sitzen).
- Zentrifuge bei 10.000-14.000 x g bei 0 ° C für 30 min.
- Entfernen Sie den Überstand.
- Hinzuzufügen 1.000 ul RT 95% Ethanol zu der Probe und zu invertieren 10-15 mal.
- Zentrifuge bei 10.000-14.000 × g bei 4 ° C für 10 min.
- Entfernen Sie den Überstand und der Luft trocknen lassen, die auf Bank für 10-20 min.
- Resuspendieren der DNA-Pellets in einem geeigneten Volumen Nuklease freiem H 2 O (in der Regel 100-200 ul). Hinzufügen von mehr als 200 & mgr; l werden in der Regel machen die Probe zu stark verdünnt zur Verwendung in Kinetik Experimente.
- Messen Sie die resuspendierten DNA mit einem Spektralphotometer nach dem mAnweisungen ersteller von.
- Digest kommen Tore mit Nicking-Enzym Nb.BsrDI bei 65 ° C für 1 h unter Verwendung von 4 Einheiten des Enzyms pro 1 ug Plasmid (siehe Tabelle 8); verdauen Gabel Tore mit Nicking-Enzym Nt.BstNBI bei 55 ° C für 1 h unter Verwendung von 8 Einheiten des Enzyms pro 1 ug Plasmid (siehe Tabelle 9).
Hinweis: Schritt 4.2 entfernt Enzymverdau Puffer und hilft zu konzentrieren die Tore für Kinetik Experimente. Schritt 4.2 übersprungen für Join Toren, weil beide Restriktionsenzym PvuII-HF und Nicking-Enzym Nb.BsrDI den gleichen Verdauungspuffer zu teilen. In Schritt 4.2.8 wird Nuklease freiem H 2 O anstelle von TE verwendet, da EDTA ist ein Chelatbildner für zweiwertige Kationen und Restriktionsenzyme, die diese Ionen müssen funktionieren hemmen.
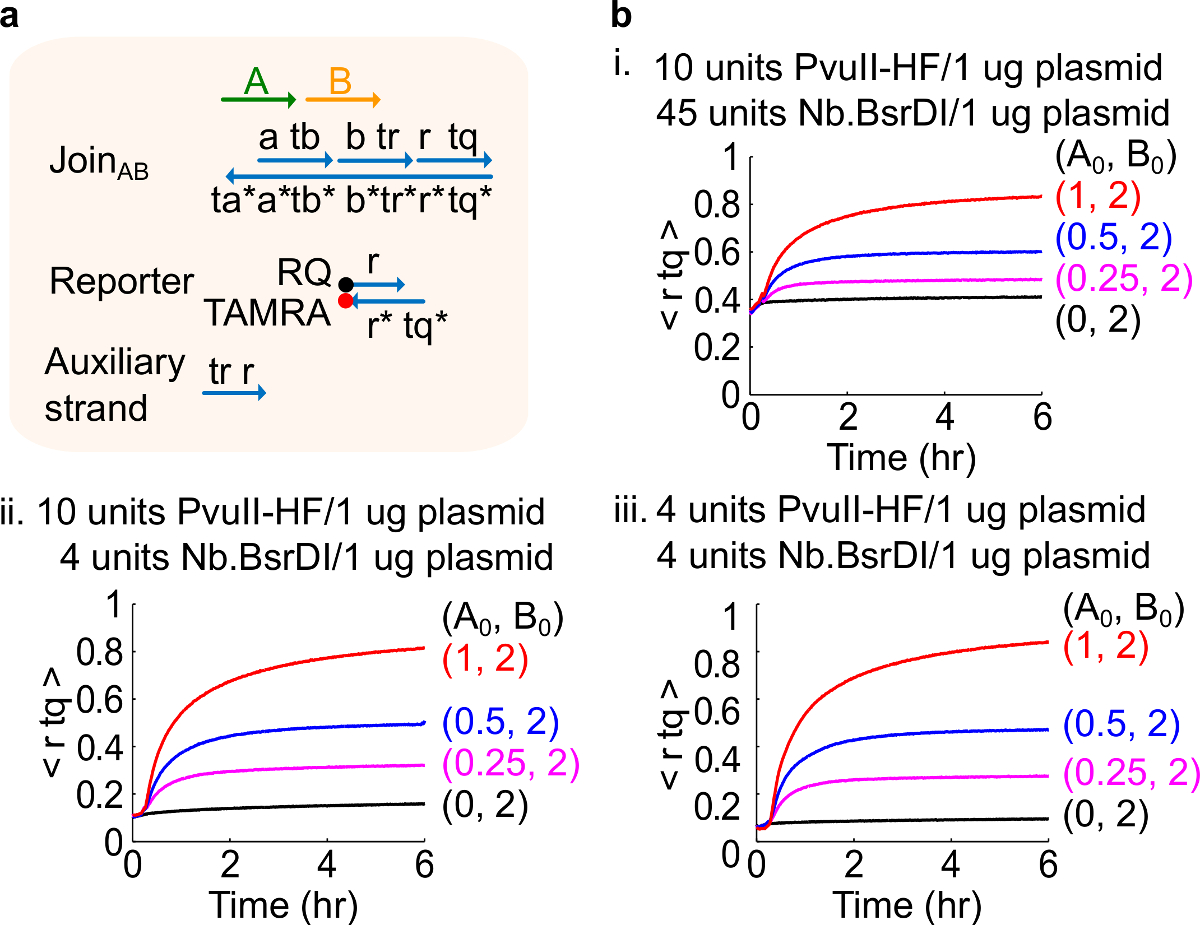
Anmerkung: Die Zugabe von überschüssigen Mengen an Enzymen können hohe Mengen an Ausgangsschaltung Leckage (Figur 4), was höchstwahrscheinlich durch Überverdauung 46 verursacht wird, führen. Dieses Problem can durch die Optimierung der Enzymmengen behandelt werden (siehe Abbildung 4). Typische Reichweite von Enzymen ist von 1 bis 10 Einheiten / 1 ug Plasmid.
5. Herstellung von einzelsträngigen Oligonukleotiden
Hinweis: Dieser Abschnitt beschreibt das Protokoll für die Resuspendierung und Quantifizieren der chemisch synthetisierte einzelsträngige DNA (ssDNA), die für die Signalstränge und Nebenstränge verwendet werden. Für Strangsequenzen siehe Tabelle 10. Beachten Sie, dass das folgende Protokoll ist ein Beispiel für die Vorbereitung 10 uM ssDNA. Andere Konzentrationen von ssDNA kann in ähnlicher Weise hergestellt werden.
- Nach dem Empfang von der DNA-Oligos Hersteller, drehen die Rohre DNA enthaltend 10.000-14.000 × g für 1 min, um sicherzustellen, daß alle getrockneten DNA ist an der Unterseite des Rohres.
- Resuspendieren der DNA mit 1x Tris Ethylendiamintetraessigsäure (EDTA) Puffer (TE-Puffer: 10 mM Tris und 1 mM EDTA, pH 8,0) auf eine Endkonzentration von 100 uM zu erreichen. FürBeispiel resuspendieren 8 nmol DNA in 80 ul TE-Puffer.
- Mischungs 10 ul der DNA in 100 & mgr; M mit 90 ul molekulares Wasser in ein Mikrozentrifugenröhrchen, das eine Endkonzentration von 10 uM zu erreichen, sollten.
- Messen die genaue Konzentration der DNA-Probe unter Verwendung eines Spektrophotometers nach Herstelleranweisungen. Das folgende Protokoll zeigt ein Beispiel, wie die DNA-Konzentration gemessen werden kann.
- Blank das Spektralphotometer mit 2 & mgr; molekular Wasser.
- Die Absorption bei 260 nm (A 260) der DNA-Probe. Verwenden Sie die folgende Gleichung, um die Lager-Konzentration zu berechnen.
Hinweis: Die Probenkonzentration ist M = A 260 / Extinktionskoeffizienten. Der Extinktionskoeffizient kann über die Spezifikation Datenblatt durch die DNA-Herstellers.
6. Herstellung von Fluorescent Reporter
Hinweis: Dieser Abschnitt beschreibt dieProtokoll für die Vorbereitung Reporter C können andere fluoreszierende Reporter in ähnlicher Weise zusammengebaut werden.
- Bestell-Hochleistungs-Flüssigkeits-Chromatographie (HPLC) gereinigt Oligonukleotide ROX- (der obere Strang der Reporter C) und -RQ (der untere Strang der Reporter C) von DNA-Hersteller (siehe Tabelle 10 für Sequenzen ).
- Nach dem Empfang der synthetisierten Oligonukleotide, resuspendieren und quantitativ Proben wie in Schritt 5 beschrieben.
- Mischen Sie die Reporter oberen und unteren Stränge (dh ROX- und -RQ) in 1x Tris-Acetat-EDTA (TAE) mit 12,5 mM Mg 2+ (siehe Tabelle 11 für die detaillierte Rezept ). Man beachte, dass hier 30% Überschuss Quencher markierten Strang -RQ wird zugesetzt, um den Reporter, der dafür sorgt, dass alle mit einem Fluorophor markierte Stränge auch mit unvollkommenen Stöchiometrie abgeschreckt zubauen.
- Annealing der Reporter-C-Komplex unter Verwendung eines Thermocyclers, Abkühlen von 95 ° C bis 20 ° C bei einer Rate von 1 ° C / min. Die Proben können bei 4 ° C gelagert werden.
7. Fluoreszenzmessungen
Hinweis: Der Abschnitt beschreibt ein allgemeines Protokoll für die Fluoreszenz-Messungen der Kinetik (siehe 5 für die Versuchsdurchführung), und dieses Protokoll wird in den Schritten 8, 9 verwendet werden, und 10 auch beachten, dass dieses Protokoll für die Verwendung eines Spektrofluorimeters. Alternativ könnten diese Experimente auch in einem Plattenlesegerät durchgeführt werden, obwohl die Empfindlichkeit, wohl auch Variationen und mangelnde Temperaturregelung in Langzeitversuchen kann ein Problem sein.
- Stellen Sie den Temperaturregler auf 25 ° C, und warten Sie auf die Temperatur zu stabilisieren. Verwendung einer Temperatursteuerung kann die Variabilität in dem Signal, das vom Temperaturänderung führen kann, zu vermindern.
- Setzen Sie die richtigen Parameter for kinetische Messungen in der Datenerfassungssoftware des Spektrofluorimeters. Detaillierte Beispieleinstellungen sind wie folgt:
- Stellen Sie die Schlitzbreite auf 2,73 nm für beide Anregungs- und Emissions Monochromatoren.
- Stellen Sie die Integrationszeit auf 10 Sekunden für jeden 60 sec Zeitpunkt. Stellen Sie die Gesamtmesszeit bis 24 Stunden.
- Festlegen der Anregungs- / Emissionswellenlängen, die in dem Experiment verwendeten Fluorophore entsprechen. Beispiel Wellenlängen sind wie folgt: ROX (588 nm / 608 nm) und TAMRA (559 nm / 583 nm).
- Hinzufügen Nuklease freiem H 2 O und 10-fach Tris-Acetat-EDTA-Puffer, enthaltend 125 mM Mg 2+ (10x TAE / Mg 2+), um ein synthetisches Quarzzelle. Siehe Tabellen 12, 13 und 14 beispielsweise Volumina zu benutzen.
- Hinzufügen polyT Stränge, um eine Endkonzentration von ~ 1 & mgr; M (siehe Tabelle 12, 13 und 14 für Volumen) zu erreichen, und dann vortexen synthetischenQuarz-Zellen für 10-15 sec. Im Allgemeinen werden Pipettenspitzen unspezifisch binden DNA. Zugabe hoher Konzentrationen von polyT Strängen können diese nicht-spezifischen Bindungsfehler zu verringern.
- In Reportern und Nebenstränge. Siehe Tabelle 12, 13 und 14 beispielsweise Volumina zu benutzen. Beachten Sie, dass für die Reporter-Kalibrierung, keine Hilfsstränge benötigt werden.
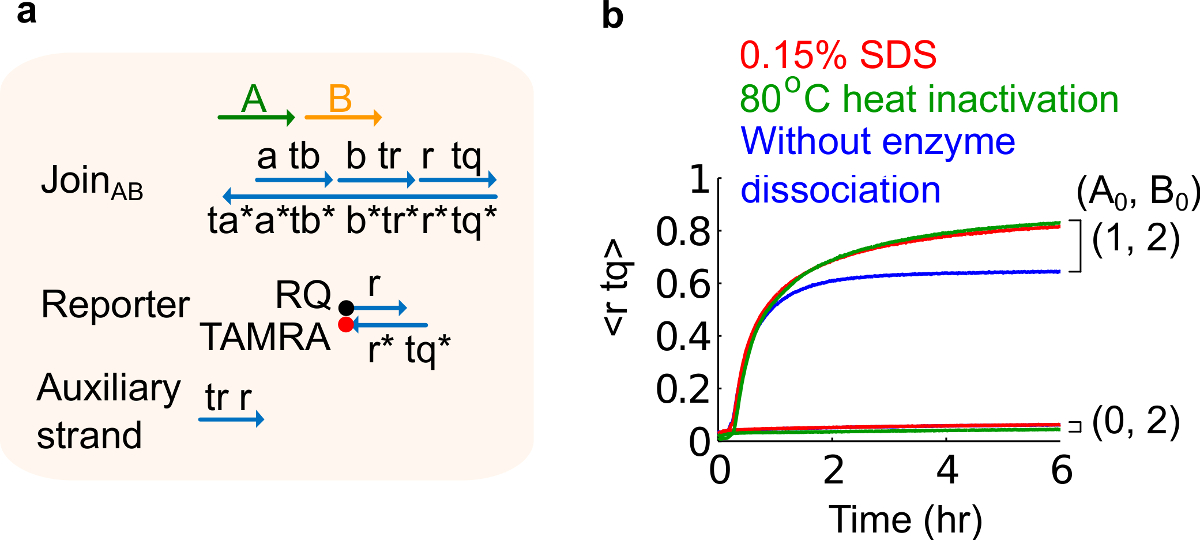
- Zugabe von 10% Natriumdodecylsulfat (SDS) in einer Endkonzentration von 0,15% SDS erzielen. Anmerkung: SDS verwendet wird, um Enzyme aus dem Plasmid stammenden Gates zu distanzieren, weil Enzyme mit Strangverdrängungsreaktion (siehe Abbildung 6) eingreifen. SDS wird hier stattdessen der Hitzedenaturierung von Enzymen empfohlen, um die Dissoziation und Rekombination der falschen Gate Stränge, die sich nachteilig auf die Schaltungsfunktion zu vermeiden.
- [Diesen Schritt für Reporter Kalibrierung überspringen.]
- In beizutreten und Gabel Tore (siehe Tabelle 13 und 14 für Volumen)dem synthetischen Quarzzelle und mischen Sie die Lösung durch Pipettieren es nach oben und unten für mindestens 20 Mal (die Küvette nicht vortexen, weil Vortex-Lösungen mit SDS kann in Blasen, die Fluoreszenz-Kinetik-Messungen beeinflussen führen).
- Auch für die folgenden Messschritten bewegen so schnell wie möglich, weil das Leck Reaktion initiiert unmittelbar nach der Zugabe des beizutreten und Gabel Toren der synthetischen Quarzzelle.
- Legen Sie die synthetischen Quarzzellen in die Kammer eines Spektrofluorimeter.
- Starten Sie das Kinetik-Messung.
- Nach 5 Minuten der Messung Addiereingang Stränge (siehe Tabelle 12, 13 und 14 für die Volumina) auf dem synthetischen Quarzzelle und mischen die Reaktion durch Pipettieren von oben und unten mindestens 20 mal. Beachten Sie, dass die Probe sollte vorsichtig gemischt werden, um Luftblasen zu vermeiden. Führen Sie diesen Schritt, während das Datenerfassungsprogramm wird angehalten, um zu vermeiden Messsignale von externen ausgelöstLicht.
- Nehmen Sie die Reaktionskinetik, bis sie stabilen Zustand erreicht. Die Reaktionskinetik auf dem Computer angezeigt wird.
8. Kalibrieren Fluorescent Reporter
Hinweis: Dieser Abschnitt beschreibt das Protokoll für die Herstellung von Eichkurven von fluoreszierenden Reportern. Kalibrierungskurven verwendet werden, um willkürliche Fluoreszenzeinheiten zur molaren Konzentration Signal umzuwandeln.
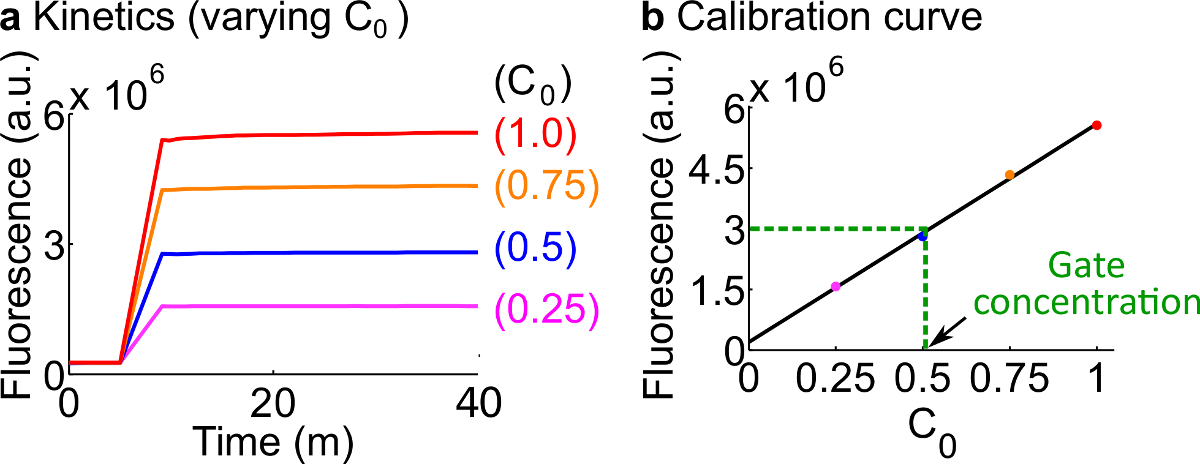
- Kalibrieren Sie fluoreszierenden Reportern im Anschluss an die in Schritt 7. Verwenden Sie die Volumina der Reaktionspartner und Puffer beschriebenen Protokoll, wie in Tabelle 12 zusammengefaßt Die Standardkonzentration für dieses Beispiel ist 50 nM (1x). Reporter sind bei 3x; Eingang ist 1x. Für Fälle, in denen das Eingangs sind bei 0,25x, 0,5x, 0,75x, das Volumen der Nuclease freie H 2 O entsprechend anpassen, um die endgültigen Volumen von jeder Reaktion zu halten, um 600 & mgr; l zu sein. Ein Beispiel Daten sind in 7A gezeigt.
- Machen Sie eine Kalibrierungskurve desReporter C durch eine lineare Anpassung der endgültigen Fluoreszenzwerte gegen die Anfangskonzentration des Signals C (ein Beispiel Eichkurve ist in Figur 7B gezeigt). Diese Kalibrierungskurve kann zur willkürlichen Fluoreszenzeinheiten zu seinem entsprechenden Signalkonzentration umzuwandeln.
9. Quantifizierung der Konzentration von Plasmid stamm ndsDNA Tore
Anmerkung: jeweils unabhängig verarbeiteten Charge von Plasmid stamm ndsDNA Gates ergibt eine Ausbeute von verschiedenen Funktionsgatter, und dieser Abschnitt beschreibt ein Protokoll für die Quantifizierung der Konzentration von Plasmid stamm ndsDNA Toren.
- Quantifizierung der Konzentration von Plasmid stamm ndsDNA Toren nach der in Schritt 7. Verwenden Sie die Volumina der Reagenzien beschrieben, wie in Tabelle 13 zusammengefaßt Protokoll Hinweis:. Tabelle 13 beschreibt ein Beispiel Rezept Fork BC Quantifizierung beitreten. AB und andere Tore kann in ähnlicher Weise durchgeführt werden, aber unter Verwendung von unterschiedlichen Eingangssträngen, Hilfs Stränge und Reportern.
- Wandeln die in diesem Versuch in einer Konzentration von Signal C unter Verwendung der Kalibrierungskurve aus Schritt 8.2 gemessenen endgültigen Fluoreszenzwert. Dann zurück-Berechnung der ndsDNA Gate-Konzentration. Beispielsweise einen endgültigen Fluoreszenzwertes für die Gate-Quantifizierung Experiment entspricht 25 nM Signal C (0,5x), basierend auf der Kalibrierungskurve in 7B. Da der Bestand an Fork BC wird 40 Mal in dieser Reaktion verdünnt, der Aktienkonzentration der Fork BC Tor ist 1 & mgr; M.
10. Kinetik Messungen für die Reaktion A + B-> B + C
Anmerkung: Dieser Abschnitt beschreibt ein Protokoll zum Testen der DNA Realisierung eines formalen chemischen Reaktion unter Verwendung von Fluoreszenz kinetische Messungen.
- ProForm Kinetikmessung nach der in Schritt 7. Verwendung von Reagenzien und Volumina Puffer beschriebenen Protokoll, wie in Tabelle 14 zusammengefasst.
Ergebnisse
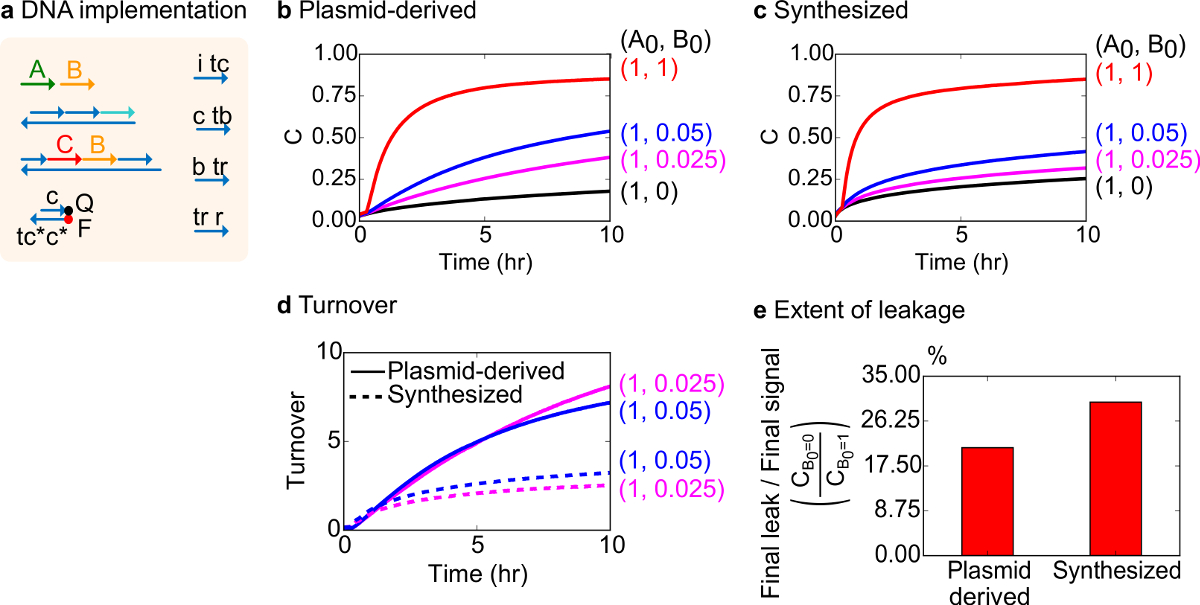
Für einen Funktionstest wurde ein DNA-Umsetzung des bimolekularen katalytische Reaktion (dh A + B-> B + C) erzeugt. Die Leistung von Plasmid stammende Gates mit den Gates von synthetischer DNA zusammengesetzt, verglichen. Katalytische Reaktionen sind ein guter Test für Gate Reinheit, da eine fehlerhafte Gate irreversibel Falle ein Katalysator, was zu einer überproportionalen Einfluss auf die Produktmenge hergestellt 18,19. Zur gleichen Zeit wird eine kleine Leckage Reaktion resultiert in der nicht ausgelösten Freisetzung des katalytischen Signal linear verstärkt werden, was zu einer überproportionalen Fehlersignal. Versuchsdaten auf Plasmid abgeleitet synthetisiert sind in Abbildung 8B und 8C gezeigt. In den Experimenten wird die Konzentration der Signalstrang A fixiert, während die Menge des katalytischen Signal B variiert. Signal C wird verwendet, um den Fortschritt der Reaktion ohne Unterbrechung des katalytischen ausgelesenFahrrad fahren. Katalyse in den Daten festgestellt werden, da Reaktionen nähern Abschluß selbst bei Mengen von Katalysator B viel kleiner als die Menge von A. Da SDS wurde nicht für Experimente mit der synthetisierten System erfolgen aufgenommen wird die Reaktionsgeschwindigkeit (die durch die Zugabe von SDS betroffen sein könnten) nicht verglichen und die analytische Schwerpunkt liegt stattdessen auf Katalyse (detailliert wie folgt).
Die weitere Analyse der katalytischen Umsetzung dieser Reaktion wurde durchgeführt. Umsatz wird als der für jeden Katalysator B zu einer gegebenen Zeit gebildeten Menge Signal C definiert. Insbesondere wurde Umsätzen aus experimentellen Daten durch Dividieren der Leck subtrahierte Signal C durch die anfängliche Menge an Katalysator B zugesetzt berechnet. Für eine ideale Katalysatorsystem sollte diese Umsatzzahl linear mit der Zeit zu und unabhängig von der Menge an Katalysator zu sein, solange das Substrat nicht einschränkend. In einem realen System kann fehlerhafte Toren cat deaktivieren talysatoren, und der Umsatz wird einen Maximalwert, auch wenn nicht alle verfügbaren Substrat zu Produkt umgewandelt zu erreichen. Die maximale Umsatzwert gibt an, wie viele Substrate (Signal A) ein Katalysator (Signal B), bevor er inaktiviert konvertieren. Hier ist zu beobachten, dass das synthetisierte System von der idealen linearen Anstieg des Umsatzes viel früher als die Plasmid-abgeleitete System abweicht tut, was anzeigt Sequestrierung des Katalysators durch eine unerwünschte Nebenreaktion (8D). Der Umsatz Vergleich nur für niedrige Konzentrationen, weil bei hohen Konzentrationen von Katalysatoren gezeigt, werden alle Gatter ausgelöst wird und Freigabesignal C. Die Schaltung Leckage wird auch verglichen, und es wird beobachtet, daß das Verhältnis der Lecksignal unter Verwendung von Plasmid stammende Gates etwa 8% unter Verwendung von synthetisierten Gatter nach 10 h Reaktion (8E).
iles / ftp_upload / 53.087 / 53087fig1.jpg "/>
Abbildung 1. (A) CRNs dienen als normative Programmiersprache. DNA Reaktionsnetzwerke können manipuliert werden, um die Dynamik eines formalen CRN nähern (B) DNA Umsetzung Beispiel chemische Anweisung:. A + B-> B + C. DNA-Stränge sind als Linien mit Pfeilen an dem 3'-Ende gezogen und * Komplementarität. Alle Signalstränge A ( a>, grün), B (, orange) und C ( c>, rot) werden von einem Brückenkopf-Domäne bestand (bezeichnet als ta, tb und tc) und eine Identität Domain (A, B, und C beschriftet). Die bimolekulare Reaktion A + B-> B + C benötigt zwei Mehrstrang-Komplexe Registriert AB und BC Fork und vier Hilfsstränge , , und . Die Reaktion verläuft über sieben Schritte der Strangverdrängung, wo jeder Schritt Starts mit toehold bindend. (C) Reporter-Strategie. Die Reaktion wird unter Verwendung eines Reporter in denen die unteren Strang mit einem Fluorophor (roter Punkt) markiert und mit dem oberen Strang ist mit einem Quencher (schwarzer Punkt) befestigt ist, gefolgt. Aufgrund der Co-Lokalisierung von Fluorophor und Quencher wird Reporterfluoreszenz im intakten Reporter abgeschreckt. Das Signal C kann den oberen Strang des Reporters zu ersetzen, was zu einer Erhöhung der Fluoreszenz. (Dieser Wert wurde aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2. (A) NdsDNA Toren aus bakteriellen Plasmid-DNA hergestellt. Mehrere Kopien des doppelsträngigen ndsDNA Gate Vorlage werden in ein Plasmid kloniert. Die klonierten Plasmide sind dien in E. verwandelt coli-Zellen und Kolonien auf der Platte sind Sequenz verifiziert. Sobald die Sequenz bestätigt ist, wird die Plasmid-DNA amplifiziert und extrahiert. Schließlich wird die doppelsträngige Plasmid in die gewünschten ndsDNA Gates durch enzymatische Verarbeitung. (B) Die enzymatische Verarbeitung ndsDNA Toren. Das Restriktionsenzym PvuII wird verwendet, um das Tor aus dem Plasmid freizusetzen. Die freigesetzten Toren weiterverarbeitet mit Nicking-Enzyme: Nb.BsrDI wird verwendet, um Kerben zu generieren für Sie Mitglied AB (Panel I); Nt.BstNBI wird verwendet, um Kerben für Fork BC (Panel II) zu erzeugen. Beschränkung und Nicking Websites werden als farbcodierte Boxen angezeigt. (C) Sequence Blick auf die Gate-Vorlage von Join AB (Panel I) und Fork BC (Panel II). Die PvuII-Restriktionsstelle (in lila Box markiert) ist an beiden Enden der ndsDNA Toren. Die Nb.BsrDI und Nt.BstNBI Nicking Standorte sind in roten und schwarzen Kästchen hervorgehoben sind. Die Standorte der Schnitt sind mit Pfeilspitzen markiert. Sequenz N irgendein Nukleotid ist. (Dieser Wert wurde mit Genehmigung aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Figur 3 (A) PCR einer DNA-Gate-Vorlage. Ein DNA-Gate Vorlage enthält die ndsDNA Gate-Sequenzen in der Mitte (a blauen Bereich) und Spacer-Sequenzen an beiden Enden (schwarze Bereiche, wobei diese beiden End-Sequenzen sind orthogonal). Primer können an die Spacer-Sequenzen der Template-Gatter binden und erzeugen vier überlappenden DNA-Fragmenten durch PCR (überlappende Sequenzen sind farbcodiert, in der Figur). (B) Gibson Montage. Die vier amplifizierte DNA-Fragmentierungts werden dann in eine linearisierte Plasmid-Backbone durch Gibson Montageverfahren 43 montiert. (Dieser Wert wurde mit Genehmigung aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 4. Schaltungsleistung mit unterschiedlichen Enzymmengen. (A) eine vereinfachte Darstellung der Gate-reporterHilfsStränge und die Signalstränge für die entsprechenden Experimente verwendet. (B) Kinetik Experimente mit Plasmid stamm beitreten AB mit verschiedenen Enzymmengen verarbeitet . ich. 10 Einheiten PvuII-HF und 45 Einheiten von Nb.BsrDI pro 1 ug Plasmid; ich ich. 10 Einheiten PvuII-HF und 4 Einheiten Nb.BsrDI pro 1 ug Plasmid; iii. 4 Einheiten PvuII-HF und 4 Einheiten Nb.BsrDI pro 1 ug Plasmid. Alle Hilfsstränge wurden mit 2x (1x = 10 nM). Die Gate-Komplex war 1,5fach, und die Experimente wurden bei 35 ° C in 1 × TAE / Mg 2+ durchgeführt wird. (Dieser Wert wurde mit Genehmigung aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

. 5 Flussdiagramm der Kinetik Experimente Blue:. Materialien, um Küvetten hinzufügen (0,875 ml synthetischen Quarzzelle). > B + C - Referenztabelle 14 für bestimmte Mengen für kinetische Experiment der A + B hinzuzufügen. Grün: Anleitung eines Spektrofluorimeter (als SPEX beschriftet). Rot: Mischanleitung.53087fig5large.jpg "target =" _ blank "> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 6. Enzym Dissoziation und Schlussverhalten. (A) eine vereinfachte Darstellung der Gate, reporter, Hilfsstränge und Signalstränge für die entsprechenden Experimente verwendet. (B) Kinetik Experimente der Plasmid stamm Registriert AB mit 80 ° C Wärme Inaktivierung (grün Spuren), 0,15% Natriumdodecylsulfat (SDS) (rot) und eine Kontrolle ohne Hitzeinaktivierung oder Zugabe von SDS (blau). Die Standardkonzentration 1x = 10 nM, und alle Hilfs Stränge und Eingang B waren 2x. Die Gate-Komplex war 1,5fach, und die Experimente wurden bei 35 ° C in 1x Tris-Acetat-EDTA-Puffer, enthaltend 12,5 mM Mg 2+ (1x TAE / Mg 2+ durchgeführt wird ). (Dieser Wert wurde aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 7. Reporter Kalibrierung. (A) Reporter C Kinetik. Der Reporter-Konzentration wurde auf 3x (1x = 50 nM), und die Anfangskonzentration des Signals C ist in der Figur angegeben. (B) Die Fluoreszenzpegel des Signals C an der Messendpunkt (40 min) zeigt eine lineare Beziehung mit der Anfangskonzentration von Signal C. In einer Quantifizierung Beispiel Fork BC-Gate (grüne gestrichelte Linie), die Fluoreszenz-Wert von Fork BC war Maßnahmed als 3 × 10 6 (au), die mit 25 Nm (0,5-fach), basierend auf der Eichkurve entspricht. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 8. Bimolekulare katalytische Reaktionskinetik (A + B-> B + C). (A) eine vereinfachte Darstellung des Tores, reporter, Hilfsstränge und Signalstränge für die entsprechenden Experimente verwendet. Die Experimente wurden in 1x Tris-Acetat-EDTA-Puffer, der 12,5 mM Mg 2+ (1x TAE / Mg 2+) laufen. Alle Tor-Komplexe wurden bei 75 nM-Konzentration (1,5-fach) und Hilfs Stränge wurden bei 100 nM Konzentration (2x). Kinetik-Daten für Plasmid stamm Gates und Daten für synthetisiert Gates in (B) gezeigt, und (C) sind. Signal wurde bei 50 nM (1x). Verschiedene Mengen Signal (Katalysator) wurden in das System eingeführt, und die Reaktion wurde bei 35 ° C getestet. (D) Plasmid stamm Gates zeigte höheren Umsatz als synthetisierte DNA-Gates bei niedrigen Aufwandmengen von Eingangs zugegeben. (E) Umfang der Leckage. Das Balkendiagramm zeigt das Verhältnis des endgültigen Leckage nach Endsignal (C B0 = 0, / C B0 = 1) an den Endpunkten (10 h). (Dieser Wert wurde aus Lit. 29 modifiziert worden ist.) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}
| Tor-Vorlagen | Sequenzen | Länge (nt) |
| JoinAB | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCTACATTGCTTCTACGAGTCATCCTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 173 |
| FORKBC | TCTAGTTCGATCAGAGCGTTATTACCAGTAGTCGATTGCTCAGCTGCCATCATAAGAGTCACCATACCCACATTGCCACATCGAGTCCCTTTTCCACCATTGCACCTTAGAGTCCGAATCCTACCATTGCTTAACCGAGTCTCACAACCAGCTGTCATTATGGACTTGACACACAGATTACACGGGAAAGTTGC | 194 |
Tabelle 1. Die Sequenzen der ndsDNA Gate-Vorlagen.
| Tor | Strand | Länge des unteren Strang (nt) |
| JoinAB | JoinAB-Bottom, , , | 87 |
| ForkBC | ForkBC-Bottom, , , , | |
| 108 |
Tabelle 2. Strands umfasst Registriert AB und Gabel BC. (Diese Tabelle wurde von Ref 29 geändert wurde.)
| Domain | Sequenz | Länge (nt) |
| ta | CTGCTA | 6 |
| tb | TTCCAC | 6 |
| tc | TACCCA | 6 |
| tr | TCCTAC | 6 |
| tq | AACCAG | 6 |
| ein | CATTGCTTCTACGAGTCATCC | 21 |
| b | CATTGCACCTTAGAGTCCGAA | 21 |
| c | CATTGCCACATCGAGTCCCTT | 21 |
| r | CATTGCTTAACCGAGTCTCAC | 21 |
| ich | CTGCCATCATAAGAGTCACCA | 21 |
| Primerstrang | Sequenzen | Länge (nt) |
| Forward-Primer-1 | AAGAGAGACCACATGGTCCTTCTTGAGTTTGTAACAG CGTTATTACCAGTAGTCGATTGC | 60 |
| Reverse-Primer-1 | ACTACTATTTACTAATCCCATTGCGTGTTCTTATT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward-Primer-2 | AATAAGAACACGCAATGGGATTAGTAAATAGTAGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse-Primer-2 | GCGAAACTAGCTTGTGGTGATATTGTCTCGTGTGT TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward-Primer-3 | ACACACGAGACAATATCACCACAAGCTAGTTTCGC CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse-Primer-3 | ACATTGTACGCCTAAATCATCAAGAATAATTGTTG TAATCTGTGTGTCAAGTCCATAATG | 60 |
| Forward-Primer-4 | CAACAATTATTCTTGATGATTTAGGCGTACAATGT CGTTATTACCAGTAGTCGATTGC | 58 |
| Reverse-Primer-4 | GAGCGCAGCGAGTCAGTGAGCGAGGAAGCCTGCAG TAATCTGTGTGTCAAGTCCATAATG | 60 |
Tabelle 4. Die Primersequenzen für die PCR von ndsDNA Gate Vorlagen.
| Reagens | Volume für 1x Reaktion (ul) |
| High-Kopie Plasmidrückgrat (~ 300 ng / ul) | 10 |
| PvuII-HF (20.000 Einheiten / ml) | 2 |
| PstI-HF (20.000 Einheiten / ml) | 2 |
| 10x Cut intelligente Puffer | 2 |
| H 2 O | 4 |
| Volle Lautstärke | 20 d> |
Tabelle 5. Protokoll zur Plasmidrückgrat verdauen.
| Reagens | Volume für 1x Reaktion (ul) |
| DNA-Vektor (~ 50 ng / & mgr; l) | 1 |
| PCR amplifizierte Fragment 1 (~ 50 ng / & mgr; l) | 1 |
| PCR amplifizierte Fragment-2 (~ 50 ng / & mgr; l) | 1 |
| PCR amplifizierte Fragment-3 (~ 50 ng / & mgr; l) | 1 |
| PCR amplifizierte Fragment-4 (~ 50 ng / & mgr; l) | 1 |
| 2x Gibson Assembly Master Mix | 5 |
| Volle Lautstärke | 10 |
| Reagens | Volume für 1x Reaktion (ul) |
| Plasmid-DNA (~ 1 & mgr; g / & mgr; l Konzentration) | 1000 |
| PvuII-HF (20.000 Einheiten / ml) | 200 |
| 10x Cut intelligente Puffer | 133,3 |
| Volle Lautstärke | 1.333,3 |
Tabelle 7. Protokoll zur ndsDNA Toren eingesetzt Plasmid mit dem Restriktionsenzym PvuII-HF verdauen.
| Reagens | Volumen (ul) |
| Registriert Toren (~ 5 ug / ul-Konzentration) | 150 |
| Nb.BsrDI (10.000 Einheiten / ml) | 300 |
| 10x Cut intelligente Puffer | 50 |
| Volle Lautstärke | 500 |
Tabelle 8. Protokoll für die Join-Gates verdauen mit Nicking-Enzym Nb.BsrDI.
| Reagens | Volumen (ul) |
| Gabel-Gatter (~ 5 ug / ul Konzentration) | 150 |
| Nt.BstNBI (10.000 Einheiten / ml) | 600 |
| 10x NEB-Puffer 3.1 | 83,3 |
| Volle Lautstärke | 833,3 |
Tabelle 9 Protokoll für Gabeltore verdauen mit Nicking-Enzym Nt.BstNBI.
. Tabelle 10 Strand-Sequenzen für die Durchführung der chemischen Reaktion A + B -.> B + C (. Diese Tabelle wurde von Ref 29 modifiziert wurden)
| Reagens | Volumen (ul) | Endkonzentration |
| ROX- 100 & mgr; M | 10 | 10 uM (1x) |
| -RQ bei 100 μ; M | 13 | 13 & mgr; M (1.3x) |
| 10x TAE mit 125 mM Mg 2+ | 10 | 1x TAE mit 12,5 mM Mg 2+ |
| H 2 O | 67 | - |
| Volle Lautstärke | 100 | 10 uM (1x) |
Tabelle 11. Protokoll zur Montage Reporter C.
| Reagens | Volumen (ul) | Endkonzentration |
| H 2 O | 514 | - |
| 10x TAE mit 125 mM Mg 2+ | 60 | 1x TAE mit 12,5 mM Mg 2+ |
| PolyT bei 300 μ; M | 2 | 1 & mgr; |
| Reporter C mit 10 & mgr; M | 9 | 150 nm (3x) |
| 10% SDS | 9 | 0,15% |
| auf 5 & mgr; M | 6 | 50 nM (1x) |
| Volle Lautstärke | 600 | - |
Tabelle 12. Protokoll für die Kalibrierung der Reporter C. Die hier angegebenen Mengen ist für eine Gesamtreaktionsvolumen von 600 ul (entsprechend der Verwendung eines 0,875 ml synthetischen Quarzzelle), kann jedoch eingestellt werden, um unterschiedliche Größen Zellen arbeiten.
| Reagens | Volumen (ul) | Finale conZentrierung |
| H 2 O | 493 | - |
| 10x TAE mit 125 mM Mg 2+ | 60 | 1x TAE mit 12,5 mM Mg 2+ |
| bei 300 & mgr; M polyT | 2 | 1 & mgr; |
| Reporter C mit 10 & mgr; M | 9 | 150 nm (3x) |
| bei 100 & mgr; M | 3 | 10x |
| bei 100 & mgr; M | 3 | 10x |
| bei 100 & mgr; M | 3 | 10x |
| 10% SDS | 9 | 0,15% |
| Fork BC bei ~ 1 & mgr; M (Konzentration unbekannt) | 15 | ~ 0,5x |
| bei 100 & mgr; M | 3 | 10x |
| Volle Lautstärke | 600 | - |
Tabelle 13. Protokoll für die Kalibrierung der Gabel BC. Die hier angegebenen Mengen ist für eine Gesamtreaktionsvolumen von 600 & mgr; l, kann aber eingestellt werden, um unterschiedliche Größen Zellen arbeiten.
| Reagens | Volumen (ul) | Endkonzentration | |
| H 2 O | 407,2 | - | |
| 10x TAE mit 125 mM Mg 2+ | 52,8 | 12,5 mM Mg 2+ | |
| bei 300 & mgr; M polyT | 2 | 1 & mgr; | |
| Reporter C mit 10 & mgr; M | 9 | 150 nm (3x) | |
| bei 10 uM | 6 | 100 nM (2x) | |
| bei 10 uM | 6 | 100 nM (2x) | |
| bei 10 uM | 6 | 100 nM (2x) | |
| 6 | 100 nM (2x) | ||
| 10% SDS | 9 | 0,15% | |
| Registriert AB bei 1 uM | 45 | 75 Nm (1,5-fach) | |
| Fork BC bei 1 uM | 45 | 75 Nm (1,5-fach) | |
| bei 10 uM | 3 | 50 nM (1x) | |
| bei 10 uM | 3 | 50 nM (1x) | |
| Volle Lautstärke | 600 | - | |
Tabelle 14 Protokoll für eine chemische Reaktion A + B-> B + C. Die hier angegebenen Mengen ist für eine Gesamtreaktionsvolumen von 600 & mgr; l, kann aber eingestellt werden, um unterschiedliche Größen Zellen arbeiten.
| Synthetisiert Tore | Plasmid stammTore | ||||
| Beschreibung | Kosten | Registriert Tore | Fork Tore | ||
| PAGE gereinigten langen Strang (100 nt, diente als die unteren Stränge eines Gate) | ~ $ 75 | Beschreibung </ strong> | Kosten | Beschreibung | Kosten |
| PAGE gereinigten kurzen Strang (~ 30 nt, diente als Top-Stränge eines Gate) | ~ $ 185 | Gate-Vorlage | ~ $ 100 | Gate-Vorlage | ~ $ 100 |
| Gesamt | ~ $ 260 | Plasmid-Extraktionskit | ~ $ 26 | Plasmid-Extraktionskit | ~ $ 26 |
| Restriktionsenzym (PvuII-HF) | ~ $ 11 | Restriktionsenzym (PvuII-HF) | ~ $ 11 | ||
| Nicking-Enzym (Nt.BsrDI, Begleiten Tore) | ~ $ 29 | Nicking-Enzym (Nt.BstNBI, Gabel Tore) | ~ $ 62 | ||
| Gesamt | ~ $ 166 | Gesamt | ~ $ 199 |
fo:.. Halten-with-previous.within-page = "always"> Tabelle 15 Kostenvergleich zwischen Plasmid stamm Toren und synthetisiert Gates (. Diese Tabelle wurde von Ref 29 modifiziert wurden)
| Synthetisiert Tore | Plasmid stammTore | ||
| Verarbeitung | Bearbeitungszeit | Verarbeitung | Bearbeitungszeit |
| Glühen | 1 Stunde | Cloning | 5 Stunden |
| PAGE Reinigung | 2 Stunden | Plasmid-Extraktion | 2 Stunden |
| Gesamt | 3 Stunden | Zwei Schritte der Enzymverdauung | 0,5 Stunden |
| Ethanol-Fällung | 1 h | ||
| Gesamt | 8,5 h | ||
Tabelle 16. Bearbeitungszeit Vergleich zwischen Plasmid stamm Toren und synthetischen Tore. (Diese Tabelle wurde von Ref 29 geändert wurde.)
Diskussion
Dieser Artikel beschreibt ein Verfahren zum Ableiten ndsDNA Gates aus hochreiner Plasmid-DNA. Darüber hinaus wird ein Protokoll zur Charakterisierung Gate Leistung durch einen Fluoreszenz-Assay-Kinetik dargestellt. Experimentelle Daten zeigen, dass das Plasmid stammSystem trifft sein synthetisches Gegenstück, selbst wenn das synthetische System ist aus Strängen mit Polyacrylamid-Gelelektrophorese (PAGE) gereinigt, montiert. Wahrscheinlich ist die verbesserte Leistung von Plasmid stammende Gates vor allem auf die hohe Reinheit der biologischen DNA. Synthetische DNA enthält eine Vielzahl von Fehlern, insbesondere Deletionen, die Oligonukleotide mit einer Länge von n-1, und solche Nebenprodukte entstehen, werden in der Regel nicht vollständig PAGE oder Hochleistungs-Flüssigkeits-Chromatographie (HPLC) Reinigungsverfahren entfernt. Ähnliche Verbesserungen bei den hier berichteten wurden in einer früheren Studie von einer katalysierten Haarnadel-Verstärker, der aus biologischen Quellen 21 abgeleitete DNA verwendet beobachtet.
Aber auch die Verwendung von Plasmid-abgeleitete Gatter können nicht vollständig beseitigen Fehler in der Gate Leistung, für die es mindestens zwei Gründe: Erstens über Verdauung oder Mangel an Schnittgenauigkeit können die Gates mit zu vielen führen Einschnitte oder Kerben in den falschen Positionen. In jedem Fall sind Gates eher in unerwünschter Reaktionen teilzunehmen. Solche Probleme können durch eine Optimierung der Menge des verwendeten Enzyms (siehe Abbildung 4) gelockert werden. Zweitens, in diesen Experimenten am Ein- und Hilfsstränge wurden synthetische DNA und enthielt Deletionen und Mutationen bei. Prinzipiell sind alle Einzelstrang Antriebs- und Hilfs Stränge könnte auch aus Phagemid-DNA durch eine Nicking-Enzym-Verdauung des vorcodierten m13 Virusgenom 26 erhalten werden. Vielleicht die Leistungsfähigkeit der Schaltung kann ferner unter Verwendung von ssDNA aus dem Bakteriengenom abgeleitet verbessert werden.
Während die Verwendung von Plasmid-abgeleitete Gattern wurde gefunden, daß die Schaltungsleistung zu verbessern, eine Analyseder Kosten und Bearbeitungszeiten ergeben, dass, während die Herstellung von Plasmid stammende Gates etwas günstiger ist (Tabelle 15), dauert es 2-3 mal längere Verarbeitungszeit im Vergleich zur Montage und Reinigung von Gattern aus kommerziell synthetisiert Oligos (Tabelle 16). Die primären Kosten von Plasmid stammende Gates Gensynthese und die Verwendung von Restriktionsenzymen. 300 pmol von Gattern (ausreichend für 15 Reaktionen bei 30 nM), die geschätzten Kosten für die Join-Gatter ist ungefähr $ 170 und $ 200 für Gabel Tore, die Kostendifferenz wobei aufgrund der Verwendung von unterschiedlichen Nicking-Enzyme. Im Gegensatz dazu ist die chemische Synthese der Stränge für die gleiche Gate kostet rund 260 $ einschließlich einer PAGE Reinigungsgebühr. Die primären Zeitkosten für Plasmid stamm Toren ist in der Klonierung, die, genau wie die DNA-Synthese, kann zu einer Gensynthese Unternehmen ausgelagert werden. Haben jedoch nach der Montage Plasmid stamm Gatter den Vorteil, dass der Host-Plasmide können leicht zu einem replizierendennd in Form von bakteriellen Glycerinstamm gespeichert werden. Dadurch ist es möglich, um die Gates mehrfach wieder verwenden.
Wir freuen uns, könnte die verbesserte Leistung von Plasmid stamm Toren einen wesentlich größeren Dynamikbereich als Verhaltensweisen sind experimentell bisher mit DNA CRNs gezeigt zu ermöglichen. Zum Beispiel schlug den letzten theoretischen Arbeit 47,48, dass selbstorganisierte räumliche Muster auf der Makroskala kann mit DNA CRNs durch eine Reaktionsdiffusionsmechanismus realisiert werden. Die hier vorgestellte Methode bietet eine praktikable Weg zum Aufbau der zugrunde liegenden molekularen Komponenten für solche Selbst Strukturieren DNA Materialien. Obwohl anspruchsvoll, die Entwicklung Makromaßstab Morphologien in einer programmierbaren Weise würde erhebliche Auswirkungen in Bereichen wie Biomaterialforschung an der regenerativen Medizin haben.
Offenlegungen
The authors declare no competing financial interests.
Danksagungen
Figuren 1, 2, 3, 4, 6, 8 und den Tabellen 2, 3, 10, 15, 16 sind aus Lit. 29 modifiziert. Diese Arbeit wurde von der National Science Foundation (NSF gewähren CCF-1.117.143 und NSF-CCF 1.162.141 auf GS). Y.-JC wurde vom taiwanesischen Regierung Stipendien unterstützt. SDR wurde von der National Science Foundation Graduate Research Fellowship Program (GRFP) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity PCR Master Mix with HF Buffer | NEB | M0531S | |
| PvuII-HF | NEB | R3151L | |
| PstI-HF | NEB | R3140S | |
| Gibson Assembly Master Mix | NEB | E2611S | |
| Terrific Broth, Modified | SIGMA-ALDRICH | T0918-250G | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAGEN Hispeed Maxi-prep Kit | QIAGEN | 12662 | |
| Nb.BsrDI | NEB | R0648L | |
| Nt.BstNBI | NEB | R0607L | |
| NanoDrop 2000c | Thermo Scientific | ||
| Double-stranded Genomic Blocks | IDT | ||
| Horiba Jobin-Yvon Spex Fluorolog-3 Fluorimeter | Horiba/Jobin Yvon | ||
| Synthetic Quartz Cells | Starna | 23-5.45-S0G-5 | |
| QIAGEN Gel Extraction Kit | QIAGEN | 28706 | |
| Plasmid Backbones | BioBrick | E0240-pSB1A2 | High copy number plasmid with Ampicillin resistance. Sequence can be found from http://parts.igem.org |
Referenzen
- Zhang, D. Y., Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 3, 103-113 (2011).
- Krishnan, Y., Simmel, F. C. Nucleic acid based molecular devices. Angew. Chem. Int. Ed. Engl. 50, 3124-3156 (2011).
- Zhang, D. Y., Winfree, E. Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303-17314 (2009).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475, 368-372 (2011).
- Qian, L., Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 332, 1196-1201 (2011).
- Zadegan, R. M., Jepsen, M. D., Hildebrandt, L. L., Birkedal, V., Kjems, J. Construction of a fuzzy and boolean logic gates based on DNA. Small. 11, 1811-1817 (2015).
- Seelig, G., Soloveichik, D., Zhang, D. Y., Winfree, E. Enzyme-free nucleic acid logic circuits. Science. 314, 1585-1588 (2006).
- Zadegan, R. M., et al. Construction of a 4 zeptoliters switchable 3D DNA box origami. ACS Nano. 6, 10050-10053 (2012).
- Andersen, E. S., et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 459, 73-76 (2009).
- Zhang, D. Y., Hariadi, R. F., Choi, H. M., Winfree, E. Integrating DNA strand-displacement circuitry with DNA tile self-assembly. Nat. Commun. 4, (1965).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406, 605-608 (2000).
- Green, S. J., Lubrich, D., Turberfield, A. J. DNA hairpins: fuel for autonomous DNA devices. Biophys. J. 91, 2966-2975 (2006).
- Venkataraman, S., Dirks, R. M., Rothemund, P. W., Winfree, E., Pierce, N. A. An autonomous polymerization motor powered by DNA hybridization. Nat. Nanotechnol. 2, 490-494 (2007).
- Green, S. J., Bath, J., Turberfield, A. J. Coordinated chemomechanical cycles: a mechanism for autonomous molecular motion. Phys. Rev. Lett. 101, 238101 (2008).
- Omabegho, T., Sha, R., Seeman, N. C. A bipedal DNA Brownian motor with coordinated legs. Science. 324, 67-71 (2009).
- Turberfield, A. J., et al. DNA fuel for free-running nanomachines. Phys. Rev. Lett. 90, 118102 (2003).
- Dirks, R. M., Pierce, N. A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U. S. A. 101, 15275-15278 (2004).
- Seelig, G., Yurke, B., Winfree, E. Catalyzed relaxation of a metastable DNA fuel. J. Am. Chem. Soc. 128, 12211-12220 (2006).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318, 1121-1125 (2007).
- Yin, P., Choi, H. M., Calvert, C. R., Pierce, N. A. Programming biomolecular self-assembly pathways. Nature. 451, 318-322 (2008).
- Chen, X., Briggs, N., McLain, J. R., Ellington, A. D. Stacking nonenzymatic circuits for high signal gain. Proc. Natl. Acad. Sci. U. S. A. 110, 5386-5391 (2013).
- Phillips, A., Cardelli, L. A programming language for composable DNA circuits. J. R. Soc. Interface. 6, S419-S436 (2009).
- Lakin, M. R., Youssef, S., Polo, F., Emmott, S., Phillips, A. Visual DSD: a design and analysis tool for DNA strand displacement systems. Bioinformatics. 27, 3211-3213 (2011).
- Lakin, M. R., Youssef, S., Cardelli, L., Phillips, A. Abstractions for DNA circuit design. J. R. Soc. Interface. 9, 470-486 (2012).
- Zhang, D. Y., Winfree, E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 38, 4182-4197 (2010).
- Ducani, C., Kaul, C., Moche, M., Shih, W. M., Hogberg, B. Enzymatic production of 'monoclonal stoichiometric' single-stranded DNA oligonucleotides. Nat. Methods. 10, 647-652 (2013).
- Lin, C., et al. In vivo cloning of artificial DNA nanostructures. Proc. Natl. Acad. Sci. U. S. A. 105, 17626-17631 (2008).
- Bhatia, D., et al. Icosahedral DNA nanocapsules by modular assembly. Angew. Chem. Int. Ed. Engl. 48, 4134-4137 (2009).
- Chen, Y. J., et al. Programmable chemical controllers made from DNA. Nat. Nanotechnol. 8, 755-762 (2013).
- Arkin, A., Ross, J. Computational functions in biochemical reaction networks. Biophys. J. 67, 560-578 (1994).
- Érdi, P., Tóth, J. . Mathematical models of chemical reactions: theory and applications of deterministic and stochastic models. , (1989).
- Magnasco, M. O. Chemical kinetics is Turing universal. Phys. Rev. Lett. 78, 1190 (1997).
- Oishi, K., Klavins, E. Biomolecular implementation of linear I/O systems. IET Syst. Biol. 5, 252-260 (2011).
- Senum, P., Riedel, M. Rate-independent constructs for chemical computation. PLoS One. 6, (2011).
- Soloveichik, D., Cook, M., Winfree, E., Bruck, J. Computation with finite stochastic chemical reaction networks. Natural Computing. 7, 615-633 (2008).
- Soloveichik, D., Seelig, G., Winfree, E. DNA as a universal substrate for chemical kinetics. Proc. Natl. Acad. Sci. U. S. A. 107, 5393-5398 (2010).
- Tyson, J. J., Chen, K. C., Novak, B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 15, 221-231 (2003).
- Cardelli, L. Two-domain DNA strand displacement. Math. Struct. Comput. Sci. 23, 247-271 (2013).
- Angluin, D., Aspnes, J., Eisenstat, D. A simple population protocol for fast robust approximate majority. Distrib. Comput. 21, 87-102 (2008).
- Cardelli, L., Csikasz-Nagy, A. The cell cycle switch computes approximate majority. Sci. Rep. 2, 656 (2012).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170-173 (2011).
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. J. Vis. Exp. , (2012).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 6, 343-345 (2009).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. , e253 (2007).
- Lessard, J. C. Transformation of E. coli via electroporation. Methods Enzymol. 529, 321-327 (2013).
- Nasri, M., Thomas, D. Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res. 15, 7677-7687 (1987).
- Dalchau, N., Seelig, G., Phillips, A. Computational design of reaction-diffusion patterns using DNA-based chemical reaction networks. DNA Computing and Molecular Programming. , 84-99 (2014).
- Scalise, D., Schulman, R. Designing modular reaction-diffusion programs for complex pattern formation. Technology. 2, 55-66 (2014).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten