
Method Article
Rad51 を介した DNA 鎖交換反応のリアルタイム観察
要約
蛍光共鳴エネルギー移動に基づくリアルタイム観察システム DNA のストランドの Rad51 を介した交換反応が開発されました。形成反応中間体と酵素反応速度も分析中の製品への変換を検出することができるここに示すプロトコルを使用しております。
要約
Rad51 を介した DNA 鎖交換反応は、相同組換えの重要なステップです。この反応で Rad51 は単一座礁させた DNA の核タンパク質フィラメントを形成し、非具体的に相同の調査に二本鎖 DNA (dsDNA) をキャプチャします。相同性が発生して後、は、Rad51 は、dsDNA の相補的な鎖 ssDNA のペアリングを仲介する DNA 鎖交換を触媒します。この反応は生体内で多数アクセサリー蛋白質によって厳しく規制されています。中間体の形成と最終製品にその進行の速度論的解析のために挑戦的な証明されていますこのようなアクセサリ蛋白質の in vitroの役割を調べるための従来の生化学的アッセイの採用が正常にされているが、反応中間体の不安定および一時的な性質。手順をソリューション、蛍光共鳴エネルギー移動 (FRET) に直接反応を観察する-リアルタイム観測のこの反応のシステムが確立されました。リアルタイム観測の速度論的解析を示します中間三本鎖 DNA の形成、この中間体の成熟とから ssDNA のリリースを含む 3 段階反応モデルを介した Rad51 DNA 鎖交換反応に従う成熟した中間値。Swi5 Sfr1 複合体、真核生物で保存されているアクセサリー蛋白質は強くこの反応の 2 番目と 3 番目の手順を強化します。ここで紹介するフレットに基づく試金はどの再結合を介してアクセサリー蛋白質 Rad51 DNA 鎖交換活性を刺激する分子メカニズムを明らかにする私たちを有効にします。このプロトコルの主な目的は、分裂酵母、以外の種からの蛋白質で働く特にそれら相同組換えの分野の研究者が利用できるテクニックのレパートリーを強化するように、ここに記載した所見の進化的保存を決定ことができます。
概要
相同組換え (HR) は、2 つの異なる DNA 分子間の遺伝情報のシャッフルを促進します。HR は 2 つの基本的な生命現象に不可欠な: 有糸分裂の区切り (Dsb)2が配偶子形成1遺伝的多様性の生成と DNA 二重鎖の修理。Dsb は DNA 損傷の最も深刻な形で、染色体の破損を構成します。がん3の両方の特徴である豊富な染色体再配列とゲノム不安定性、Dsb の不適切な修理があります。
DNA 鎖交換反応は、HR の中央のフェーズです。リコンビナーゼの非常に節約された RecA 型家族のメンバーは、Rad51 タンパク質は真核生物4,5でこの反応を触媒する重要なタンパク質です。この反応で Rad51 に一本鎖 DNA (ssDNA) DSB 末修飾アミノ酸処理によって生成される、フォーム複雑なヘリカル蛋白と呼ばれるシナプス前繊維。このフィラメントをキャッチそのまま二本鎖 DNA (dsDNA) 特異的相同シーケンスを検索します。フィラメントには、相同のシーケンスが検出されると、3 本鎖 DNA を含む中間反応の形成し、Rad51 フィラメントがこの構造6,7,8鎖交換を仲介します。この反応を効率的に行うためには、Rad51 BRCA1 と BRCA2、乳房がん感受性遺伝子9,10の製品などアクセサリ蛋白質のいくつかの種類が必要です。
理解アクセサリー要因は、Rad51 を調節する方法で腫瘍ゲノム不安定性の原因を暴くに不可欠なステップです。多くの研究はシナプス前繊維形成と安定性11,12,13,14,15,16, これらの要因の影響にかかわっているが、3 つの鎖の中間と最終製品に処理の形成にこれらの要因の貢献はまだ不明です。3 つの鎖の中間体は不安定でサンプルの除たんぱくなど一般的な実験操作によって崩壊しやすいために、従来の生化学的な実験を通してこれらの反応の手順を観察することは非常に困難または電気泳動。
この問題を克服するために我々 は DNA 鎖交換反応蛍光共鳴エネルギー移動 (FRET) を使用して 2 つの開発済みのリアルタイム観測システム、適応: DNA 鎖のペアリングおよび DNA 鎖変位試金17、 18 (図 1)。ペアリングのアッセイは、Rad51 フォーム フルオレセインを用いたシナプス前繊維 dna 鎖交換反応を開始する amidite (FAM) - ラベル ・ ssDNA、相同カルボキシ-x-ローダミン (ロックス) - dsDNA のラベルが追加されます。フィラメントは ROX ラベル dsDNA をキャッチし、3 つの鎖の中間体を形成する、2 つ同時が近くに来るし、ROX (図 1 a)、FAM の蛍光性の放出を急冷します。DNA 鎖の変位アッセイでラベルのない一本鎖 Dna 上に形成されたシナプス前繊維は FAM とロックス二重ラベル dsDNA と培養です。鎖交換が完了し、一本鎖 Dna を標識 FAM が 3 つの鎖から解放される中間、FAM の増加の排出 FAM はロックス (図 1 b) に近くないので。これらの試金は 3 つの鎖の中間および最終製品に処理の形成を観察することを有効にする反応に乱れる事なくリアルタイム。
このリアルタイム観測システムを使用して、Rad51 を介した DNA 鎖交換反応が最初反応中間体 (C1) の形成を含む、最初の中間の第二の中間に移行中で 3 ステップで進むことがわかった(C2) と C219から ssDNA のリリース。また分裂酵母 (S. 酵母) がわかった Swi5-Sfr1、進化上保存された Rad51 アクセサリ蛋白質複雑な13,16,20,21,22である刺激C1 C2 の移行、Rad5119によって ATP 加水分解に依存している方法で C2 から ssDNA のリリース。
これらの調査結果は進化的保存されているかどうかは不明のまま。HR は、特にこれらの作業s.pombe、以外の生物からの蛋白質の分野の研究者がどの程度を決定するこれらの技術を適用されることを期待してこのプロトコルを提供 Rad51 駆動の分子機構鎖交換は保存されています。さらに、これらの手法は, 分裂酵母Swi5 Sfr1 の役割を決定する上で非常に成功した証明しました。したがって、これらの技術は HR アクセサリー等の精密な役割を暴くで貴重なできない合理的な予測です。
プロトコル
1. タンパク質と Dna の準備
- 以前に報告された13,21(クマシー染色によって判断される)、同質性にs.pombe Rad51 と Swi5 Sfr1 の蛋白質を浄化します。
- 表 118に掲げるオリゴヌクレオチド dna を準備します。
注: オリゴヌクレオチドを購入した (材料の表を参照してください)、HPLC グレードの合成します。ペアリング反応、オリゴヌクレオチド DNA の繊維の 16FA(-) 16 a (-) _40bp ・ 16AR (+) _40bp が必要。DNA の変位の試金、オリゴヌクレオチドのため 16A(-)、16FA (-) _40bp と 16AR (+) _40bp が (図 1と表 1) が必要です。このプロトコルのすべての DNA 濃度は塩基濃度ではなく濃度のフラグメントを参照してください。 - ドナー dsDNA を形成するには、薄肉 PCR チューブ内の補足の繊維の等モル量のアニーリング バッファー (10 mM トリス塩酸 pH 7.5、100 mM の NaCl、10 mM MgCl2) を混ぜて、この上 prechilled 金属ラックでミキシングを行います 20 μ L より大きい容量を確保(2 ° C および 4 ° C) の間氷します。
注: ペアリングの試金のためのオリゴヌクレオチドの組み合わせが 16 a (-) _40bp ・ 16AR (+) _40bp。変位試金のためのアニール オリゴヌクレオチド 16FA (-) _40bp ・ 16AR (+) _40bp。 - 5 分間 90 ° C で熱処理の混合物を熱し、PCR 機械を使用して 30 の ° c 3 時間以上冷やします。-20 ° C で熱処理した DNA を保存します。
2. DNA ストランド ペアリング及び変位測定
- Dna 分析をペアリングを実行します。
- 1.6 mL の反応バッファー A の準備 (30 mM HEPES 島 pH 7.5、0.0075% polyoxyethylenesorbitan monolaurate、0.1 mg/mL ウシ血清アルブミン [BSA] 0.25 mM ATP 15 mM MgCl21 mM ジチオトレイトール [DTT]) 36 を含む nM 2.0 mL マイクロ遠心分離機の 16FA(-)プラスチック チューブ (ポリプロピレン) 前 5 分の 37 ° C で孵化させなさいと。
- SsDNA Rad51 フィラメントを形成するには、前培養反応バッファーに 1.5 μ M の最終的な集中に Rad51 タンパク質を加えるし、5 分の 37 ° C でそれを孵化させなさい。
- 0.15 μ M の最終濃度に混合物に Swi5 Sfr1 タンパク質を加えるし、さらに 5 分の 37 ° C でそれを孵化させなさい。
- 混合物の 1.5 mL を取ると磁性攪拌器を含む 1.0 × 1.0 cm 石英キュベットにそれを転送し、蛍光にキュヴェットを設定します。分光光度計 37 ° C のペルチェ温度コント ローラーを構成し、マグネチックスターラーを注入されたサンプルの急速な混合を確実に 450 rpm にセットします。
- 525 でファムの蛍光性の放出の測定を開始 nm (帯域幅: 20 nm) 493 で励起による nm (帯域幅: 1 nm)。毎秒データを収集します。
- 100 測定開始後 s、36 の最終的な集中に ROX ラベル ドナー dsDNA の注入でさらに 30 分の 1 秒間隔で発光のシリンジとメジャー変更使用混合物に nM。
- DNA ストランド変位測定を実行します。
- 1.6 ml 反応バッファー A の 36 を含む nM 16A(-) 2.0 mL マイクロ遠心分離機プラスチック チューブ前 5 分の 37 ° C で孵化させなさいと。
- 2.1.2 の手順で説明するよう、37 ° C で Swi5 Sfr1 の存在下で ssDNA Rad51 フィラメントを形成します。2.1.3。
- 混合物の 1.5 mL を取るし、磁性攪拌器を含む石英キュベットに転送 2.1.4 の手順で説明するように、分光光度計にキュヴェットを設定します。
- 蛍光発光を測定を開始前後 100 s、2.1.6 の手順で説明するようにドナー dsDNA の FAM と ROX ラベルを挿入します。さらに 30 分の 1 秒の間隔で蛍光性の放出の変化を測定します。
3. ペアリングから実験データの分析及び変位測定
- 最大の FRET 効率を推定します。
- 16AR 熱処理した 16FA(-) を準備 (+) _40bp と 16FA 16AR と焼鈍 (-) _40bp (+) _40bp 1.3 と 1.4 の手順で説明する同じ手順を使用して。
- 130 μ A の反作用バッファーを準備 36 を含むいずれかの 16FA(-) の nM、16FA(-) 16AR 熱処理した (+) _40bp、16FA 16AR と焼鈍 (-) _40bp (+) _40bp または 16FA 0.2 × 1.0 cm 石英キュベットで (-) _40bp。
- キュベットを蛍光にセットし、5 分の 37 ° C で孵化させなさい。
- 500 から 600 の蛍光スペクトルを測定 493 で励起による nm nm。
- Rad51 の FAM の放出と ROX でファムの消炎に及ぼす影響をテストするための混合物に 1.5 μ M の最終的な集中に Rad51 を追加し、5 分の 37 ° C で孵化させなさい。
- 500 から 600 の蛍光スペクトルを測定 493 で励起による nm nm。
- 最大 FRET 効率 (E最大) に以下の式を使って計算します。
E最大= (525 で蛍光強度 FAM と ROX ラベル dsDNA の nm)/(525 で蛍光強度 FAM の nm ラベル ssDNA)
- 変位測定から実験データを分析します。
- 最終的な製品量の変化を変位測定で観測された蛍光性の変更を変換は、以下の式を使用してこの分析から得られた生の実験データを正規化、生F はどこからの蛍光強度生データと F正規化された次の式によって計算される蛍光が変化するです。
正規化されたF = ([F生時 x]-[F生時 0])/(([F生時 0]/E最大)-[F生時 0])
F生時間 0 では、最初の 5 監視平均蛍光 (すなわち、混合キュヴェットに、乳液を導入後に必要な時間) のデッドタイム後 s。 - 退色と自発的な量の影響を除外するには、F正規化から F正規化されたこのアッセイで最終的な製品量の変化である FDを取得するサンプルの蛋白質がなければを減算します。
FD = [F正規化されたサンプルの] - [F正規化された蛋白質がなければ]
- 最終的な製品量の変化を変位測定で観測された蛍光性の変更を変換は、以下の式を使用してこの分析から得られた生の実験データを正規化、生F はどこからの蛍光強度生データと F正規化された次の式によって計算される蛍光が変化するです。
- ペアリングの試金からの実験データを分析します。
- ここで生F は raw データから F正規化蛍光強度は次の式によって計算される蛍光の変化、以下の方程式を使ってペアリングの分析から得られた生の実験データを正規化します。
正規化されたF = (F生時 x)/(F生時 0)
F生時 0 は最後の 20 の監視平均蛍光 dsDNA 基板を挿入することにより反応を開始する前に s。 - 基板の量の変化に蛍光性の変更を変換と退色と自然ペアリングの影響を除外、F正規化された以下の方程式を使用してサンプルの正規化、どこ FPは発生量の変化この試金の基板。
FP = 1 - (([F正規化タンパク質なし] - [F正規化されたサンプルの])/[1 E最大]) - DNA 鎖交換反応速度を調べる解析プログラム23を用いたペアリング反応の非線型の最後広場回帰分析を行う (材料の表を参照してください)。
- FPの経時変化データを含む .txt 形式のファイルを準備します。
- プログラムを起動し、プログラムのウィンドウに補足のコード ファイルにスクリプトを貼り付けます。
- 最後の広場の非線型回帰分析を開始します。この分析の結果は、同じウィンドウに表示されます。
- ここで生F は raw データから F正規化蛍光強度は次の式によって計算される蛍光の変化、以下の方程式を使ってペアリングの分析から得られた生の実験データを正規化します。
結果
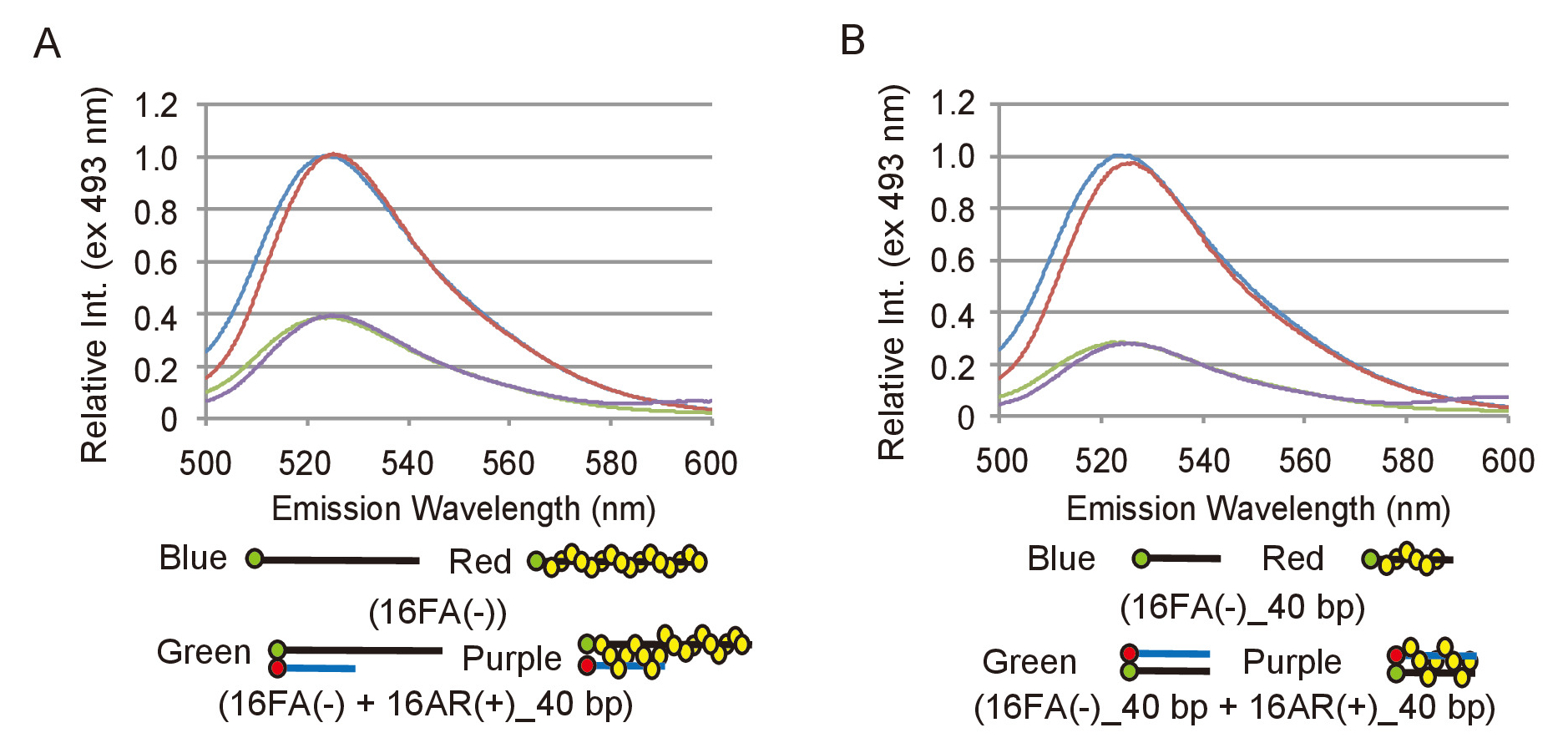
ペアリングと変位の測定から実験的データを効果的に分析するために、FAM の蛍光性の放出の変化が dna の製品への変換に対応するかを定義する必要です。これを達成するため、蛍光強度の適切な範囲を決定する必要があります。ペアリングの試金のため ssDNA 基板に対応する、16FA(-) の蛍光性の放出は熱処理した 16AR 16FA(-) の排出量と比較して (+) _40bp (図 2 a) この反作用の最終製品に対応します。これは最大の FRET 効率に相当し、はそれゆえの蛍光強度の最大の削減、すべて ssDNA 基板は、dsDNA 製品に変換されたかどうかに予想されます。変位試金、16FA の排出のため熱処理した 16AR (-) _40bp (+) 基板に対応する、_40bp は 16FA の排出量と比較して (-) _40bp 最終製品 (図 2 b) に対応します。この場合、FAM の蛍光強度の最大の増加は、dsDNA 基板のすべては ssDNA 製品に変換されますシナリオを伝えています。S.pombe Rad51 はファムや焼入れ両測定法 (図 2) で ROX でファムの効率の蛍光性の放出には影響しなかった.オリゴヌクレオチドのラベリングの効率に依存している最大の FRET 効率オリゴヌクレオチドの各の新しい準備と再測定はずです。
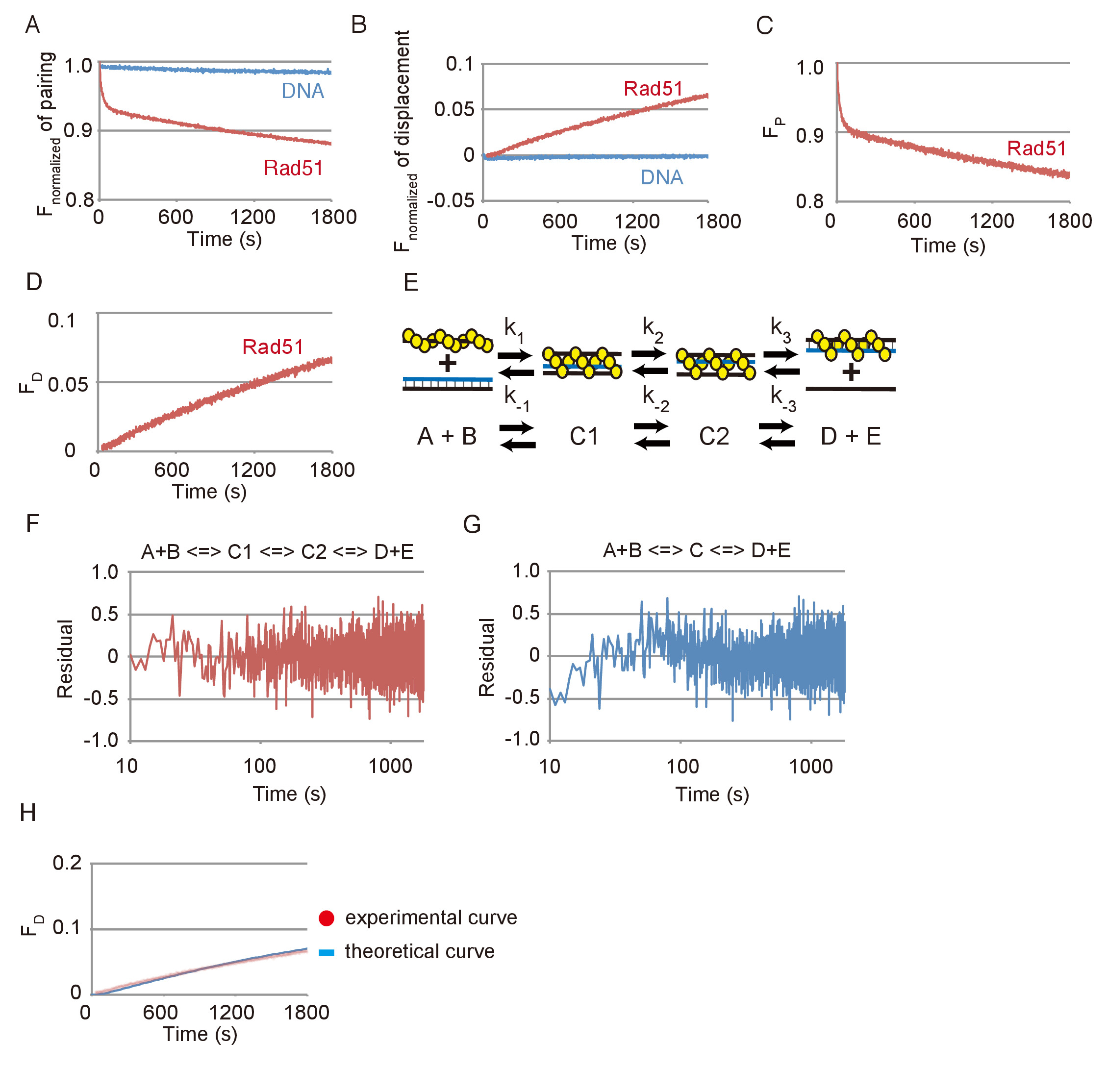
DNA 鎖のペアリングと変位の反応の代表的なデータは、図 3のとおりです。Rad51 Rad51 見て相当な変化と比較することがなく FAM の排出に見られるごくわずかな変更によって明らかにされた基板 DNAs と退色の自発的反応の効果は両方のアッセイで小さかった (図 3 aと図 3 b)。図 3 aまたは3 b 図で示されているデータに基づいて、蛍光の変化に変換された基板 (FP) または最終製品 (FD) の量の変化、それぞれ手順 3.2.2 又は 3.3.2 (で説明した方程式を使用して図 3と図 3 D)。
ペアリングの反応は、順次 3 段階反応モデルでは、最初の 3 つの鎖の中間 (C1) の形成から成る、第二の中間に最初の中間の移行を使用してシミュレートしました (C2 に C1) と ssDNA のリリース(D + E) の 2 つの製品を形成する第二の中間から (図 3E)。シーケンシャルの 3 段階反応モデル フィットに実験データを用いたシミュレーション実験データ分析とシミュレーションによって得られた理論曲線をペアリング DNA 鎖の間の残差だったかどうかをテストするのには (図 3 f) を計算します。さらに、ペアリングの反応とシーケンシャル 2 段階反応モデルを使用して生成される理論曲線と残差がまた計算される (図 3) があった。ペアリングの反応、2 段階モデルの残差は、ペアリングの反応、3 段階モデルの残差は、このような偏差を表示しないに対し、早期の系統的偏差を表示します。これは 3 段モデルがペアリングの反応をシミュレートするための 2 段階モデルよりもより良いフィットであることを示します。
ペアリングのシミュレーションから得られた速度論的パラメーターを使用して置換反応の理論曲線を生成した 3 段モデルは DNA 鎖交換後半のステップを検出する置換反応と一致しているかどうかをテストするのには反応図 3に示すように、 3 D 図(図 3 H) で示されている置換反応の実験データと比較しました。理論曲線に合わせて変位測定法の実験データ。これらの結果から 3 段階モデルを用いたシミュレーションは Rad51 を介した DNA 鎖交換反応、合理的に評価することができることが分かった.
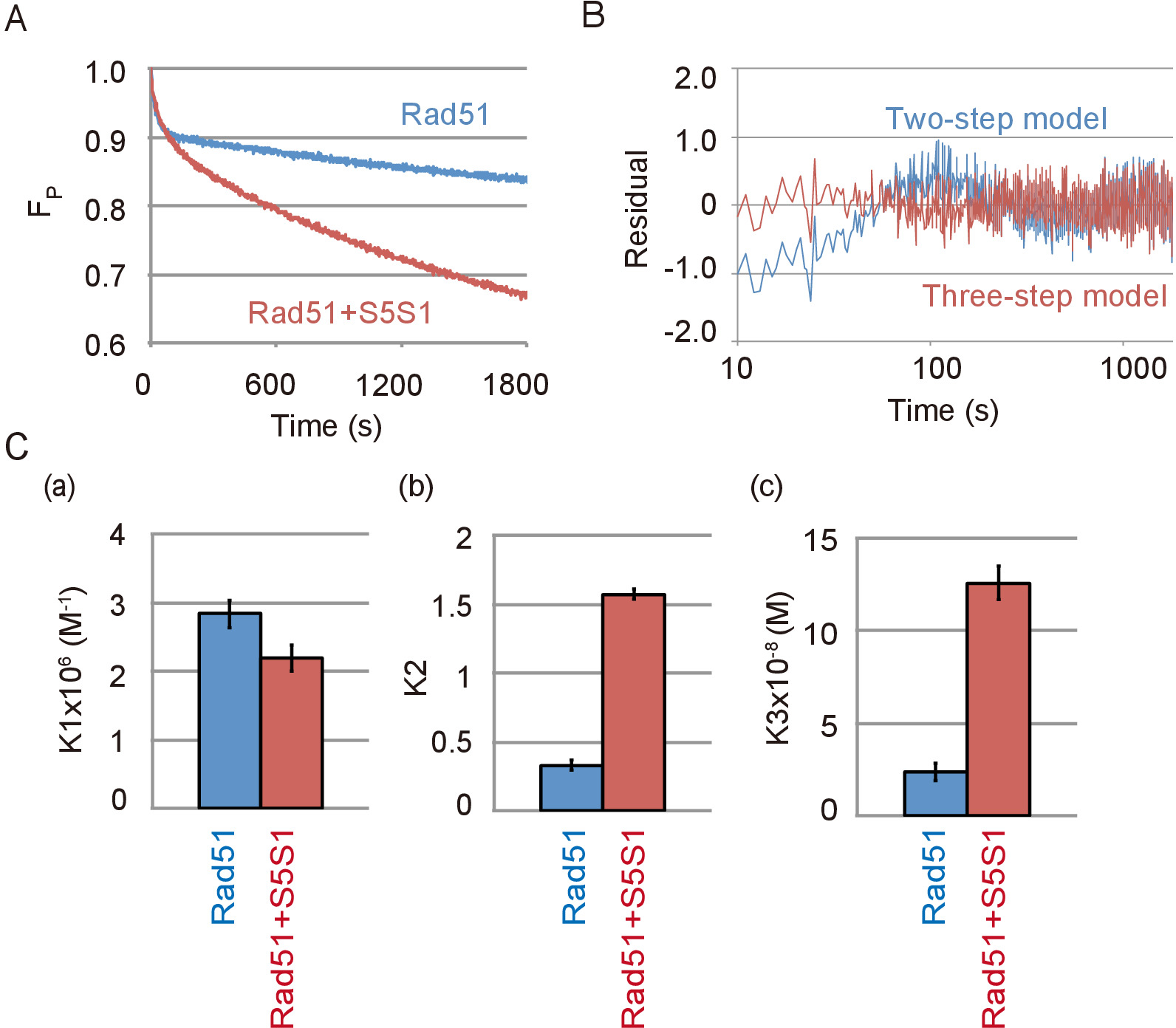
Swi5 Sfr1 複合体、Rad51 のアクセサリー蛋白質 Rad51 を含む反応をペアリング DNA 鎖の代表的なデータは、図 4 aに表示されます。Swi5 Sfr1 複合体は、Rad51 のペアリングの活動を強く刺激。Swi5 Sfr1 の不在で見られたようにペアリングの反応は 3 段階モデルを Swi5 Sfr1 (図 4 b) の存在下での 2 段階モデルよりもより良いフィット。3 段階のモデルを用いた反応のシミュレーションにより各反応ステップ Swi5 Sfr1 の有無の反応平衡定数を算出しました。Swi5 Sfr1 複合体が最初の反応ステップを刺激しない反応の平衡定数が示されます (図 4、パネル、) の 3 つの鎖の中間が形成されるが、C1 C2 の移行 (を刺激して強く図 4、パネル b) と C2 中級 (図 4パネル c) から ssDNA のリリース。

図 1: DNA の実験的なデザインのストランドのペアリングと変位のアッセイ。ペアリング (A) ストランドの DNA の模式図と変位 (B) 試金。黄色の円は、Rad51 モノマーを表しています。緑色の円盤の含んでいる"F"、"R"を含んでいる赤い円はフルオレセイン amidite (FAM) とカルボキシ-X-ローダミン (ロックス) をそれぞれ表します。黒 dna は同一シーケンスの青の DNA 鎖に相補的です。表 1に示すように、矢印で細い黒線は各オリゴヌクレオチドの名前をポイントします。この図は、伊藤らから適応されています。19と変更されました。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 2: ペアリングと変位の試金の最大の FRET 効率の測定。(A) ssDNA 基板、16FA(-)、dsDNA 製品の蛍光スペクトルの比較、16FA(-) 16AR とペアになって (+) ペアリング アッセイの _40bp。青と赤の線は、Rad51 と基板の蛍光スペクトルをそれぞれ表します。緑と紫の線はそれぞれ Rad51 と最終製品の蛍光スペクトルを示しています。(B) 16FA dsDNA 基板間の蛍光スペクトルの比較 (-) _40bp 16AR とペアになって (+) _40bp、および ssDNA 製品 16FA (-) _40bp、変位測定の。青と赤の線は、Rad51 と最終製品の蛍光スペクトルをそれぞれ表します。緑と紫の線はそれぞれ Rad51 と基板の蛍光スペクトルを示しています。この図は、伊藤らから適応19と変更されました。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 3: Rad51 を介した DNA 鎖のペアリングと変位の反応。(A) ペアリング反応 Rad51 の有無の正規化された蛍光の時間コース。(B) 置換反応 Rad51 の有無の正規化された蛍光の時間コース。(C)Rad51 とペアリングの反応の基質の量の変化の時間コース。(D) Rad51 と置換反応の基質の量の変化の時間コース。(E) A シーケンシャル 3 段階反応モデルの模式図。A と B は、シナプス前のフィラメントとドナー dsDNA に対応します。C1 は、最初の (未熟な) 3 つの鎖の中間に相当します。C2 は 2 番目 (成熟した) 3 つの鎖の中間に相当します。D と E は、ヘテロ二本鎖と ssDNA C2 からリリースに対応します。(FおよびG) を 3 つのステップ (F) または (G) の 2 段階モデルを用いたシミュレーションによって得られた実験データ分析と理論曲線をペアリング DNA 鎖の間の残差。(H) 赤い点線は Rad51 + 青パネルに示すように、行は最終製品の理論曲線を示しますと置換反応の実験データ。理論上の曲線は、パネルCに示すようにペアリングの分析から得られる反応速度定数を用いたシミュレーションによって生成されました。この図は、伊藤らから適応19と変更されました。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 4: Swi5 Sfr1 は、2 番目を刺激し、DNA の 3 番目のステップ鎖交換反応。(A) ペアリング反応 Swi5 Sfr1 (S5S1) の有無で基板量の変化の時間コース。Swi5 Sfr1 の試金 DNA ストランド ペアリングの実験データの間 (B) 残差と 2 段階 (青線) または 3 つのステップ (赤ライン) モデルを用いたシミュレーションによる理論曲線が得られます。4 a を図に示すように (C) ペアリング反応模擬分析プログラム23を使用して、3 つのステップ モデル (材料の表を参照してください)。各反応のステップ、K1 の反応の平衡定数 (、)、(b)、K2、K3 (c) シミュレーションから得られました。この図は、伊藤らから適応19と変更されました。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
| ペアリングの試金 dna のオリゴヌクレオチド | ||||
| 16FA(-) | 5'-[ファム] - AAATGAACATAAAGTAAATAAGTATAAGGATAATACA AAATAAGTAAATGAATAAACATAGAAAATAAAGTAAAGGATAT AAA-3' | |||
| 16A (-) _40bp | 5'-AAATGAACATAAAGTAAATAAGTATAAGGATAATACAAAA-3' | |||
| 16AR (+) _40bp | 5 '- TTTTGTATTATCCTTATACTTATTTACTTTATGTTCATTT-[ロックス]-3' | |||
| DNA のオリゴヌクレオチド鎖変位測定 | ||||
| 16A(-) | 5'-AAATGAACATAAAGTAAATAAGTATAAGGATAATACAAAATA AGTAAATGAATAAACATAGAAAATAAAGTAAAGGATAT AAA-3' | |||
| 16FA (-) _40bp | 5'-[ファム] - AAATGAACATAAAGTAAATAAGTATAAGGATAATACAAAA-3' | |||
| 16AR (+) _40bp | 5 '- TTTTGTATTATCCTTATACTTATTTACTTTATGTTCATTT-[ロックス]-3' | |||
表 1: オリゴヌクレオチド DNA 鎖のペアリングと変位試金で使用されるの一覧。フルオロ (フルオレセイン amidite、FAM; カルボキシメチル-x-ローダミン、ROX) の位置に示されている該当する場合は、かっこを正方形します。
ディスカッション
ここでは、Rad51 主導 DNA 鎖交換の実時間を計測するフレットを利用して詳細なプロトコルを説明しました。重要なは、これらの測定は、反応速度の決定を許可します。上記説明は、私たちの公開されている結果を再現するための十分なこのセクションで説明するいくつかの重要なポイントがあります。さらに、利点と DNA 鎖交換留学フレット ベースの方法論の欠点は、後ほど、DNA 代謝の他の側面を勉強するような技術のアプリケーションと一緒に。
すべての生化学的 reconstitutions 確実に高純度の基質反応すべて不可欠です。クマシー染色によって判断される蛋白質の準備の明白な純度にもっぱら基づいて汚染活動の不在を仮定する過失があります。特に、微量核酸またはヘリカーゼの存在は徹底的にペアリングと変位のアッセイの結果に影響を与えます。したがって、タンパク質の新しいバッチを精製するたびにこのような活動のテストをおすすめします。さらに、ネイティブ ポリアクリルアミドゲル電気泳動法によって合成された DNA の純度を確認することをお勧めします。にもかかわらず、多くの企業は、オリゴヌクレオチドの純度を保証する、独自のテスト合成 DNA の純度はバッチによって異なりますを通して見えてきた頻繁。
石英キュベットで実験を行う場合次の 2 つの点を考慮することが重要です。まず、いくつかのタンパク質は、無指定石英キュベットに結合する傾向があります。このカウンターは、BSA と polyoxyethylenesorbitan monolaurate は反応バッファーに含まれます。第二に、温度は、反応速度と蛍光強度の抜本的な影響を及ぼします。、この効果を最小限に抑える石英キュベットは前を使用する前に 37 ° C で培養する必要があります。
従来の生化学的アッセイは、DNA 鎖交換の勉強に非常に有用されている、いくつかの欠点がある.代表的な時間コース実験で反応はある特定の温度で培養した、因数は必要な縦長で撤回、洗剤と反応を終了するプロテアーゼ処理による除タンパクしました。時間コースを修了すると、サンプル、製品から dna を分離する電気泳動にさらされます。ここで説明する方法の主な利点は、反応妨害なしのリアルタイムで観察できるということです。反応中に任意の timepoint を自体反応を伴わず検査し、サンプルを deproteinize または電気泳動の潜在的に破壊的な力にそれらを服従する必要はありません。不安定な DNA の構造を監視しているとき、これは特に関連します。
にもかかわらず、従来の試金をこれらの強みは、ここで説明したメソッドがいくつかの欠点。オリゴヌクレオチド DNA の鎖交換用簡単結果の解釈になりますが、このような基板がセルの人事に関与する dna に似ていないことを覚えていることが重要です。いくつかの従来のアッセイを利用プラスミド サイズ DNA 基板は塩基数交換で体内を反映する可能性が高い。さらに、従来の試金のサブセットの位相制約付き円形 dsDNA 基板の使用は少なくとも部分的に生理学的 DNA の張力を再作成できます。
ここで説明したメソッドのアプリケーションは、Rad51 主導 DNA 鎖交換機構の解明し始めています。それにもかかわらず、回答が残る多くの興味深い質問があります。Rad51 と真核生物24減数分裂固有 RecA 型シュプリンガー Dmc1 が減数分裂期人事に必要とすることの明確な証拠があります。ただし、これら 2 つのリコンビナーゼ違いの主要な生化学の不足年の分野の研究者に困惑しています。また、組換えアクセサリー要因の多数の明瞭なグループの役割は、人事の分野における研究の焦点のトピックをされています。Rad51 と Dmc1 の生化学的違いを解明に加えて、調査し、近い将来における DNA 鎖交換機構の異なる組換えアクセサリー要因の効果を比較を目指します。最後に、ここで説明したフレット ベースの方法論、DNA 鎖交換の研究に限定されないことを強調することが重要です。比較的マイナーな変更とは、さまざまな DNA 代謝25,26,27,28機能的多様性タンパク質を調査してこの技術の応用を想定します。我々 は、ここで説明する開発はさらに多くの異なる分野に属する研究者にオプション提供を願っています。
開示事項
著者が明らかに何もありません。
謝辞
この作品によって賄われていた補助金科学研究 (A) (18 H 03985) と革新的な分野 (05974 15 H) には、hi 若手研究 (B) (17 K 15061)、ba との科学的研究 (B) (18 H 02371) HT から日本学術振興会 (日本学術振興会)。
資料
| Name | Company | Catalog Number | Comments |
| 0.2 x 1.0 cm quartz cuvette | Hellma Analytics | 105-250-15-40 | |
| 1.0 x 1.0 cm quartz cuvette | Hellma Analytics | 101-10-40 | |
| adenosine triphosphate (ATP) | Sigma | A2383 | |
| DynaFit | BioKin, Ldt. | DynaFit is a program to analyze kinetics of biochemical reactions. | |
| Fluorescent labeled and non-labeled oligonucleotides | Eurofins Genomics | The sequences of oligos are listed in Table. 1. | |
| Magnetic stirrer | Aisis (Japan) | CM1609 | |
| PCR machine | TAKARA (Japan) | TP600 | TAKARA PCR Thermal Cycler Dice |
| Spectrofluorometer | JASCO | FP8300 | Contains a peltier temperature controller and magnetic stirrer system |
| Syringe | HAMILTON | 1702RN 25ul SYR (22s/2"/2) |
参考文献
- Camerini-Otero, R. D., Hsieh, P. Homologous recombination proteins in prokaryotes and eukaryotes. Annual Review of Genetics. 29 (1), 509-552 (1995).
- Cromie, G. A., Connelly, J. C., Leach, D. R. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Molecular Cell. 8 (6), 1163-1174 (2001).
- Pierce, A. J., et al. Double-strand breaks and tumorigenesis. Trends in cell biology. 11 (11), S52-S59 (2001).
- Haber, J. E. Genome Stability. , Garland Science. (2013).
- Kowalczykowski, S. C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harbor perspectives in biology. 7 (11), a016410(2015).
- Bianco, P. R., Tracy, R. B., Kowalczykowski, S. C. DNA strand exchange proteins: a biochemical and physical comparison. Frontiers in bioscience: a journal and virtual library. 3, D570-D603 (1998).
- Renkawitz, J., Lademann, C. A., Jentsch, S. Mechanisms and principles of homology search during recombination. Nature Reviews Molecular Cell Biology. 15 (6), 369-383 (2014).
- Greene, E. C. DNA Sequence Alignment during Homologous Recombination. The Journal of biological chemistry. 291 (22), 11572-11580 (2016).
- Sung, P., Krejci, L., Van Komen, S., Sehorn, M. G. Rad51 recombinase and recombination mediators. Journal of Biological Chemistry. 278 (44), 42729-42732 (2003).
- Prakash, R., Zhang, Y., Feng, W., Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor perspectives in biology. 7 (4), a016600(2015).
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. Journal of Biological Chemistry. 272 (45), 28194-28197 (1997).
- Sung, P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes & Development. 11 (9), 1111-1121 (1997).
- Kurokawa, Y., Murayama, Y., Haruta-Takahashi, N., Urabe, I., Iwasaki, H. Reconstitution of DNA strand exchange mediated by Rhp51 recombinase and two mediators. PLoS biology. 6 (4), e88(2008).
- Jensen, R. B., Carreira, A., Kowalczykowski, S. C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 467 (7316), 678-683 (2010).
- Liu, J., et al. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature. 479 (7372), 245-248 (2011).
- Lu, C. -H., et al. Swi5-Sfr1 stimulates Rad51 recombinase filament assembly by modulating Rad51 dissociation. Proceedings of the National Academy of Sciences of the United States of America. , (2018).
- Bazemore, L. R., Takahashi, M., Radding, C. M. Kinetic analysis of pairing and strand exchange catalyzed by RecA. Detection by fluorescence energy transfer. Journal of Biological Chemistry. 272 (23), 14672-14682 (1997).
- Gupta, R. C., Bazemore, L. R., Golub, E. I., Radding, C. M. Activities of human recombination protein Rad51. Proceedings of the National Academy of Sciences. 94 (2), 463-468 (1997).
- Ito, K., Murayama, Y., Takahashi, M., Iwasaki, H. Two three-strand intermediates are processed during Rad51-driven DNA strand exchange. Nature Structural & Molecular Biology. 25 (1), 29-36 (2018).
- Akamatsu, Y., Dziadkowiec, D., Ikeguchi, M., Shinagawa, H., Iwasaki, H. Two different Swi5-containing protein complexes are involved in mating-type switching and recombination repair in fission yeast. Proceedings of the National Academy of Sciences. 100 (26), 15770-15775 (2003).
- Haruta, N., et al. The Swi5-Sfr1 complex stimulates Rhp51/Rad51- and Dmc1-mediated DNA strand exchange in vitro. Nature Structural & Molecular Biology. 13 (9), 823-830 (2006).
- Argunhan, B., Murayama, Y., Iwasaki, H. The differentiated and conserved roles of Swi5-Sfr1 in homologous recombination. FEBS Letters. 591 (14), 2035-2047 (2017).
- Kuzmic, P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Analytical biochemistry. 237 (2), 260-273 (1996).
- Brown, M. S., Bishop, D. K. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harbor perspectives in biology. 7 (1), a016659(2014).
- Rudert, W. A., et al. Double-labeled fluorescent probes for 5' nuclease assays: purification and performance evaluation. BioTechniques. 22 (6), 1140-1145 (1997).
- Xiao, J., Singleton, S. F. Elucidating a key intermediate in homologous DNA strand exchange: structural characterization of the RecA-triple-stranded DNA complex using fluorescence resonance energy transfer. Journal of Molecular Biology. 320 (3), 529-558 (2002).
- Grimme, J. M., et al. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Research. 38 (9), 2917-2930 (2010).
- Algasaier, S. I., et al. DNA and Protein Requirements for Substrate Conformational Changes Necessary for Human Flap Endonuclease-1-catalyzed Reaction. The Journal of biological chemistry. 291 (15), 8258-8268 (2016).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved