
Method Article
ピペット チップを用いた液滴マイクロ流体プラットフォームに播種細胞メソッド
要約
この記事では、液滴の細胞のカプセル化効率を提供するために液滴マイクロ流体デバイスへのピペット チップを使用してセルの乏しい人口をシードするためのプロトコルを示します。
要約
様々 なマイクロ流体プラットフォーム デザイン細胞解析によく使用中液滴マイクロ流体を分離し、単一細胞レベルで細胞を細胞に及ぼす外部要因の影響を排除することによって分析のための強力なツールを提供します。微小環境。液滴の細胞のカプセル化は、各液滴の現在のセルの数と平均液滴の体積あたりの細胞数の関数としてポアソン分布によって決まります。初代培養細胞、特に免疫細胞や臨床検体は乏しいことができ、細胞の損失のないカプセル化、やりがいのままです。細胞細胞の重要な損失することがなく液滴を用いたマイクロ流体デバイスへの読み込みにピペット チップを使用する新しい方法論を提案します。様々 な細胞タイプはポアソン分布によって予測されるカプセル化効率に密接に対応する液滴の効率的な電池のカプセル化を示しています。本手法により、細胞のマイクロ流体プラットフォームへの損失のないロードと下流の単一細胞分析、例えば、異なる種類の細胞間の細胞間相互作用をデコードするために容易に適応することができます。
概要
単一セルのレベルで細胞解析の堅牢で汎用性の高いプラットフォームとしてマイクロ流体の使用は、近年、急速に増加1。これらのプラットフォームは、単一細胞および生体分子高精度と感度の非常に小さいサンプル ボリューム2,3、4を使用しての高速スクリーニングを提供します。マイクロ デザインの異なる種類の間で液滴ベースのプラットフォームは携帯電話を正確かつ正確な制御を可能にする非相溶相に囲まれた水相液滴でそれらを分離することで単一細胞を高スループット分析を有効にします。微小環境5,6。液滴を用いたマイクロ流体で、水性、両方を柔軟に分離単一または複数セルを与えるとハイドロゲル液滴タンパク質分泌や携帯電話の相互作用7,などの複雑な細胞挙動の調査で非常に貴重で、8,9します。 シグナルと免疫細胞間クロストーク微小環境の10の他の細胞との相互作用によって影響されます。液滴の単一セルの隔離は、効果的なノイズのない分析実験より効率的かつ正確な結果11,12の外部の環境因子の影響から自由を提供します。複数の入り江を持つ液滴マイクロ流体プラットフォームの設計を変更する実験細胞間相互作用を介して細胞ペアリング12,13複数のセル型のカプセル化ことができます。
液滴の細胞のカプセル化のプロセスはランダムであり細胞のカプセル化の率はポアソン分布14,15の公式を使って統計的に決定することができます。カプセル化のこのレート平均液滴接合部で細胞の到着率を考慮して推定することができますと仮定すると、各セルの到着、他の到着から独立した細胞16.独立性の仮定まばらセルの場合とみなされ、細胞数の関数として 1 つまたは複数の細胞を含む液滴の確率を予測できるにもかかわらず、独立した生産セル到着を保証できません。各液滴・液滴16,17あたりのセル数の平均値であります。液滴における細胞のカプセル化のこの推定は各液滴の現在のセルの数に依存して、1 つは入口の細胞の濃度を増加させる細胞各液滴中に存在の平均数増えることを提案することが16. したがって、単一細胞カプセル化するように、細胞濃度に削減しなければならないが空滴18の数が多い。
添付ファイル、沈降、および/または注射器で凝集、チューブ、またはによって本番デバイス読み込み中にセルの損失が予測されたカプセル化値19から実際のカプセル化の値の偏差に責任の共通の欠点.この問題を取得します彼らが既に人口の不足している、カプセル数セルだけ、はるかに低いが、予想よりもデータは提供されません十分な実験的分析のためにまれな免疫細胞や臨床検体を播種時にさらに誇張されています。形質細胞様樹状細胞 (Pdc) だけを構成するまれな免疫細胞のサブセットである約 0.2 - 0.6% 全体の白の血液細胞人口20。これらの細胞分泌する膨大な量のタイプ I のインターフェロン活性化しそれにより免疫応答21で重要な役割を果たします。水滴のような希少な細胞の細胞の挙動を勉強して、細胞播種およびカプセル化22中セル損失を防止することが不可欠です。いくつかデザイン液滴を含む液滴の生成用音響または電気の力など異なる物理的な力を利用するアクティブなカプセル化メソッドを使用して単一細胞のカプセル化を確保している関連の動きがあります。単一細胞23,24。ただし、これらのメソッドには、液滴生産16面で限界があります。
本研究では、マイクロ流体デバイスへの単一または複数のセルをロードするための従来の方法の欠点を回避する堅牢で簡単な方法を設立しました。ローらに触発され, 本手法シード重要なサンプルを失うことがなく液滴マイクロ流体プラットフォームにまれな免疫細胞の量が少ないのとは違う大きさのピペット チップを利用し、理論的に一貫した結果が得られました。予測25。この方法は簡単にすることができますと正常に液滴を用いたマイクロ流体を含むいくつかのアプリケーションに適応し、さまざまなセルタイプまたは微粒子も適用されます。
プロトコル
1. 3 入口ポリジメチルシロキサン (PDMS) デバイス作製
- エアコン ミキサーの PDMS ベース 40 g を測定カップし、追加の PDMS 硬化剤 4 g カップで基本試薬に慎重に、スポイトを使用しています。
- エアコン ミキサーのホルダーにカップを置き、ホルダーにカップの総重量を測定します。適宜ご利用いただけますミキサーに遠心バランス重量の値を設定します。
- 2 分間 2,000 rpm で泡に続いて 2 分 2,000 rpm でエアコンのミキサーでベースと硬化剤を混ぜます。
- 100 mm シリコンウエハのそれとほぼ同じサイズ約直径のアルミ艇を準備します。成形、アルミニウム ボートのレプリカの作製したシリコンウエハを置き、このセットアップは、シャーレ (直径 120 mm、高さを = = 20 mm)。
注: ペトリ皿のサイズは、si ウエハのサイズに依存します。 - カップをホルダーから外し、シリコン基板上、慎重に PDMS 事前硬化 (カップの内容)、混合物を注ぐ。
- 事前硬化 PDMS 混合物、すべての空気の泡を削除する約 20 分のデシケータによる si ウエハを含むペトリ皿を配置します。
- 20 分、削除することができます残りの空気泡のチェック後、シャーレを削除します。
- 少なくとも 3 時間の 65 ° C で設定、オーブンにペトリ皿を配置します。
- 3 h 後オーブンからペトリ皿を削除し、シリコンウエハーから硬化 PDMS の慎重に皮をむきます。
- ナイフやメスを使用してカット線に沿ってカット PDMS デバイス。入口・ 1.2 mm パンチャーを使用して各デバイスの出口の穴をパンチします。PDMS の任意のほこりや残留の部分を削除するスコッチ テープで各 PDMS デバイスをクリーニングします。
- 必要に応じて、残留 PDMS の部分を削除するために窒素ブロー。
- スライド ガラスを準備するには、石鹸水、続いてイソプロパノールと乾燥窒素でそれらを洗浄します。
- プラズマのきれいなガラスのスライドとクリーン PDMS 装置をボンド流線を閉じるにアッシャー。次の設定を使用: 力: 50 W、時間: 45 s、ブリード遅延時間: 2 s、プロセスガス: 1 ガス (空気)、ベント: 両方のバルブ、制限付きベント時間: 60 s、ポンプ スピンダウン タイム: 10 s、ベント保持時間: 0 s、ガス遮断時間: 1 s、ターボ有効にポンプ: 0. 他のすべてのガス管線を外します。
注: プラズマ アッシャーは、プラズマのブランドによって異なります使用設定アッシャーが使用。 - ソリューションを準備、シラン (1 H, 1 H, 2 H, 2 H Perfluorooctyltriethoxysilane) シランの 50 μ L を追加することによってフッ素 950 μ L にオイル。
注: シランは有毒です。ヒューム フードの下で操作してください。 - テフロン チューブに接続されている注射器に準備されたシラン ソリューションを描画します。
- デバイスのコンセントを通じて準備されたシラン ソリューションをフラッシュすることによって、デバイスを salinize します。
- オーブン、65 ° c、30 分の設定にデバイスを配置します。
- オーブンから salinized のデバイスを削除し、フッ素オイルとデバイスから余分なシランをフラッシュします。
- オーブン、接合プロセスを完了する、少なくとも 1 時間、65 ° C で設定に戻ってデバイスを配置します。
注: プロトコルはここで一時停止することができます。
2. 損失のない電池封止材
-
セルを収穫
- 再 1.0x106セル/mL、2.0x106セル/mL、4.0x106セル/mL、8.0x106セル/mL の濃度でロズウェル パーク記念研究所 (RPMI) 中 Jurkat T 細胞を中断します。pdc が存在する造血無血清培地 (例えば、X VIVO 15) 1.3x106セル/mL、3.0x106セル/mL、13.0x106セル/mL の濃度でA549 細胞のダルベッコ変更されたワシの媒体 (DMEM) 1.0x106セル/mL の濃度で。
メモ: 細胞と細胞の濃度の種類は実験に基づいて異なることができます。セルのラベルもできる実験に基づく。
- 再 1.0x106セル/mL、2.0x106セル/mL、4.0x106セル/mL、8.0x106セル/mL の濃度でロズウェル パーク記念研究所 (RPMI) 中 Jurkat T 細胞を中断します。pdc が存在する造血無血清培地 (例えば、X VIVO 15) 1.3x106セル/mL、3.0x106セル/mL、13.0x106セル/mL の濃度でA549 細胞のダルベッコ変更されたワシの媒体 (DMEM) 1.0x106セル/mL の濃度で。
-
水溶液液滴生成の先端荷重
- 3% 生体適合性界面活性剤混合物のフッ素オイルを準備するには、フッ素オイル 2 mL に界面活性剤の 3 mL を追加します。
注: フッ素オイルに追加された界面活性剤の濃度は異なる平均潜伏期のエマルジョンの安定性を決定します。界面活性剤の濃度は、特定の種類のセルで使用されるメディアによって異なります。 - シリンジ (1 mL) に油相混合の状態を描画します。シリンジから空気の泡を削除し、適切な長さのテフロン チューブに接続します。
- 注射器で図面の生体鉱物油サンプル シリンジを準備します。空気の泡を削除し、適切な長さのテフロン チューブに注射器を接続します。
- 硬化 PDMS スラブから 5 mm の直径を持つ PDMS プラグをパンチします。
注: 硬化 PDMS スラブは、1.1 に 1.9 の手順を用いて調製できます。シリコン基板ではなくプレーンのシリコンウェーハを使用します。 - 1 mm のパンチャーとプラグの中心の別の穴をパンチします。
- しっかりと収まりますように、大きい方の端からの 200 μ L ピペット チップにプラグを挿入します。
注: より大きなサンプル ボリュームの大きい細胞 1,000 μ L ピペット チップを使用します。1,000 μ L ピペット チップ、直径 5 mm と 7 mm の間で及ぶのプラグを使用できます。直径 5 mm の試料の体積は約 400 のプラグと μ はピペット チップに吸引することができます。大きい径のプラグが使用されている (7 mm) の場合より多くのサンプルの容積は (約 900 μ L) 吸気することができます。 - ピペット チップに挿入されている PDMS のプラグインで、注射器に接続されているチューブを挿入します。鉱物油と接続のピペット チップに合わせてゆっくりとシリンジのプランジャーを押してください。ピペット チップからすべての残留空気を押し出します。
- サンプル ソリューションでは、注射器に接続されているピペット チップを下げ、先端にサンプルの約 150 μ L を吸引します。
- 2.2.4 に 2.2.8 2 番目サンプルの注射器を準備する手順を繰り返します。
- シリンジ ポンプのすべての 3 つの準備された注射器を慎重に配置します。
- PDMS チップの内側 2 つの入り江で、サンプルを含む両方のピペット チップを挿入します。外側の入口で油相の混合物を含んでいる管を挿入します。
- ・ シリンジ ポンプのように流量の値を設定: 連続相解: 600 μ L/h、細胞サンプル: 100 μ L/h、各。入力し、注射器の寸法を設定します。
注: 設定は異なります直径は注射器の種類による。 - デバイスの外部チャネルを介してデバイスと油相の内部のチャネルを通じてサンプル ソリューションをフラッシュするためにポンプを起動します。
- 液滴形成が安定した液滴を収集を開始するコンセントに適切な長さのチューブを接続します。コレクションの時間は、実験によって異なります。
- ロック チューブに水滴を収集します。収集した水滴の上に RPMI 培地 (血清) なしの 200 μ L を追加し、サンプルをインキュベートします。
注: インキュベーション収集した液滴の時間変化は、実験に基づきます。水滴は、エマルジョンを壊すことによって水滴からセルを取得した後流れフローサイトメトリーによる解析や分離が実行されるロック チューブで収集されます。実験は、液滴のミクロ分析を必要とする場合、ガラス室における液滴を収集することが可能です。
- 3% 生体適合性界面活性剤混合物のフッ素オイルを準備するには、フッ素オイル 2 mL に界面活性剤の 3 mL を追加します。
-
フローサイトメトリーによる解析のための破れと携帯検索をエマルジョンします。
- 20% の準備 1 H, 1 H, 2 H, 2 H-ペルフルオロ-1-オクタノール (PFO) ソリューション (v/v) フッ素油フッ素樹脂製品オイル 10 mL に PFO の 2 mL を追加しています。
- 注射器を用いた液滴を含むコレクションの管の下部から余分な油を削除します。
- 乳剤を破るし、水相にカプセル化された細胞を解放する、エマルジョンに 20 %pfo 溶液 100 μ L を追加します。タップして一時的に混ぜます。ない渦をこの時点で行います。1-2 分間インキュベートします。
注: 追加 PFO の量は、液滴生成の量に依存します。オイルの層は完全に溶解するまで、追加の PFO を追加してください。PFO は細胞毒性、細胞死を増加高すぎる PFO 濃度または PFO にあまりにも長い潜伏をもたらすかもしれないことに注意してください。 - 30 の最も低い可能なメーカでまもなくソリューションをスピン s。
- 2% を添加した 100 mL の冷 Phosphate-Buffered 生理食塩水 (PBS) 溶液を準備胎児の子牛血清 (FCS) (98 mL の PBS で FCS の 2 mL)。
- 遠心分離後すぐに水性画分の 550 μ L をピペットおよび 2.3.5 の手順で準備として冷 PBS 溶液 2% の FCS で補われるの 500 μ L を含む新しいロック チューブに転送。任意の残油を聞かせて新しいロック チューブの下にシンク。
- 任意の残油を吸引せず、慎重にこのロック チューブから細胞を含む水相の 950 μ L を吸引し、ソリューションを新しいロック チューブに転送します。
- 10 分の新しいロック チューブに細胞をスピンします。
- 2.3.5 の手順で準備として再冷 PBS 溶液 2% の FCS で補われるの 300 μ L のセルを中断します。
注: セルも再中断できる実験によってメディアなど他の適切なソリューションに。フローサイトメトリーを用いた解析、実験に基づく細胞を染色します。
3. セルのペアリング
-
セル収穫と染色
- Jurkat T 細胞、培養用フラスコからをカウントし、1,500 rpm 5 分で細胞をスピンします。
- 上澄みを除去し、1 mL の PBS 1.0x106セル/mL の濃度を得るために 1.0x106セルを再度中断します。追加 PBS のセル数によって決まります。
- 3.1.1 に 3.1.2 同じ細胞濃度 Jurkat T 細胞の 2 番目のサンプルを準備する手順を繰り返します。
- 5 分間 1,500 rpm で 1 mL の PBS で 2 回両方のサンプルを洗います。
- 再 1.0x106セル/mL の細胞濃度 1.25 μ M carboxyfluorescein 染色エステル (CFSE) 色素を 1 つのセルのサンプルと 1.25 μ M の遠赤染料または 1.25 μ M の細胞増殖の色素とその他の細胞サンプルを中断します。染色液 1 mL 1.0x106セルです。
注: セルは流れの cytometer またはけい光顕微鏡で使用できるフィルターによって異なる染料でラベルすることができます。 - 37 ° C で 10 分間の色素を持つ細胞サンプルをインキュベートします。
- 10 分後に 1 mL の氷冷 FCS を追加することによって染色の反応を停止します。
- 5 分間 1,500 rpm で 1 mL の PBS で 2 回細胞サンプルを洗います。
- 再 RPMI メディア 10.0x106セル/mL、各色の濃度で細胞サンプルを中断します。
-
Agarose ハイドロゲル液滴セル ペアリングのための生産の先端荷重
注: セル agarose のゲルの液滴を用いたペアリング システム 27 ° C と、ゲルのゲル化を防ぐために、細胞生存率9を保証する液滴発生・収集プロセス全体で 37 ° C 間の温度を維持します。- 75 ° C まで PBS の濃度 4% (w/v) で加熱することによって超低ゲル化温度の agarose を溶かし、20 分間混合物をかき混ぜます。
- 2% (w/v) の agarose の集中をもたらす細胞標識 Jurkat T でアガロース溶液を混ぜます。さまざまなラベルのセルで他のサンプルについては、この手順を繰り返します。
- 界面活性剤の 20 mL を 30 mL フッ素オイル (油相混合物) に追加することによって 2% 界面活性剤混合物のフッ素オイルを準備します。
- 2.2.2 - 2.2.14 の手順に従います。
注: 低融点アガロースと安定した液滴の生産を確保するための粘性の性質のための値を設定流量シリンジ ポンプで次のように: 石油相混合物: 2,000 μ L/h、細胞サンプル: 200 μ L/h. を入力し、注射器の寸法を設定。 - ロック チューブに水滴を収集し、60 分のための 4 ° C で水滴を孵化させなさい。
-
エマルジョン破壊と FACS 解析アガロース ビーズ検索
- 60 分の水滴の後、注射器を用いた液滴を含むロック チューブから余分な油を削除します。
- 水滴から油の相間を削除する PFO の 200 μ L を追加します。
注: チューブに追加 PFO の量は、液滴生成の量に依存します。オイルの層は完全に溶解するまで、追加の PFO を追加してください。 - 1 mL の 10 分間 1,500 rpm で遠心分離によって油を完全に削除する冷 PBS で 2 回収集アガロース ビーズを洗浄します。
- フローサイトメトリーを使用して収集したアガロース ビーズを分析します。
注: 蛍光顕微鏡下でビーズを観察することが可能ですも。
結果
実験の結果, 25 ミクロン (図 1) の高さを持つ 3 入口 PDMS ベース マイクロ流体デバイスを使用しました。このデバイスのセットアップでは、細胞懸濁液またはメディア、水性相をフラッシュする界面活性剤や 2 つの内部の入り江と油を洗い流すことのため外側の入口を使用しました。世代とコレクション、フローサイトメトリーを用いた下流解析前にチップ オフ時間のカップルのため水滴に孵化します。潜伏期間中に血清成分媒体で現在は不安定になり、崩壊する液滴を発生し、界面活性剤対話できます。したがって、フッ素オイルに最適化された界面活性剤濃度を追加する重要です。フッ素系油の界面活性剤の濃度の異なる 2% 血清を添加した造血の無血清培地を含む単分散液滴の安定性をテストしました。それは推論することができますこれらの単分散液滴が高度図 2から少なくとも 24 時間の安定した界面活性剤 3% 追加油相に。同様の結果は、10% の FCS (データは示されていない) 添加の有無にかかわらず RPMI メディアが得られました。したがって、液滴の安定性は、培地、血清成分の異なるソースを使用する場合最適な界面活性剤濃度依存性です。
我々 のアプローチのカプセル化効率を実証するには、最初播種細胞 (図 3A) のほとんどの従来のアプローチである、注射器に接続されているチューブを用いた細胞を播種しました。我々 は 1.0x106セル/mL、2.0x106セル/mL と 4.0x106セル/mL の濃度で Jurkat T 細胞を収穫し、予測値 (図 3B) より低かったカプセル化効率を得られます。1.0x106セル/mL、1 つのセルに含まれる液滴の分数だった 2.5% は、高い細胞濃度を使用してさらに増加しなかった。セル読み込みの効率を高めるため我々 は、高い三脚を半分の長さでチューブをマウントして PDMS 装置 (図 4A) に接続されていた半分の細胞懸濁液を読み込む手法を変更します。このアプローチを使用して、我々 は 1.0x106セル/mL、2.0x106セル/mL と 4.0x106セル/mL、濃度の異なる Jurkat T 細胞とも 1.0x106セル/mL の濃度でまれな Pdc をカプセル化2.0x106セル/mL と 12.0x106セル/mL。我々 はこの方法でセルの堆積を防止することによりカプセル化の改善率を予想しました。ただし、テストすべての濃度で実験結果が予測されたポアソンの値 (図 4Bと図 4C) がはるかに低かった。
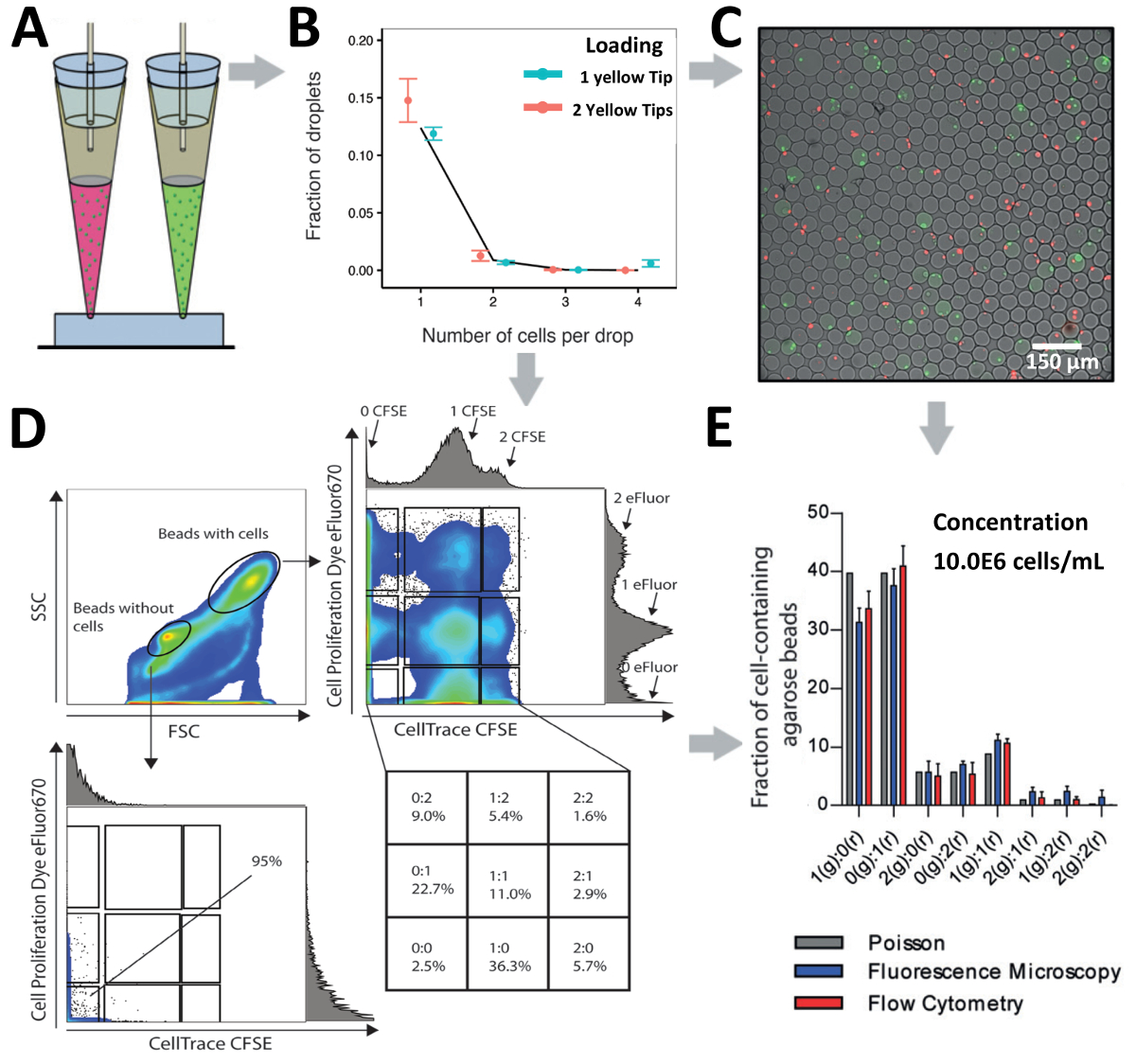
統計的に予測値 (図 5A) に一貫した実験結果を得るため私たちの細胞カプセル化率を最適化の先端荷重アプローチを使用してください。Jurkat T 細胞の異なる濃度の得られたカプセル化効率はすべて濃度 (図 5B) 当社計算値を一致します。驚くことに、A549 腫瘍細胞塊を形成する傾向があるのような付着性のセルがあっても、1.0x106セル/mL (図 5C) の細胞濃度で若干改善されたカプセル化効率を観察しました。1.0x106セル/mL、3.0x106セル/mL と 13.0x106セル/mL (図 5D) の異なる細胞濃度でより少なく利用でき、不足している Pdc をカプセル化するためにシステムの有効性を検討しました。200 μ L を超える可能性がある大きなボリュームのロードを容易にするには、など、細胞またはより豊かな主な免疫細胞を扱う場合も調べた細胞カプセル化効率 1000 μ L ヒント (青) を使用。200 μ L のヒント (イエロー) (図 5E) と比較して似たようなカプセル化効率、これらの 1,000 μ L のヒントを与えたデモンストレーションを行った。
チップ設計と研究手での質問に依存して、細胞の不均一性にプロービングのための 1 つの入口または細胞間相互作用をデコードするための並列で複数の入り江をロードセルに私たちの技術をロードのヒントを使用できます。Jurkat T 細胞の 2 つの異なるラベル集団 1 つの入口から (10.0x106セル/mL の濃度) で Jurkat T 細胞の読み込みを行った (10.0 の結合された濃度で x106セル/mL) 2 つの入り江 (図 6からAと図 6B)。超低ゲル化温度の agarose を使用しておよびそれに続く下流の分析を介して顕微鏡とフローサイトメトリー (図 6 を許可フォーム agarose ゲル ビーズに製造後ゲル化、カプセル化の中に水滴が生成されました。 Cと図 6D)。顕微鏡による解析では、携帯のペアリングは、ハイスループット細胞 (図 6C) のペアリングを示すさまざまな組み合わせで達成されたことを明らかにしました。さらに、フローサイトメトリーによるハイドロゲル ビーズの同じ人口の分析は、ビーズから細胞ビーズをセルに基づいて異なる転送 (FSC、サイズ)、脇 (SSC、粒度) と分けることことを明らかにした散布パターン (図 6 D)。セルなしのビーズの人口にゲーティング蛍光信号の有無によってカプセル化細胞の欠如を確認しました。さらに、細胞ビーズ人口のゲートは異なるラベル Jurkat T 細胞のカプセル化の複数のサブ集団指標の存在を明らかにしました。効率的な電池の組み合わせに基づいて顕微鏡の両方達成することができますとフローサイトメトリー法による解析し、ポアソン予測 (図 6E) と比較して若干増加したカプセル化効率を示した示した。

図 1.3 つの入口と一つの出口の PDMS による液滴マイクロ流体デバイス。デバイスはそれぞれ連続油相の細胞培養媒体、細胞懸濁液の 3 つの入り江の構成します。生成された液滴は、出口で収集されます。サンプルの流れいるカプセル水滴の流れ中心の接合部に laminarly。入口、フィルター構造は、タンパク質のような大きな粒子を保持または細胞凝集塊のバックアップします。フィルター構造のギャップの直径は、青い線で示されます。生産ノズルでチャネルの直径は、赤い線で示されます。チップ全体の高さだった 25 μ mこの図の拡大版を表示するのにはここをクリックしてください。
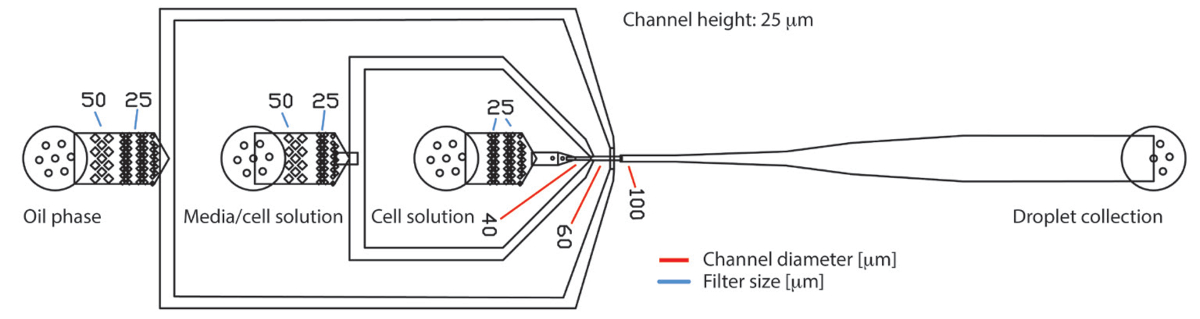{kind=link}

図 2.液滴安定 24 時間以上です。グラフは、液滴の界面活性剤濃度の異なる 3 つの時間の経過とともに造血無血清培地 + 2% 血清を含む領域を表示A) 0.5% B) 3% C) 5%。この図の拡大版を表示するのにはここをクリックしてください。
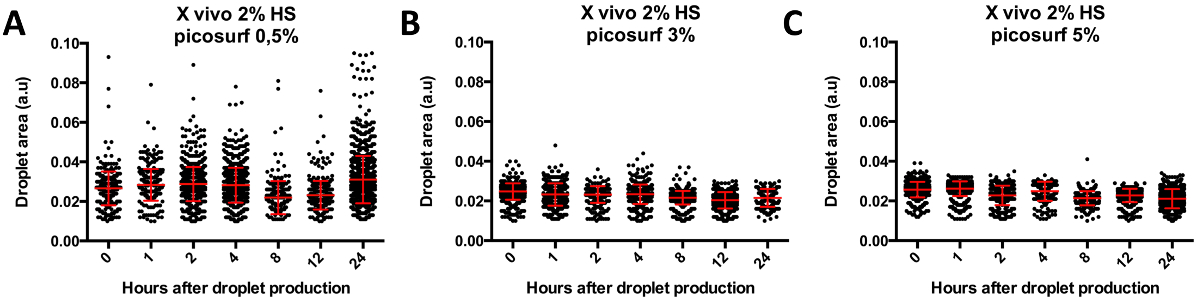{kind=link}

図 3.チューブは、イーガーローディングのアプローチのセルを基づいています。Jurkat T 細胞は注射器のチューブに接続されているを使用してデバイスに異なる濃度で読み込まれます。 A) 図は、実験のセットアップを示していますB)細胞カプセル化率光学顕微鏡検査によって決定されます。ドット: 測定値;線を閉じる: ポアソン分布。エラー バーは、平均値の標準誤差を表します。この図の拡大版を表示するのにはここをクリックしてください。
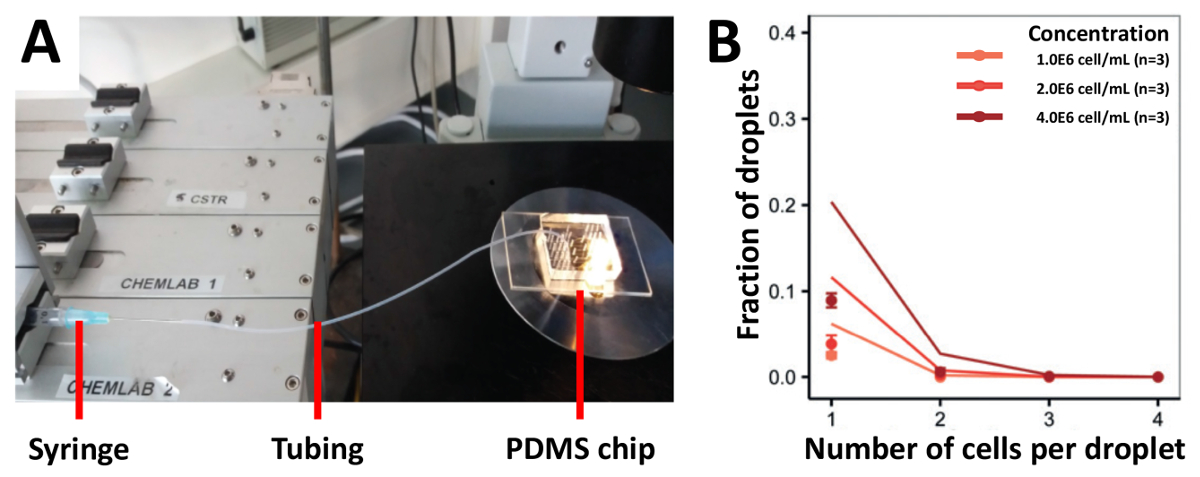{kind=link}

図 4.垂直円管内のイーガーローディングのアプローチを使用して異なる濃度で種々 の細胞のカプセル化します。Jurkat T 細胞と (濃度) の Pdc 携帯カプセル化の効率を判断する内部に閉じ込められました。A)図管内イーガーローディングのアプローチの実験のセットアップを示しています。B)セル Jurkat T のカプセル化効率を示します。C)グラフは、Pdc のカプセル化効率を示しています。ドット: 測定値;線を閉じる: ポアソン分布。エラー バーは、平均値の標準誤差を表します。この図の拡大版を表示するのにはここをクリックしてください。
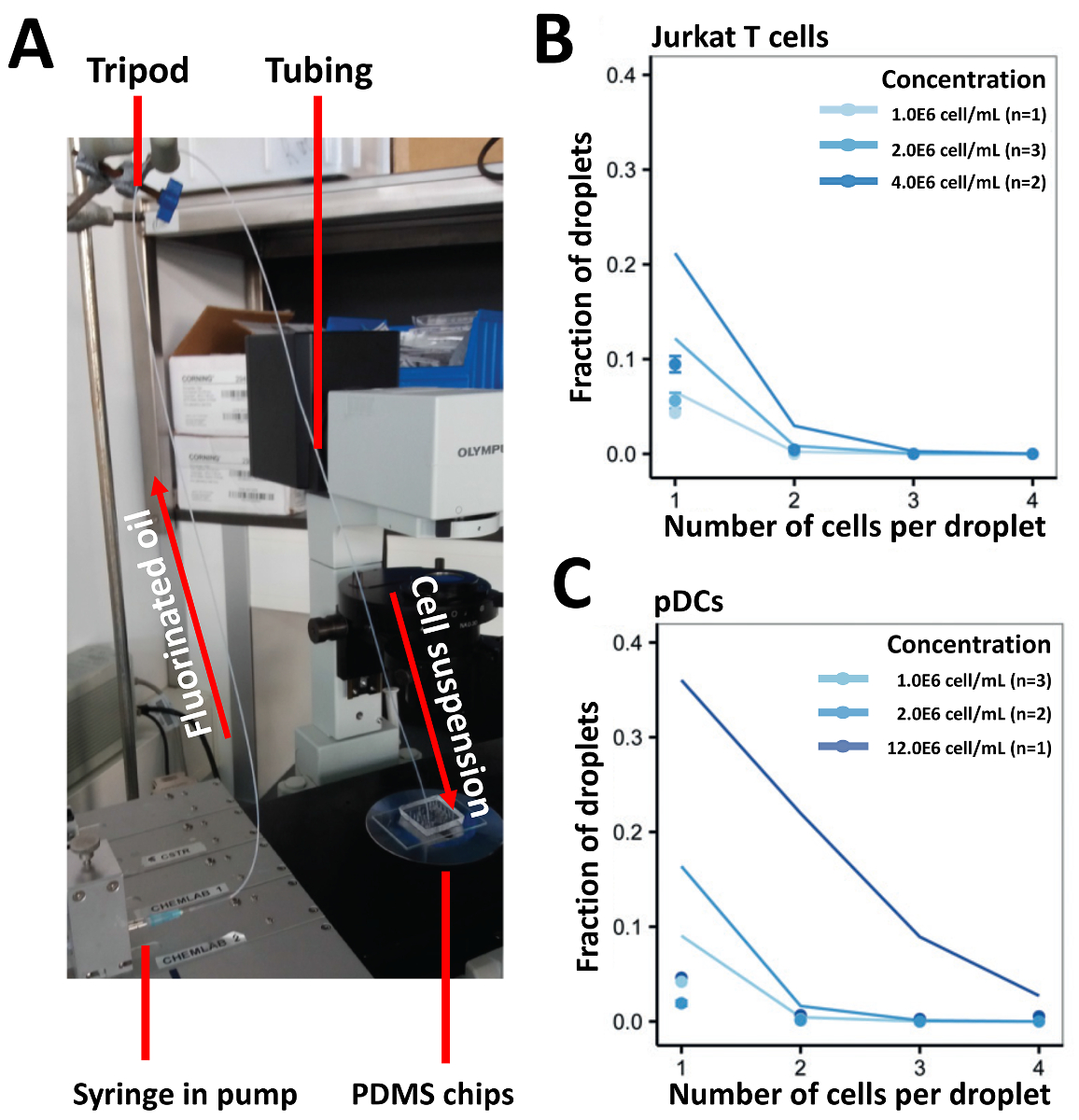{kind=link}

図 5.別のセルの種類をカプセル化するアプローチをロードをヒントします。A)技術の読み込みヒントの模式図。B) Jurkat 細胞のカプセル化効率を示します。C)グラフは、A549 細胞のカプセル化効率を示しています。D)グラフは、Pdc のカプセル化効率を示しています。E) 200 μ L ピペット チップ (黄色) および 1,000 μ L ピペット チップ (青) を使用してセル Jurkat T のカプセル化効率を示します。ドット: 測定値;線を閉じる: ポアソン分布。エラー バーは、平均値の標準誤差を表します。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 6.液滴のペアリングのセルです。A)液滴 2 入口から明瞭な細胞のペアリングのためのアプローチの読み込みヒントの模式図。B)グラフ 1 つの入口または並列で 2 つの入り江を使用してセル Jurkat T のカプセル化を示しています。1 つの先端の細胞濃度は 2.0x106セル/mL、2 つの細胞濃度のヒント両方 1.0x106セル/mL。ドット: 測定値;線を閉じる: ポアソン分布。C)ハイドロゲル ビーズと Jurkat T 細胞の蛍光顕微鏡オーバーレイ。D)グラフは agarose のゲルのビーズで対セル フローサイトを示しています。プロットは、前方散乱と脇の散布の両方を示します。E) agarose のゲルの細胞数の比較ビーズ蛍光顕微鏡とフローサイトメトリーによって検出されました。バー: 平均値;平均、n ≥ 4 のひげ: 標準誤差です。エラー バーは、平均値の標準誤差を表します。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
ディスカッション
このプロトコルでは、ロード、高スループット、単一細胞解析のためのセルをカプセル化と制御細胞の細胞間相互作用研究のためのペアリングを実行する効率的かつ簡単な方法を説明してきました。さらに、ロードセル マイクロ流体デバイスへのいくつかの従来の方法と比較して私たちのイーガーローディングのアプローチのヒントは他の方法と比較してより効率的な手法であることを示した。
勉強して臨床検体や珍しい携帯型マイクロ流体の液滴ベースで数で不足しているいくつかの固有の課題を持っています。行ったような細胞傾向にある注射器やチューブの表面の堆積物により、予測される値に適合するように細胞のカプセル化を防止します。この問題を回避するするには、いくつかのグループは、注射器で攪拌棒を使用します。ただし、レアな限定セル人口を使用して、総細胞体積も限られていることにより、大きな注射器の使用を制限してバーを攪拌します。さらに、我々 もテフロン被覆管の細胞接着を防ぐために一般的に使用されるチューブを交換が、このメソッドは結果を改善しなかったし、細胞付着の問題が (データは示されていない) を悪化させる場合はチューブが長すぎます。また、チューブでは大きな注射器の容積の細胞の損失を防ぐために注射器ではなく、細胞が読み込まれたしてイーガーローディングのアプローチ垂直管を使用しました。この手法を使用すると、少量のサンプル量で細胞、読み込まれるレアな限定である Pdcなど。また、チューブからサンプルが垂直方向にセル沈降を防ぐためにデバイスに読み込まれます。細胞播種用チューブは小型をマイクロ チャネルと比較することができます。チューブ内の流れは圧力駆動型を次の放物線速度のプロフィール26。これは、最大流速は管の中心には、最低速度は27のチューブの端を意味します。チューブ内のセルの人口を洗い流すときは、どこが落ち着く、境界で速度がゼロに近いのでエッジに向かってプッシュする細胞が発生速度勾配。沈降または管の細胞の決済により、予測モデルと実験のデータが一致しなかった、代表の結果に示すようにカプセル化効率が低下します。
液滴マイクロで働いている科学者によって使用される別の一般的適応ソリューションは、注射器19細胞沈降を防ぐために Iodinaxol などの試薬を一致する密度の添加による細胞培養媒体の密度を増やすことです。しかし、密度試薬に一致することができます携帯電話の動作に影響を与えるし、悪影響でセル (データは示されていない)28のサイトカイン分泌に影響を与える。
にもかかわらず、従来のセル技術の読み込みにいくつかの大小様々 な変更は、カプセル化効率のわずかな改善を示した実験結果まだ一致しませんでした理論計算。ただし、先端のイーガーローディングのアプローチで私達はポアソン統計による支配以前の方法とカプセル化効率の限界克服できた。この手法は浮遊細胞をロードするために有利なだけでなく、プライマリ ケラチノ サイトやマイクロ流体チップに A549 などの付着性細胞をロードするためにも適用できます。豊富な細胞、たとえば A549 K562等を使用する場合より大きなサンプル ボリュームを使用できます。したがって、サンプル量に応じて異なるサイズのピペット チップを使用もできます、この単純な手法は、単一セルのカプセル化とカプセル化セル複数合わせることができます。
低細胞濃度は、滴の単一セルのカプセル化を確保するため必要があるときは、各液滴セル ペアリングに関連する研究のためにカプセル化細胞の平均数を増やす細胞濃度が高いが望まれる.マイクロ流体チップの微細加工 nanowells29,30,31ペア免疫細胞に以前記載されている単一セル方法がいくつかあります。液滴マイクロ ポアソン統計によって決まりますその 1:1 セルを 2 つの異なる細胞型のペアリングは、最適な細胞濃度で実現できます。ポアソンの予測に基づいて、またある液滴が他の組み合わせを含むかもしれない確率。1:1 セルのペアリングは、単一セルのレベルで高められた細胞の理解の結果細胞間相互作用を検討することが望ましいことができますが、複数セルのペアリングの主要な利点があります。それは、他の細胞型の 1 つのセル型の複数のセルの影響を理解することができます。異なる免疫細胞間のクロストークは、いくつかの感染症や病原体に対する効果的な免疫応答を生成するヘルプし、も堅牢性を私たちの免疫システムの32に追加します。など、セルラー通信はな異なるコンテキスト、例えば1:1、2:1、1:2、2:2、3:1 などの高精度で尋問することができます。どのように単一の収量増加理解またはセルのペアは、免疫反応の誘導を制御します。これは研究など順次、それぞれの標的細胞を殺すナチュラル キラー細胞や細胞傷害性 T 細胞の能力に特に興味深い。
前述、水滴、セルの複数のカプセル化細胞濃度が高いが望まれます。ただし、セルのカプセル化のための 1 つの入口から細胞をロードするときセルのサンプルの高濃度は細胞を可能性が入口に集計します。これは、結果、カプセル化率が低い、理論値から偏差が高い。この問題を回避するするには、は、同様に 2 つの独立した入り江からセルをロードできます。理論的には、細胞の x 数の平均が保証されて電池封止材のさらに高いレベルを達成するために複数の入り江その他マイクロ流体デバイスを開発することが可能ででしょう。本研究では Jurkat T 細胞から読み込まれる 1 つの入口および同じ濃度を使用して 2 つの入り江と似たようなカプセル化効率を得られるカプセル化効率を調べた。この変更は、チップ上のペアの異なる細胞型の研究者をことができます。
このメソッドは、セル セルの重要な損失なしマイクロ流体デバイスへの読み込みでエイズ、念頭に置いておく必要があるある特定の注意があります。鉱物油とピペット チップの細胞サンプルを吸引、注射器を入力時に気泡の混入を避ける必要があり、システム全体をする必要があります無料の空気。また、鉱物油をサンプルと混同しないで心に保つことが重要です。含む試料、ピペット チップを漏れとさらに気泡の混入を防ぐために最大限の注意と、マイクロ流体デバイスの入り江にしっかりと挿入します。要約すると、チップの読み込みはコスト効果の高い方法で細胞の重要な損失することがなく細胞カプセル化を通じて細胞挙動の高スループット分析のためことができます簡単です、まだ、堅牢な技術です。ピペット チップを持つセルの読み込み、このアプローチは非常に柔軟性と異なる種類の細胞、特に珍しいの主な免疫細胞のカプセル化効率をすぐに取得するために合わせることができる入口に最適な試料濃度で使用する場合予測モデル。
開示事項
何を開示する必要があります。
謝辞
寛大な支援のアイントホーフェン工科大学に感謝いたします。
資料
| Name | Company | Catalog Number | Comments |
| 1H,1H,2H,2H-Perfluoro-1-octanol | Sigma-Aldrich | 171468-5G | |
| 1H,1H,2H,2H-Perfluorooctyltriethoxysilane | Fluorochem/UK | S13150 | Silane (toxic) |
| Agarose (Ultra-low Gelling Temperature) | Sigma-Aldrich | 9012-36-6 | |
| BD Wegwerpspuiten met Luer-Lok-punten | Fisher Scientific | 10630694 | Syringe |
| Biopsy Punch 1.2 mm | Harris Uni-Core | ||
| Cell Proliferation Dye eFluro 670 | eBioscience | 65-0840-85 | |
| CellTrace CFSE | Invitrogen | C34554 | |
| CellTrace Far Red Cell | Invitrogen | C34564 | |
| Eppendorf Tubes | Eppendrof Tubes | Safe-Lok tubes 1 mL and 2 mL | |
| Glass Slide | Sigma Aldrich | CLS294775X38-72EA | Corning microscope slides, plain L × W 75 mm × 38 mm |
| Harvard Pumps | Harvard Apparatus | C-400750; C-400727 | Syringe pumps |
| HFE-7500 3M Novec Engineered fluid | Fluorochem/UK | 51243 | Flourinated oil |
| Kai Biopsy Punch 5 mm | Amstel Medical | 1980130 | |
| Luer stub | Instechlabs/USA | LS20S | Luer stub, 20ga (pink) x 0.5in (12mm), non-sterile |
| Mineral oil (Light) | Sigma Aldrich | M8410-1L | |
| Phosphate buffered saline | Sigma-Aldrich | P4417-50TAB | Tablets |
| Pico-Surf 1 (5%in Novec 7500) | Sphere Fluidics | 020317-09 | Surfactant |
| Plasma Asher | Emitech | K1050X | Plasma asher |
| RPMI 1640 Medium | Gibco | 11875093 | |
| Silicone Elastomer Base 184 | Sylgard | 9355218 | PDMS base |
| Silicone Elastomer Curing Agent | Sylgard | 9355218 | Curing Agent |
| Stainless steel catheter coupler | Instechlab/USA | SC20/15 | 20ga x 15mm, non-sterile |
| TFE Teflon Tubing | Sigma-Aldrich | 58696-U | PTFE Tubing L × O.D. × I.D. 50 ft × 1/16 in. × 0.031 in. |
| Thinky mixer ARE-250 | EX-4025F | Conditioning mixture |
参考文献
- Yin, H., Marshall, D. Microfluidics for single cell analysis. Current Opinion in Biotechnology. 23 (1), 110-119 (2012).
- Meyvantsson, I., Beebe, D. J. Cell Culture Models in Microfluidic Systems. Annual Review of Analytical Chemistry. 1, 423-429 (2008).
- Yi, C., Li, C. W., Ji, S., Yang, M. Microfluidics technology for manipulation and analysis of biological cells. Analytica Chimica Acta. 560, 1-23 (2006).
- Wang, H. Y., Bao, N., Lu, C. A microfluidic cell array with individually addressable culture chambers. Biosensors and Bioelectronics. 24, 613-617 (2008).
- Zhang, Y., et al. A programmable microenvironment for cellular studies via. microfluidics-generated double emulsions. Biomaterials. 34 (19), 4564-4572 (2013).
- Teh, S. -. Y., Lin, R., Hung, L. -. H., Lee, A. P. Droplet microfluidics. Lab on a chip. 8 (2), 198-220 (2008).
- Hu, H., et al. Efficient cell pairing in droplets using dual-color sorting. Lab Chip. 15 (20), 3989-3993 (2015).
- Brouzes, E., et al. Droplet microfluidic technology for single-cell high-throughput screening. Proceedings of the National Academy of Sciences. 106 (34), 14195-14200 (2009).
- Chokkalingam, V., et al. Probing cellular heterogeneity in cytokine-secreting immune cells using droplet-based microfluidics. Lab on a Chip. 13 (42), 4740 (2013).
- den Haan, J. M. M., Arens, R., van Zelm, M. C. The activation of the adaptive immune system: Cross-talk between antigen-presenting cells, T cells and B cells. Immunology Letters. 162 (2), 103-112 (2014).
- Shah, G. J., Ohta, A. T., Chiou, E. P. -. Y., Wu, M. C., Kim, C. -. J. EWOD-driven droplet microfluidic device integrated with optoelectronic tweezers as an automated platform for cellular isolation and analysis. Lab on a Chip. 9 (12), 1732 (2009).
- Griffiths, A. D., Tawfik, D. S. Miniaturising the laboratory in emulsion droplets. Trends in Biotechnology. 24 (9), 395-402 (2006).
- Lagus, T. P., Edd, J. F. High-throughput co-encapsulation of self-ordered cell trains: cell pair interactions in microdroplets. RSC Advances. 3, 43 (2013).
- Moon, S., Ceyhan, E., Gurkan, U. A., Demirci, U. Statistical modeling of single target cell encapsulation. PLoS ONE. 6 (7), (2011).
- Abate, A. R., Chen, C. -. H., Agresti, J. J., Weitz, D. A. Beating Poisson encapsulation statistics using close-packed ordering. Lab on a Chip. 9 (18), 2628 (2009).
- Collins, D. J., Neild, A., deMello, A., Liu, A. -. Q., Ai, Y. The Poisson distribution and beyond: methods for microfluidic droplet production and single cell encapsulation. Lab Chip. 15 (17), 3439-3459 (2015).
- Kemna, E. W. M., et al. High-yield cell ordering and deterministic cell-in-droplet encapsulation using Dean flow in a curved microchannel. Lab on a Chip. 12 (16), 2881 (2012).
- Köster, S., et al. Drop-based microfluidic devices for encapsulation of single cells. Lab on a Chip. 8 (7), 1110 (2008).
- Mazutis, L., et al. Single-cell analysis and sorting using droplet-based microfluidics. Nature Protocols. 8 (5), 870-891 (2013).
- Sun, P., et al. Functional characterization of ex vivo. blood myeloid and plasmacytoid dendritic cells after infection with dengue virus. Virology. 383 (2), 207-215 (2009).
- Tel, J., et al. The chemotherapeutic drug oxaliplatin differentially affects blood DC function dependent on environmental cues. Cancer Immunology, Immunotherapy. 61 (7), 1101-1111 (2012).
- Wimmers, F., et al. Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nature Communications. 9 (1), 3317 (2018).
- Gong, J., Kim, C. -. J. All-electronic droplet generation on-chip with real-time feedback control for EWOD digital microfluidics. Lab on a Chip. 8 (6), 898 (2008).
- Demirci, U., Montesano, G. Single cell epitaxy by acoustic picolitre droplets. Lab on a Chip. 7 (9), 1139 (2007).
- Rho, H. S., Yang, Y., Veltkamp, H. -. W., Gardeniers, H. Direct Delivery of Reagents from a Pipette Tip to a PDMS Microfluidic Device. Chips and Tips. , (2015).
- Paul, P. H., Garguilo, M. G., Rakestraw, D. J. Imaging of Pressure- And Electrokinetically Driven Flows through Open Capillaries. Analytical Chemistry. 70 (13), 2459-2467 (1998).
- Whitesides, G. M., Stroock, A. D. Flexible methods for microfluidics. Physics Today. 54, 42 (2001).
- Mita, A., et al. Anti-proinflammatory Effects of Iodixanol (OptiPrep)-Based Density Gradient Purification on Human Islet Preparations. Cell Transplant. 19 (12), 1537-1546 (2013).
- Dura, B., et al. Profiling lymphocyte interactions at the single-cell level by microfluidic cell pairing. Nature Communications. 6 (1), 1-13 (2015).
- Dura, B., et al. Longitudinal multiparameter assay of lymphocyte interactions from onset by microfluidic cell pairing and culture. Proceedings of the National Academy of Sciences. 113 (26), 3599-3608 (2016).
- Yamanaka, Y. J., et al. Single-cell analysis of the dynamics and functional outcomes of interactions between human natural killer cells and target cells. Integrative Biology. 4 (10), 1175 (2012).
- Satija, R., Shalek, A. K. Heterogeneity in immune responses: From populations to single cells. Trends in Immunology. 35 (5), 219-229 (2014).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved