
Method Article
χCRACを用いたin vivoでのタンパク質-RNA相互作用ダイナミクスの高時間分解能モニタリング
Erratum Notice
要約
cDNAの速度論的架橋と解析は、生細胞におけるタンパク質-RNA相互作用のダイナミクスを高い時間分解能で調べることができる方法です。ここでは、酵母細胞の増殖、UV架橋、収穫、タンパク質精製、次世代シーケンシングライブラリ調製ステップなど、プロトコルについて詳しく説明します。
要約
RNA結合タンパク質(RBP)とそのRNA基質との間の相互作用は、流動性と複雑さを示します。その寿命の中で、単一のRNAは、その産生、安定性、活性、および分解を調節する多くの異なるRBPによって結合することができます。そのため、これら2つのタイプの分子間に存在するダイナミクスを理解するために多くのことが行われてきました。特に重要なブレークスルーは、「cross-lインクとimmunoprecipitation」(CLIP)の出現でした。この技術により、どのRNAが特定のRBPによって結合しているかを厳密に調査することができました。要するに、目的のタンパク質はin vivoでそのRNA基質にUV架橋され、非常にストリンジェントな条件下で精製され、タンパク質に共有結合的に架橋されたRNAはcDNAライブラリに変換され、配列決定されます。その構想以来、CLIPを特定の研究分野に従順にするために、多くの派生技術が開発されてきました。しかし、紫外線を用いた架橋は非効率的であることで有名です。その結果、曝露時間が長くなり、RBP-RNA相互作用の時間的研究が不可能になります。この問題を克服するために、私たちは最近、大幅に改良されたUV照射および細胞収穫装置を設計および構築しました。これらの新しいツールを使用して、生細胞におけるRBP-RNA相互作用を高い時間分解能で時間分解解析するためのプロトコルを開発しました:キネティックCROSSリンクとCDNAのAナリシス(χCRAC)。私たちは最近、この技術を使用して、栄養ストレス適応における酵母RBPの役割を研究しました。この原稿は、χCRAC法の詳細な概要を提供し、Nrd1 RBPで得られた最近の結果を示しています。
概要
RNAはその機能を発揮するためにRBPに依存することが多く、これらの分子間のダイナミクスを理解することに大きな関心が寄せられています。多くのRBPは、多種多様な生物で同定されています。しかし、生体内でRBP-RNA相互作用を研究することは常に困難であることで有名です。このような相互作用の研究における大きなブレークスルーは、CLIP1の出現によってもたらされました。この方法は、紫外線(UV、254 nm)照射を利用して、RBPとそれらの直接結合RNAとの間の共有結合を誘導します(すなわち、ゼロ距離架橋)。続いて、目的のRBPをストリンジェントな条件下で免疫精製し、タンパク質に共有結合的に架橋されたRNAのみが同定されるようにします。次に、結合したRNAはRNaseで部分的に消化され、その後、シーケンシング用のcDNAライブラリに変換されます。高い精製ストリンジェンシーは、タンパク質とRNA回収の特異性を大幅に高めるために重要であり、架橋リボ核タンパク質(RNP)複合体のSDS-PAGE精製によってもさらに強化されます。CLIPおよび関連メソッドは、シーケンシングライブラリの調製中に、RNAに架橋したアミノ酸が逆転写酵素を頻繁に終結させるか、酵素がこの部位に変異を導入する原因となるため、タンパク質結合部位へのヌクレオチド分解能の洞察も提供します1,2,3。
導入以来、オリジナルのCLIPプロトコルは驚くほど多様な派生方法論を生み出してきました。特に重要なブレークスルーは、ハイスループットシーケンシングとCLIPアプローチ3をマージするHITS-CLIP(またはCLIP-seq)の開発でした。それ以来、これはすべてのCLIPベースの方法論で採用されています。iCLIPは、RBP結合部位のより正確なマッピングを容易にするRNase媒介トリミングおよびアダプターライゲーション技術の改善を導入しました4。PAR-CLIPは、4チオウリジン/ウラシル標識と365 nmでの架橋を組み合わせたもので、T-C置換を分析することで架橋部位のマッピングを可能にしました5。CRAC、尿素-iCLIP、dCLIP、およびuvCLAPは、アフィニティー樹脂へのバックグラウンド結合をさらに減少させ、タンパク質捕捉の特異性をさらに高める変性条件と二重アフィニティー精製ステップを導入しました2,6,7,8,9。さらに、CRAC、uvCLAP、およびdCLIPは、目的のRBPにアフィニティータグでタグ付けすることを導入したため、特異的抗体を生成する必要性が克服されました。
CLIP手法を迅速化するために、いくつかの最適化も行われました。元のCLIPプロトコルは、SDS-PAGE後のRBP-RNA複合体を可視化するために、捕捉されたRNAの放射性標識を利用していました。しかし、放射能の使用は、そのような作業のために設置されていない実験室にとって問題になる可能性があります。irCLIPには、赤外線イメージング10 による可視化を容易にする蛍光色素結合アダプターが組み込まれており、sCLIPは、ストレプトアビジン結合HRP11を介して可視化するために、捕捉されたRNAのビオチン化を利用しています。さらに、eCLIPはRNA標識を完全に放棄します。代わりに、タンパク質は既知のサイズ12のみに基づいて切除されます。ストレプトアビジンベースの精製は、ビオチン化3'アダプターがRNA上にライゲーションされ、逆転写および環状化後の精製を可能にするために使用されるFAST-iCLIPのライブラリ調製プロセスをスピードアップするためにも使用されています13。iCLIPプロトコルのさらなる機能強化により、ライブラリの複雑さも大幅に増加しました4。
最後に、CLIPは、異なる細胞サブコンパートメントからのRBPの捕捉を可能にするように変更されました14,15、光活性化可能なリボヌクレオシド5,16,17のパルス誘導を使用して新たに転写されたRNAを視覚化し、メチル化RNAを捕捉する18,19,20、RNA-RNA相互作用を調べる21,22、および3'末端をマッピングする23,24.
RBPとRNAの間の相互作用の理解を支援するCLIPベースの技術の大きな貢献にもかかわらず、UV架橋の非効率性によって制限されてきました。単層で増殖した培養細胞は一般に比較的容易に架橋できますが、これは溶液中の組織または細胞でははるかに困難です。組織は、必要な細胞層に浸透するために複数回のUV曝露を必要とする可能性がありますが、微生物細胞は、芳香族のUV吸収化合物を含むリッチ培地で増殖することがよくあります25。実際、最大30分のUV照射時間は、そのようなサンプルについてRBPとそれらの結合RNAとの間の十分な架橋を生成するために使用されてきた26、27、28。この長時間のUV曝露は、UV誘発性DNA損傷などの細胞内のストレス応答を誘発し、一部のアプリケーションでは最終データを汚染する可能性があります。
CLIP研究の大部分は、細胞内の特定のタンパク質-RNA相互作用の単一の「スナップショット」を生成することに焦点を当てています。しかし、タンパク質-RNA相互作用は、特に細胞が環境の変化にさらされる場合、本質的に動的です。これには、必須栄養素の利用可能性の突然の低下や急激な温度変化が含まれます。そのため、ストレス時のRBPの役割を真に理解するには、ストレス時のRBPターゲットの全スペクトルをキャプチャし、選択したRBPがストレス応答のどの段階でアクティブであるかを判断できるため、時間分解分析を実行するのが最善です。特に、酵母の研究では、適応の最初の数分が生存に絶対に不可欠であり、細菌のRNA半減期は数分から数秒まで変化する可能性があることが示されました29、30、31、32、33。したがって、このような時間分解解析は、理想的には高い時間分解能で実行する必要があります。しかし、架橋時間が長いため、初期段階の適応応答の研究は特に困難です。
これらの問題を克服するために、我々は最近、微小な時間スケールで細胞を架橋および回収できる改良された方法を開発しました。当社のχCRAC法は、RBP-RNA相互作用の動的変化を、これまでにない分解能で定量的に測定することができます。この方法にとって重要なのは、溶液中の酵母および細菌に必要な架橋時間を約10倍短縮し、RBP-RNA相互作用を瞬時に効果的に凍結する新しいUV照射装置32 の開発であった。また、UV照射後の細胞を迅速に回収するために、0.5L培養で指数関数的に増殖する酵母を約30秒32で回収できる真空ろ過装置を開発しました。これらの技術革新により、RBP-RNAダイナミクスを微小スケール分解能で研究することができます。さらに、実用性を高めるために、元のCRACプロトコル2 にいくつかの最適化を導入しました。
χCRACを用いて、我々は最近、グルコース欠乏に応答する酵母核RBP、Nab3の標的ームを研究した。出芽酵母では、Nab3はRrd1、RBP、およびRNAヘリカーゼSen1と複合体を形成してNNS複合体を形成することができます。RNAポリメラーゼおよび新生転写産物に結合するNNSは、転写終結を誘発することができる34。この複合体は、主に不可解な非コードRNA転写物の除去に関与していますが、タンパク質をコードする遺伝子の発現を制御することも示されています。この研究では、わずか1分間のストレスの後、Nab3のノンコーディングおよびコーディング転写物への異なるターゲティングが示されました32。我々は、Nab3による共転写終結が、従来のCLIPベースのアプローチでは検出が困難であったレトロトランスポゾン遺伝子の非常に一過性のパルス様発現をもたらすことを実証した。さらに、当社のUV架橋剤のUV照射時間が短いため、短寿命のノンコーディングRNAの回収率も大幅に増加しました32。χCRACは、RBPがストレスに対する反応を即時の時間スケールでどのように形成するかだけでなく、応答のライフサイクル全体におけるRBPの役割の変化を解明する上で重要なツールとなる可能性があります。この原稿は、χCRACプロトコルのすべてのステップの詳細な概要を提供します。説明の目的で、この方法は、転写終結およびRNA崩壊に関与する酵母Nrd1タンパク質35,36、および多数の時点にわたるグルコース欠乏に応答するそのRNA標的体を研究するために使用された。最後に、当社のUV照射ユニットがHeLa細胞のRBPとRNAを迅速に架橋できることを実証し、接着細胞でも高分解能の時間分解解析を行うことができることを実証しました。
プロトコル
| TN150 |
| 50 mM トリス pH 7.8 |
| 150 mM 塩化ナトリウム |
| 0.1% NP-40 |
| 1Xプロテアーゼ阻害剤 |
| TN1000 |
| 50 mM トリス pH 7.8 |
| 1M NaCl |
| 0.1% NP-40 |
| NP-PNK |
| 50 mM トリス塩酸塩 pH 7.8 |
| 10 mM マグネシウムCl2 |
| 0.1% NP-40 |
| 5 mMβ-メルカプトエタノール |
| 5 x PNK |
| 250 mM トリス塩酸塩 pH 7.8 |
| 50 mM マグネシウムCl2 |
| 50 mMβ-メルカプトエタノール |
| WB I |
| 50 mM トリス塩酸塩 pH 7.8 |
| 300 mM ナトリウム |
| 10 mMイミダゾール |
| 6Mグアニジン塩酸塩 |
| 0.1% NP-40 |
| 5 mMβ-メルカプトエタノール |
| WB II |
| 50 mM トリス塩酸塩 pH 7.8 |
| 50 mM ナトリウム |
| 10 mMイミダゾール |
| 0.1% NP-40 |
| 5 mMβ-メルカプトエタノール |
| 溶出バッファー |
| 50 mM トリス pH 7.8 |
| 50 mM ナトリウム |
| 250 mMイミダゾール |
| 0.1% NP-40 |
| 5 mMβ-メルカプトエタノール |
| プロテアーゼKバッファー |
| 50 mM トリス |
| 0.1% NP-40 |
| 5 mM β-メルカプトエタノール |
| 1% SDS |
| 5 mM EDTA |
| 50 mM NaCl2 |
| 哺乳類溶解バッファー |
| 50 mM トリス塩酸塩 pH 8 |
| 100 mM 塩化ナトリウム |
| 0.5% V/V トライトン X-100 |
| 0.25% w/v Na-デオキシコール酸 |
| 0.1% w/v SDS |
| 5 mM EDTA |
| 1 mM DTT (フレッシュ追加) |
| 1Xプロテアーゼ阻害剤 |
表1:χCRACに必要なバッファーとその組成。
1. UV架橋とライセート製造
- 溶液中の微生物
- 3.5 Lの所望の培地に、一晩培養から0.05の開始OD600 まで酵母を接種します。180rpmで連続振とうしながら30°Cで成長させる。
- 成長中に、他の必要な材料を準備します。
- 液体窒素の容器を用意します。
- ストレス誘発培地3Lを準備し、水浴中で30°Cに温めます。
- フィルター装置をセットアップし、架橋剤(図2A)をオンにして、50 mLコニカルチューブに各タイムポイントに1つずつラベルを付けます。
- 細胞が目的のOD 600に達したら、500 mLの細胞を架橋剤に直接注ぎ、250 mJの254 nmのUVをUV照射します。架橋剤の使用の詳細については、図2Aおよび図3Aを参照してください。
注:UV照射エネルギーは、目的のタンパク質ごとに慎重に最適化する必要があります。詳細については、ディスカッションを参照してください。 - 架橋後、真空ろ過装置の1つを使用して細胞をろ過します(図2B、C)。濾過した細胞でメンブレンを巻き上げ、t = 0(時間ゼロ)50mLコニカルチューブに入れ、液体窒素中でフラッシュフリーズする。
- 残りのセルを 6 つの異なるフィルターでフィルター処理します。採取した細胞を、膜を培地に滴下し、ストライプで50秒間激しく混合することにより、以前に加温したストレス誘発培地3 Lに再懸濁します。50秒後、t = 1サンプルを採取する準備をします。
- 1分後、500 mLの細胞を架橋し、ステップ1.1.3〜1.1.4のようにろ過して回収します。2、4、8、14、20分後、または必要に応じて別の時点から繰り返します。
- セルを含む円錐管を-80°Cで保管します。 リン酸緩衝生理食塩水(PBS)を4°Cで一晩セットします。
- 翌日、架橋サンプルを含む各円錐管を取り、激しく振とうして細胞を25 mLの冷たいPBSに再懸濁します。
- 細胞懸濁液を新しいコニカルチューブに移し、4,600 x gで4°Cで5分間スピンします。
- PBSを注ぎ、すぐに再び回転させて残留PBSを収集し、残りの液体をピペットでデカントします。
- 空のチューブと比較して、チューブ内のペレットの重量を計算します。
- 2つのペレット容量の氷冷TN150、60 μLのDNase 1、および10 μLのRNase阻害剤を追加します。氷上で30分間インキュベートします。
- たとえば、400 mgの細胞の場合、800 μLの氷冷TN150を追加します。
- DNaseの添加は、ほとんどの可溶性タンパク質に必須ではありませんが、RNAポリメラーゼなどのクロマチン結合タンパク質を研究する際には非常に重要です。さらに、細菌溶解物の粘度を低下させます。正確に2つのペレット容量の溶解バッファーを使用することが非常に重要であり、そうしないと溶解効率が低下する可能性があります。
- 3つのペレット容量(mL単位)のジルコニアビーズを細胞懸濁液に加えます。酵母の場合は直径0.5 mmのビーズを使用し、細菌の場合は0.1 mmを使用します。
- 例えば、400 mgの細胞の場合、1.5 mLチューブで1.2 mLのジルコニアビーズを測定し、溶解バッファーに再懸濁した細胞に加えます。
- 細胞懸濁液を1分間ボルテックスし、次いで氷上に1分間置く。合計5回繰り返します。
- 2つのペレットボリュームのTN150バッファーとボルテックスを激しく加えて混合します。
- コニカルチューブ内の懸濁液を4,600 gでベンチトップ遠心分離機で4°Cで20分間遠心分離します。
- 遠心分離後、将来のウェスタンブロット解析のために上清の50 μLサンプルを採取し、全細胞タンパク質発現を調べます。
- 上清を1.5 mLチューブに移し、微量遠心機でライセートを4°Cで20,000 x g で20分間回転させます。
- または、5 mLチューブを使用する場合は、13,000 x g で20分間遠心分離します。
- 遠心分離後、将来のウェスタンブロット分析のために上清の50 μLサンプルを採取し、タンパク質の可溶性発現を調べます。
- RBPキャプチャ(セクション2)に進みます。
- 培養接着細胞
- UV架橋の24時間前に十分な接着細胞をペトリ皿に播種し、翌日80%のコンフルエントに達するようにします。37°Cの細胞培養インキュベーター内で所望の培地中で一晩増殖させ、5%CO2。
注:石英ペトリ皿を使用する場合は、播種の2.5時間前に培養器具をポリ-D-リジン(70,000〜140,000 wt)とウシ胎児血清(FCS)で処理することにより、細胞の接着を促進することが有益です。増殖表面全体を覆うのに十分なポリ-D-リジンを加え、室温(RT)で5分間インキュベートします。次に、石英ペトリ皿を水で十分にすすぎ、細胞培養インキュベーター内で2時間または完全に乾くまで乾燥させます。その後、成長表面を完全に覆うのに十分なFCSを加え、インキュベーターに少なくとも30分間入れます。細胞を播種する前に、FCSを完全に除去する必要があります。 - 細胞が80%のコンフルエントに達したら、培地を取り出し、15 mLの氷冷PBSで洗浄します。次に、残っている液体をすべて完全に取り除き、すぐに次の手順に進みます。
- ペトリ皿を接着細胞用トレイに移し(図3B)、300mJの254nmのUVをUV照射する。架橋剤の使用の詳細については、図 2A および 図3B を参照してください。
注:UV照射エネルギーは、目的のタンパク質ごとに慎重に最適化する必要があります。詳細については、ディスカッションを参照してください。 - 架橋後すぐに、ペトリ皿を氷の上に置き、10 mLの氷冷PBSを加えます。掻き取って細胞を回収し、15 mLのコニカルチューブに移します。300 x g で4°Cで5分間遠心分離してペレット化します。
- PBSを取り出し、細胞ペレットを1 mLの氷冷PBSに再懸濁し、1.5 mLの微量遠心チューブに移します。4°Cで300 x g で5分間の遠心分離により細胞を再度ペレット化した。
- PBSを取り出し、セルペレットをドライアイス上で急速凍結します。セルペレットは、必要になるまで-80°Cで保管してください。
- 各時点について、手順 1.2.3–1.2.6 を繰り返します。
- 細胞ペレットを1 mLの溶解バッファーに再懸濁し、15 mLのコニカルチューブに移します。その後、1 mLの溶解バッファーを加えて合計2 mLにします。
- 5 μLの哺乳類RNase阻害剤を追加します。
- 10アンペアで氷上で10秒間5倍超音波処理します。超音波処理ラウンドの間に30秒待ちます。
- 各サンプルのタンパク質濃度を計算し、最低濃度に正規化します。
- 1.98 mLのライセートを2 mLチューブに移します。
- 10 μLのDNase Iを加え、1,200 rpmで振とうしながら37°Cで5分間インキュベートします。
- ライセートを16,000 x g で4°Cで20分間遠心分離します。
- 遠心分離後、将来のウェスタンブロット分析のために上清の50 μLサンプルを採取し、タンパク質の可溶性発現を調べます。
- RBPキャプチャ(セクション2)に進みます。
- UV架橋の24時間前に十分な接着細胞をペトリ皿に播種し、翌日80%のコンフルエントに達するようにします。37°Cの細胞培養インキュベーター内で所望の培地中で一晩増殖させ、5%CO2。
2. RBP キャプチャ
- 磁気アンチフラッグ(サンプルあたり75 μLのスラリー)またはIgGアガロース(サンプルあたり500 μLのスラリー)ビーズを5 mLのTN150で3回洗浄します。最終容量700 μLのTN150に再懸濁し、100 μLの洗浄ビーズを7本の15 mLコニカルチューブに加えます。
- 必要になるまで氷上に保管してください。
- ライセートが清澄になったら、抗FLAG/IgGビーズを含むチューブに上清を加えます。
- 4°Cで2時間ナテートする。
注:一部のプロトコルでは、ビーズを使用した一晩のインキュベーションについて説明していますが、インキュベーション時間が長いと架橋RNAの回収率が劇的に低下する可能性があるため、これは推奨されません。
3.ビーズの洗浄とタグのTEV切断
- ビーズを収穫し、ライセートを取り除きます。
- 将来のウェスタンブロット分析のために上清のサンプル50 μLを採取し、捕捉されていないタンパク質を調べます。
- ビーズを氷冷したTN1000に再懸濁し、1.5 mLチューブに移します。ナッツで10分間、4°Cで洗浄します。合計3回の洗浄を繰り返します。
- IgGアガロースビーズを使用する場合は、5 mLのTN1000で洗浄してください。アンチフラッグビーズを使用する場合は、2mLを使用してください。
- 次に、ビーズをTN150で3倍、上記と同じ量で洗浄します。
- 3回目の洗浄後、ビーズを600 μLのTN150に再懸濁します。
- 30 Uの自家製GST-TEVプロテアーゼをビーズ懸濁液に加え、RTで2時間回転させます。
注:組換えGST-TEVプロテアーゼは現在市販されていますが、このプロトコルではテストされていません。- 消化中は、サンプルごとに3つの1.5 mLチューブのカラムをセットアップして、次のステップの準備をします(つまり、7つのサンプルの場合、7つのカラムを3列に並べます)。
- チューブの最後の列に、0.4 gの塩酸グアニジウム、27 μLの5 M塩化ナトリウム、および3 μLの2.5 Mイミダゾールを加えます(pH = 8)。イミダゾールのpHは8でなければならないことに注意してください。これは、RNAの完全性を維持するために重要です。
- さらに、必要量のニッケルビーズをWB I 3xで洗浄します。サンプルあたり100μLのスラリーを使用してください。最終洗浄後、ビーズを同じ元の量のWB Iに再懸濁し、氷上に保管します。
- TEV消化が完了したら、抗FLAGビーズ用の磁気ラックまたはIgGビーズ用の遠心分離を使用して上清を収集し、以前にセットアップしたチューブの最初の列に移します。
- ウェスタンブロット分析用のTEV溶出液のサンプル50 μLを採取します。
- サーモブロックインキュベーターを37°Cに設定します。 チューブの2列目に、1 μLのRNaseカクテルを加えます(1:50希釈)。
- チューブの最初の列から550 μLのTEV溶出物を取り、2列目(RNaseカクテルを含む)に追加します。混合を確実にするために激しくピペットで。
- 最初のサンプルでこれを完了したら、すぐにチューブをサーモブロックに入れ、タイマーを開始します。後続のサンプルに進み、それぞれがずらされるようにします。
- 正確に5分間インキュベートします。完了したら、サーモブロックから最初のサンプルを取り出し、溶液を3列目のチューブ(塩酸グアニジウム粉末を含む)に移します。
注:RNaseカクテルを1:50に希釈した5分間のインキュベーションは、通常、ほとんどのタンパク質に適していますが、架橋RNAが正しいサイズ(30〜100 nt)であることを確認するために、このステップをタンパク質ごとに異なるインキュベーション時間または濃度で慎重に最適化する必要があります。 - すぐに全速力で数秒間ボルテックスしてグアニジウム粉末を溶解し、次のサンプルに進みます。
- すべてのサンプルがグアニジウム粉末に移された後、すべての粉末が完全に溶解することを確認するために再びボルテックスします。
- 洗浄したニッケルビーズ100 μLを加え、4°Cで一晩回転させます。 このインキュベーションは2時間に短縮することができます。
4.オンビーズアルカリホスファターゼ処理
- サーモブロックを37°Cに設定します。
- 精製スピンカラムを2 mLチューブに入れます(サンプルごとに1本ずつ)。ニッケルビーズをカラムに移し、上澄み液を排出させます。その後、WB Iですすぎ、カラムに塗布することにより、すべてのニッケルビーズが1.5 mLチューブから除去されたことを確認します。
- サンプルごとに6本ずつ、2 mLチューブをセットアップします(各洗浄液を回収するために1本)。流れを維持するために、カラムの外側を乾いた状態に保ちます。ビーズを500 μLのWB Iで3回洗浄し、次に500 μLのNP-PNKで3回洗浄します。
- スピンカラムの蓋を閉め、ビーズを短時間回転させて余分なバッファーを除去します。
- ストッパーをカラムに置き、カラムを1.5 mLチューブに入れ、表2に示す反応混合物を60 μL加えます。
| コンポーネント | 1倍速 | 7.5倍 |
| 5 x PNK バッファ | 12 | 90 |
| アルカリホスファターゼ | 4 | 30 |
| RNase阻害剤 | 2 | 15 |
| H2O | 42 | 315 |
| 最終巻 | 60 μL | 450 μL |
表2:アルカリホスファターゼ反応混合物。
- ビーズを37°Cで1時間インキュベートします。
- ビーズを500 μLのWB Iで1回洗浄してアルカリホスファターゼを不活性化し、次に500 μLのNP-PNKバッファーで3回洗浄します。カラムの内側をNP-PNKバッファーで十分にすすぎ、微量のグアニジウムを除去してください。
5. App-PEリンカーのRNAの3'末端へのビーズ上のライゲーション
- 残りのバッファーをスピンアウトし、 表 3 で指定されている混合物 60 μL (App-PE シーケンスについては 表 4 を参照) をカラムに加えます。反応液を25°Cで6時間インキュベートします。
| コンポーネント | 1倍速 | 7.5倍 |
| 5 x PNK バッファ | 12 | 90 |
| アプリケーション PE アダプタ (100 μM) | 0.6 | 4.5 |
| T4 RNAリガーゼ2切断型K227Q | 3 | 22.5 |
| RNase阻害剤 | 1.5 | 11.25 |
| 50% ペグ8000 | 12 | 90 |
| H2O | 30.9 | 231.75 |
| 最終巻 | 60 μL | 450 μL |
表3:App−PEリンカーライゲーション反応混合物。
| オリゴヌクレオチド名 | シーケンス (5'-3') | |||
| L5Aa | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrGrArUrCrUrNrNrUrArGrCrN-OH | |||
| L5Ab | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrGrArUrCrUrNrNrArUrUrArGrCrN-OH | |||
| L5Ac | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrCrGrCrArGrCrN-OH | |||
| L5広告 | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrCrGrCrUrUrArGrCrN-OH | |||
| L5Ba | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrGrArUrCrUrNrNrArGrCrN-OH | |||
| L5Bb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrUrGrArGrCrN-OH | |||
| L5Bc | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrCrArCrUrArGrCrN-OH | |||
| L5Bd | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrUrCrUrCrUrArGrCrN-OH | |||
| L5Ca | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrCrUrArGrCrN-OH | |||
| L5Cb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrUrGrArGrCrN-OH | |||
| L5Cc | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrCrGrArUrCrUrNrNrArCrUrCrN-OH | |||
| L5カドミウム | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrArCrUrUrArGrCrN-OH | |||
| L5Da | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrCrGrUrArUrN-OH | |||
| L5デシベル | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrCrArCrUrArN-OH | |||
| L5Dc | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrUrArGrUrGrCrN-OH | |||
| L5Dd | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrGrArUrCrUrNrNrArUrCrArCrGrN-OH | |||
| L5Ea | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrCrArCrUrGrUrN-OH | |||
| L5Eb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrUrGrArCrArN-OH | |||
| L5Ec | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrUrGrUrCrArCrN-OH | |||
| L5エド | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrArCrGrUrGrN-OH | |||
| App_PE | App-NAGATCGGAAGAGCACGTCTG-ddC | |||
表4:捕捉されたRNAの5'末端および3'末端へのライゲーションに必要なDNAおよびRNAアダプターの配列。 これらをRNaseフリーHPLCで精製した。
- ビーズを500 μLのWB Iで1回、500 μLのNP-PNKバッファーで3回洗浄します。カラムを新しいチューブに入れ、残りのバッファーをスピンアウトします。
6. RNAの5'末端のビーズ上でのリン酸化
- 表5で規定されている混合物を80 μLカラムに加えます。反応液を37°Cで40分間インキュベートします。
注:サンプルは高放射性になります。したがって、その後のすべての作業は保護スクリーンの後ろで実行し、廃棄物は地域の健康と安全の規則に従って処分する必要があります。
| コンポーネント | 1倍速 | 7.5倍 |
| 5 x PNK バッファ | 16 | 120 |
| 32名P-ATP (10 μCi/μL) | 3 | 22.5 |
| T4 PNK | 3 | 22.5 |
| H2O | 58 | 435 |
| 最終巻 | 80 μL | 600 μL |
表5:リン酸化反応混合物。
- 1 μLの100 mM ATPを加え、さらに20分間反応を進行させます。これにより、5'末端のほぼすべてがリン酸塩を有し、5'リンカーのライゲーションを容易にする。
- サンプルごとに5本の2 mLチューブをセットアップします。
- ビーズを500 μLのWB Iで1回、500 μLのNP-PNKバッファーで3回洗浄します。これらの溶出物は非常に放射性であるため、適切に廃棄する必要があることに注意してください。
- カラムを最終チューブに移動し、残りのバッファーをスピンアウトします。
7. 5'リンカーのオンビードライゲーション
注:5'リンカーには、シーケンシング後の各サンプルの識別に使用されるRNAバーコードが含まれています。したがって、どのリンカーがどのサンプルに使用されているかに注意することが絶対に重要です。
- 表6に記載の混合物を78 μLカラムに加えます。2 μLの5'アダプター(100 μM;表4を参照)を各チューブに加え、18°Cで一晩インキュベートします。
| コンポーネント | 1倍速 | 7.5倍 |
| 5 x PNK バッファ | 16 | 120 |
| ATP (10 mM) | 8 | 60 |
| RNase阻害剤 | 2 | 15 |
| T4 RNAリガーゼ | 4 | 30 |
| H2O | 48 | 360 |
| 最終巻 | 78 μL | 585 μL |
表6:5'リンカーライゲーション反応混合物。
- 翌日、ビーズ1回を500 μLのWB Iで、3回を500 μLのWB IIで洗浄し、カラムを新しい2 mLチューブに移します。
8. 溶出、SDS-PAGE、およびRNA抽出
- 遠心分離機を4°Cに設定します。 溶出用にサンプルあたり2列の1.5 mLチューブを準備します。
- カラムのボイドボリュームをニッケルビーズで素早くスピンアウトします。溶出チューブの最初の列にカラムを置き、200 μLの溶出バッファーを追加します。2分待ってから、すばやく回転させてバッファーを列に通します。
- 列をチューブの2行目に移動し、手順8.2を繰り返します。各サンプルには合計400 μLの溶出液が含まれ、2つの1.5 mLチューブに分割されます。
- すべての溶出液を採取し、一緒に5 mLチューブに移します。20 mg/mLのグリコーゲンを2 μL加えます。したがって、7つのサンプルを使用する場合、5 mLチューブに2.8 mLのプールされた溶出液があります。
- サンプルあたり100 μLのトリクロロ酢酸(TCA)を加え[例:7サンプルに対して700 μLのTCA(2.8 mLのプール溶出液)]を5 mLチューブに加え、30秒間よくボルテックスします。
- 氷上で20分間インキュベートします。
- ベンチトップ遠心分離機で、17,000 x g、4°Cで30分間遠心分離します。
- コニカルチューブから上清を慎重に取り出し、ガイガーカウンターでピペットをチェックして、ペレットが誤って除去されていないことを確認します。ある場合は、上清をチューブに戻し、さらに10分間遠心分離します。
注:上清はまだ高放射性である可能性があります。適切なシールドを使用してください。 - ペレットを2 mLの氷冷アセトンに完全に再懸濁します。
- 17,000 x g、4°Cで15分間遠心分離します。
- P1000ピペットでできるだけ多くのアセトンを取り除きます。その後、チューブを短時間回転させてアセトンの小滴を集め、P10ピペットで取り除きます。ドラフトで2分間乾燥させます。
注:アセトン上清はまだ放射性である可能性があります。適切なシールドを使用してください。 - サンプルを30 μLの1xタンパク質ローディングバッファーに再懸濁します。ペレットが適切に再懸濁されていることを確認するには、P200ピペットで溶液を取り出し、ガイガーカウンターを使用して1.5 mLチューブに残っている活性を測定して、放射能の大部分がローディングバッファーに存在し、1.5 mLチューブに残っていないことを確認します。
- サンプルを65°Cで10分間加熱します。 1 mm、4〜12%のプレキャストビストリスゲルにロードし、MOPSバッファー中で125 Vで1.5時間実行します。
- ゲルの実行が終了したら、ゲルカセットを開きます。ゲルは底板に保持する必要があります。上部を処分します。
- ゲルを粘着フィルムで包み、テープを使用して遮光カセットの内側に固定します。信号を改善するための増幅画面がカセットにあることを確認してください。
- オートラジオグラフィーフィルムをゲルに露光し、露光中はカセットを-80°Cで保管します。曝露時間は、架橋効率の異なるタンパク質によって異なります。
- フィルムを配置するときは、後続のステップで目的のバンドを切り取るために、フィルムをカセットに再調整する方法が必要です。これを確実にするために、蛍光定規を使用し、またゲルがカセットの角にあることを確認し、次にそれが一番隅に配置されたフィルムで覆われていることを確認してください。
注:経験則として、ガイガーカウンターに表示したときに少なくとも~250cpsの読み取り値を与えるローディングバッファー内の溶出物は、3時間の露光に十分な信号を与えます。それ以外の場合は、一晩暴露が行われます。
- フィルムを配置するときは、後続のステップで目的のバンドを切り取るために、フィルムをカセットに再調整する方法が必要です。これを確実にするために、蛍光定規を使用し、またゲルがカセットの角にあることを確認し、次にそれが一番隅に配置されたフィルムで覆われていることを確認してください。
- フィルムを現像します。ゲルを覆っている粘着フィルムを切り取りますが、ゲルを動かさないでください。それ以外の場合、画像はゲルからオフセットされます。
注:ゲルはおそらく非常に放射性です。ゲルスライスを切り取るときは、必ず適切なシールドを使用してください。 - フィルムをゲルの上に置き、目的のバンドを切り取ります。ゲルスライスを2 mLチューブに入れます。
- P1000ピペットチップを使用してゲルスライスを粉砕し、600 μLのプロテイナーゼKバッファーと200 μgのプロテイナーゼKを加えます(このプロトコルでは、10 μLの20 mg/mLプロテイナーゼK溶液を使用します)。激しく振とうしながら55°Cで2時間インキュベートします。
- その後、P1000チップの端をきれいなメスで切り取り、上清とゲル片を2 mLチューブに入れたスピンカラムに移します。
- カラムをRTで17,000 x g で1分間スピンし、放射性の単離されたRNAを含むフロースルーを収集します。
- フェノール:クロロホルム抽出を実行します。
- 50 μLの3 M酢酸ナトリウム、pH = 5.2、および500 μLのフェノール:クロロホルムとボルテックスウェルを加えます。17,000 x gで5分間回転します。水性の最上層を取り除き、新しい1.5 mLチューブに入れます。
- 500 μLのクロロホルムを加え、10〜15秒間激しくボルテックスします。RTで17,000 x g で5分間スピンします。 水層を取り除き、新しい1.5 mLチューブに入れます。
- 1 μLの20 mg/mLグリコーゲンと1 mLの氷冷96%エタノールを加えます。-80°Cで30分間、または-20°Cで一晩沈殿させる。
- 4°C、17,000 x gで30分間遠心分離します。上清を除去し、500 μLの70%エタノールを加え、17,000 x gで4°Cで5分間遠心分離します。すべてのエタノールを除去し、クイックスピンを実行して残留物を収集し、P10ピペットで余分なものを取り除きます。
- ペレットをドラフトで~3分間乾燥させます。20 μLのDECC処理水に再懸濁します。
- RNAを-80°Cで一晩保存するか、直ちに逆転写ステップに進みます。
9.逆転写
- 20 μLのRNAに2 μLの10 μM RTオリゴ(PE_reverse;表7を参照)と4 μLの5 mM dNTPを追加します。
| オリゴヌクレオチド名 | シーケンス (5'-3') | |||
| P5フォワード | AATGATACGGCGACCACCGAGATCTACTCTTTCCCTACACGACGCTCTCCCCCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGATGGATGGATGGATGGATGGATGGATGCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGACTGGAGATGGTGCTCTCTCCGATCCGATCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC4 | CAAGCAGAAGACGGCATACGAGATGGTCAGTGACTGGATGGTCCGATCCGATCTCTCTCCGATCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGATCCGGTCCGATCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC7 | CAAGCAGAAGACGGCATACGAGATCAGTCGTGACTGGAGATGGATCCTCCGGCTCTCCGGTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC8 | CAAGCAGAAGACGGCATACGAGATTAGCTTGTGACTGGAGATGGATGGTCTCTCCGATCCGATCTT | |||
| BC9 | CAAGCAGAAGACGGCATACGAGATGATCAGGTGACTGGAGGTCTCGTCTTCCGATCTCTCTCCGATCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
| BC10 | CAAGCAGAAGAGAGGGCATACGAGATATCAGGTGACTGGAGATGGCTCTCCGATCTCTCTCCGATCTCTCTCTCTCT | |||
| PE_reverse | CAGACGTGTGCTCTCTCCGGCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCTCT | |||
表7:PCRプライマー(バーコード配列を含む)および逆転写プライマー。
- 予熱したサーモブロックに85°Cで3分間移し、氷上で5分間スナップチルします。短時間の遠心分離でチューブの内容物を回収し、8 μLの5x逆転写酵素バッファー、2 μLの100 mM DTT、および2 μLのRNase阻害剤を加えます。
- 混合物を50°Cで3分間インキュベートした後、2 μLの逆転写酵素を添加し、50°Cで1時間インキュベートします。
- 逆転写酵素を65°Cで15分間インキュベートすることにより不活性化します。
- チューブを37°Cに予熱したサーモブロックに移し、3分間放置して順応させます。
- 2 μLのRNase Hを添加し、37°Cで30分間インキュベートします。
- SPRIビーズを使用してcDNAを単離します。
- 84 μLのビーズを2倍量加えます。15分間インキュベートします。ビーズを磁気ラックに置き、1分間放置してビーズを収穫します。
- 上清を除去して廃棄し、200μLの70%エタノールを加える。磁気ラックからビーズを取り外さないでください。ビーズをエタノールで30秒間インキュベートします。
- エタノールを除去し、洗浄ステップを繰り返します。P10チップを使用して残留エタノールをすべて取り除きます。
- ビーズをドラフトに2分間入れて乾燥させます。ビーズをラックから取り出し、12 μLの水に再懸濁してから、ビーズをラックに戻します。上清11μLを除去します。
- cDNAを-20°Cで凍結するか、直ちにPCRステップに進みます。
10. qPCR反応
- cDNAの増幅のための最終PCRの前に、定量的ポリメラーゼ連鎖反応(qPCR)を実行して、ライブラリの過剰増幅を防ぐためにcDNAを増幅するための最適なサイクル数を特定します。
- 表8に従って氷上でqPCR反応を設定します。すべてのプライマーについては表7を参照されたい。
| コンポーネント | 1倍速 |
| 2x qPCR反応マスターミックス | 5 |
| 0.1 μM P5プライマー(フォワード) | 0.8 |
| 0.1 μM BCプライマー(リバース) | 0.8 |
| cDNA(または陰性対照としての水) | 1 |
| H2O | 2.4 |
| 最終巻 | 10 μL |
表8:qPCR反応混合物。
- 増幅に必要なサイクルを適切に定量化するには、cDNAに3つのテクニカルレプリケートを使用し、3つのネガティブ(つまり水)コントロールを使用します。
- プレートを光学的に透明なフィルムで密封し、キットの製造元の指示に従ってqPCRを実行します。
- 絶対定量法でサンプルを分析し、指数関数的成長の膝に達するサイクル数(n)を特定します(例については 図4C を参照)。このサイクル数は、残りのcDNAの最終的な増幅に使用されます。
11. PCR反応とゲル抽出
- 表9に従って氷上でPCR反応をセットアップした。すべてのプライマーについては表7を参照されたい。
注:5 μLのcDNAライブラリのみが使用されます。
| コンポーネント | 1倍速 |
| 10 x 校正ポリメラーゼバッファー | 5 |
| 10 μM P5プライマー(フォワード) | 1 |
| 10 μM BCプライマー(リバース) | 1 |
| 5 mM dNTP | 2.5 |
| 校正ポリメラーゼ酵素 | 1 |
| ティッカー | 5 |
| H2O | 34.5 |
| 最終巻 | 50 μL |
表9:PCR反応混合物。
- PCRを次のように実行します:95°Cで2分間;98°Cで20秒、52°Cで30秒、72°Cで1分間のnサイクル。72°Cで5分間。χCRACライブラリを増幅するためのサイクル数(n)は、セクション10に記載されているqPCRによって決定されます。
- 1 μLのエキソヌクレアーゼIを加え、37°Cで60分間インキュベートします。
- 2倍量のビーズ(すなわち100μL)を用いて、上記のようにSPRIビーズを用いて増幅されたcDNAをクリーンアップする。11 μLで溶出します。
- 3 μLの6xローディング色素を添加し、プレキャスト6%TBEゲル上で1x TBEバッファー中で100 Vで1時間実行します。短いDNA断片の定量に適したラダーを使用してください。
- 終了したら、カセットからゲルを取り出し、ゲルを覆うのに十分な1x TBE(~50 mLなど)を備えた適切な液密容器に入れます。適量のSYBR安全色素を加える(例:50 mLの場合、10,000倍の色素を5 μL使用)
- RTで15分間穏やかに混合してゲルを染色します。 SYBRを含む1x TBEを排出し、きれいな1x TBEと交換します。RTで穏やかに振とうしながらゲルを10分間洗浄します。
- 1x TBEを排出し、ゲルを透明なフォルダーに入れます。フォルダを適切なサイズにカットします。
- 蛍光体イメージャー等の適当な手段を介してゲルを画像化する。~175 bpから~400 bpの間のDNA断片を切除します。ゲルスライスを1.5 mLチューブに入れます。
- P1000チップを使用してゲルスライスを完全に粉砕し、400 μLのH2Oを加えます。 サーモブロック中で1時間振とうしながら37°Cでインキュベートします。
- サンプルをドライアイスで10分間凍結した後、1時間振とうしながら37°Cのサーモブロックに戻します。
- フィルターカラムを取り、内部に2つのガラスマイクロファイバーフィルターを挿入して、フィルターユニットを作成します。ユニットを1.5mLチューブに入れます。
- きれいなメスでP1000チップの端を切り取り、粉砕されたTBEゲル懸濁液を取り込み、手順11.12で作成したフィルターユニットに分注します。17,000 x g で30秒間回転します。
- 1 μLのグリコーゲンを上清に加え、40 μLの酢酸ナトリウム、pH = 5.2、および1 mLの96%エタノールを加えます。-80°Cで30分間インキュベートします。
- 17,000 x g、4°Cで30分間遠心分離します。 上清を捨て、500μLの70%エタノールで洗浄する。
- 5分間回転し、エタノールを完全に除去してから、ペレットをドラフトで3分間乾燥させます。
- 10 μLのH2Oに再懸濁し、DNA濃度を測定します。
結果
χCRAC法の有効性を実証するために、HTPタグ付きNrd1タンパク質を発現する酵母株を用いて経時的実験を行った。この方法がどのように機能するかを説明する詳細な概略図を図1に示します。Nab3と同様に、Nrd1は様々なRNA転写物の核RNA崩壊に関与している37。コーデン研究室の以前の研究では、細胞がグルコース飢餓にさらされると、そのRNA標的に結合するNrd1が大幅に変化することが示唆されました28,38。そのため、グルコースを含む培地(SD-TRP)で指数関数的に増殖する細胞を、Nrd1-RNA相互作用の動的変化を監視するために、グルコースを含まない同じ培地(S-TRP)に経時的にシフトしました。サンプルを採取し、シフト前と1、2、4、8、14、および20分後にVari-Xリンカーチャンバー(図3A)で架橋しました。細胞増殖に使用された培地は、この芳香族アミノ酸によるUV吸収を減少させるために、意図的にトリプトファンを欠乏させた。培地をオートクレーブすると糖のカラメル化につながる可能性があるため、フィルター滅菌された合成培地を使用するのが最善であることに注意してください。これにより、架橋効率が低下します。
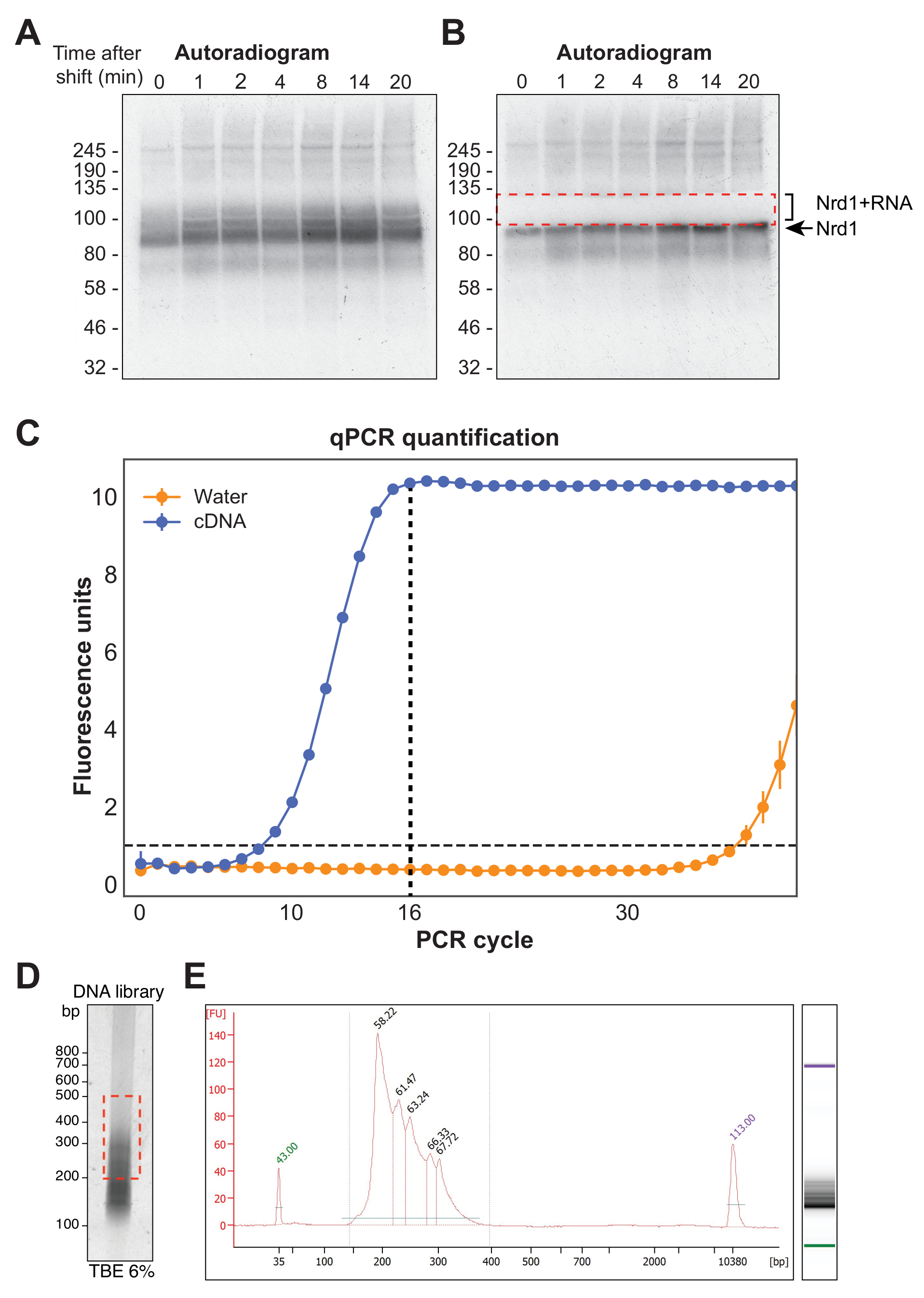
図4A は、χCRAC実験からの代表的なオートラジオグラフを示す。この例では、サンプルは一緒にプールされていないことに注意してください。代わりに、それぞれをゲル上で個別に実行しました。これは、テストされたすべての時点でタンパク質がRNAに効果的に架橋することを示すための最初の実験テストに推奨されます。RBPの予想される分子量で特に強いシグナルが観察され、シーケンシングに適さない非常に短い放射性標識RNAに結合したタンパク質を表しています。したがって、より長いRNA断片に架橋されたタンパク質であるこのバンドの上のスメアリーシグナルを単離した。断片は、タンパク質バンドのすぐ上から約30 kDaを加えたものから切断されました。 図4B は、切除後のオートラジオグラムを示しており、タンパク質はゲルに残った短いRNAに架橋され、以前はスメアリーシグナルが切除されています。
逆転写後、PCRを使用してcDNAライブラリを増幅する必要があります。ただし、ライブラリの過剰増幅は、ポリメラーゼによって優先的に増幅された配列へのバイアスを導入し、PCRアーティファクトを生成する可能性があるため、避ける必要があります。過剰増幅されたライブラリには、シーケンサーでの読み取りを浪費する多数の重複シーケンスも含まれています。最終ライブラリの増幅のための理想的なPCRサイクル数を計算するために、p5およびBCオリゴヌクレオチドを用いたqPCRを通じてcDNAのアリコートを増幅した。ライブラリがピーク蛍光に達した最初のサイクルをPCRサイクルカウントとして選択しました。 図4C は、典型的なcDNAライブラリからのqPCRの例を示しており、ピークサイクルカウントは16でした。その後、この値を最終的なχCRAC PCRに使用しました。シーケンスされたデータを処理するために、我々は、我々の研究室で以前に開発されたソフトウェア(pyCRAC)と、それに対応するパイプラインをカイネティックCRACデータの解析に使用した(Nues et al., 2017; https://git.ecdf.ed.ac.uk/sgrannem/pycrac, https://bitbucket.org/sgrann/kinetic_crac_pipeline/src/default/)。これらのオープンソースソフトウェアツールは、データの逆多重化およびトリミング、PCR重複の除去、統計的に有意なピークの同定、連続配列へのクラスタリード、および結合モチーフの同定を可能にする39。これらのツールの動作の詳細については、それぞれのWebページを参照してください。
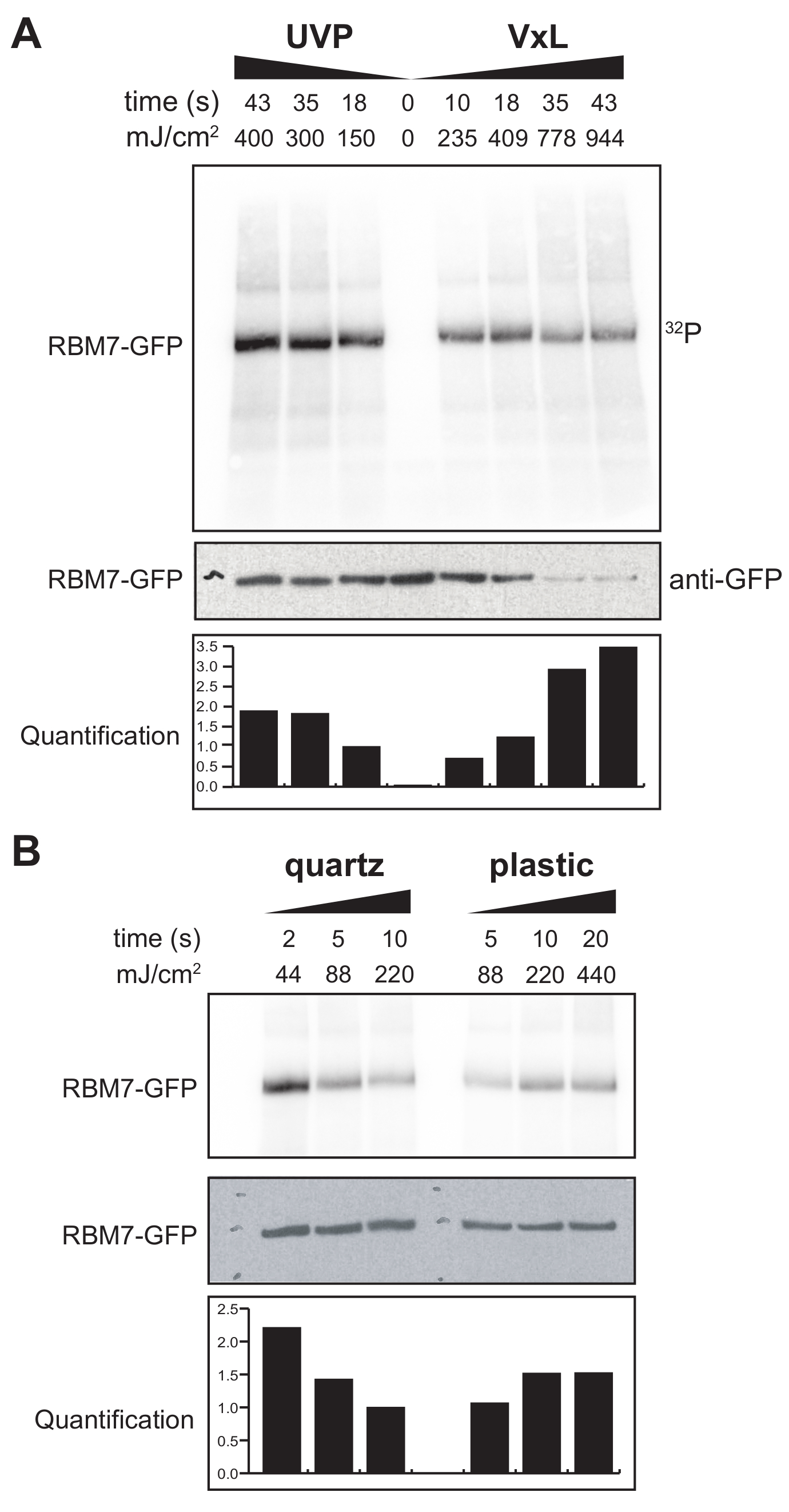
また、哺乳類細胞用のχCRACプロトコルの開発にも着手しました。哺乳類細胞株の大部分は単層として増殖しており、UV透過性バッグを備えた架橋剤のトレイは、接着細胞での実験には適していません。そこで、ペトリ皿1〜2枚(直径150mm、深さ25mm)に接着細胞をUV照射できるステージを開発しました(図3B)。最初の試験として、抗GFP抗体と従来のCLIPベースの精製を使用して、安定にタグ付けされたGFP-RBM7の架橋と捕捉を通じて、哺乳類細胞に対する架橋剤の効率を測定しました。 図5Aに示すように、架橋剤は、広く使用されているUV照射装置に匹敵する効率で254nmのUV照射を使用して、単層として増殖した哺乳動物細胞からタンパク質-RNA複合体を回収することができました。しかし、UV架橋実験に通常使用される標準的な細胞培養プラスチック製品は、254nmのUVには浸透しません。したがって、私たちの架橋剤では、細胞はUVランプの上側バンクからのみ照射を受けます。これを克服するために、我々は細胞増殖と架橋のためのUV透過性石英ペトリ皿を開発しました。石英培養器を使用すると、わずか2秒のUV照射でタンパク質-RNA複合体の堅牢な回収率を示しました(図5B)。CLIP技術などの哺乳類細胞のRBP捕捉法と組み合わせると、これらの短い架橋時間は、遺伝毒性ストレスまたはタンパク質因子の急速な枯渇に応答して、または転写または細胞周期の同期と並行して、RBPの時空間RNA結合プロファイルを回復するためのタイムコースに適しています。
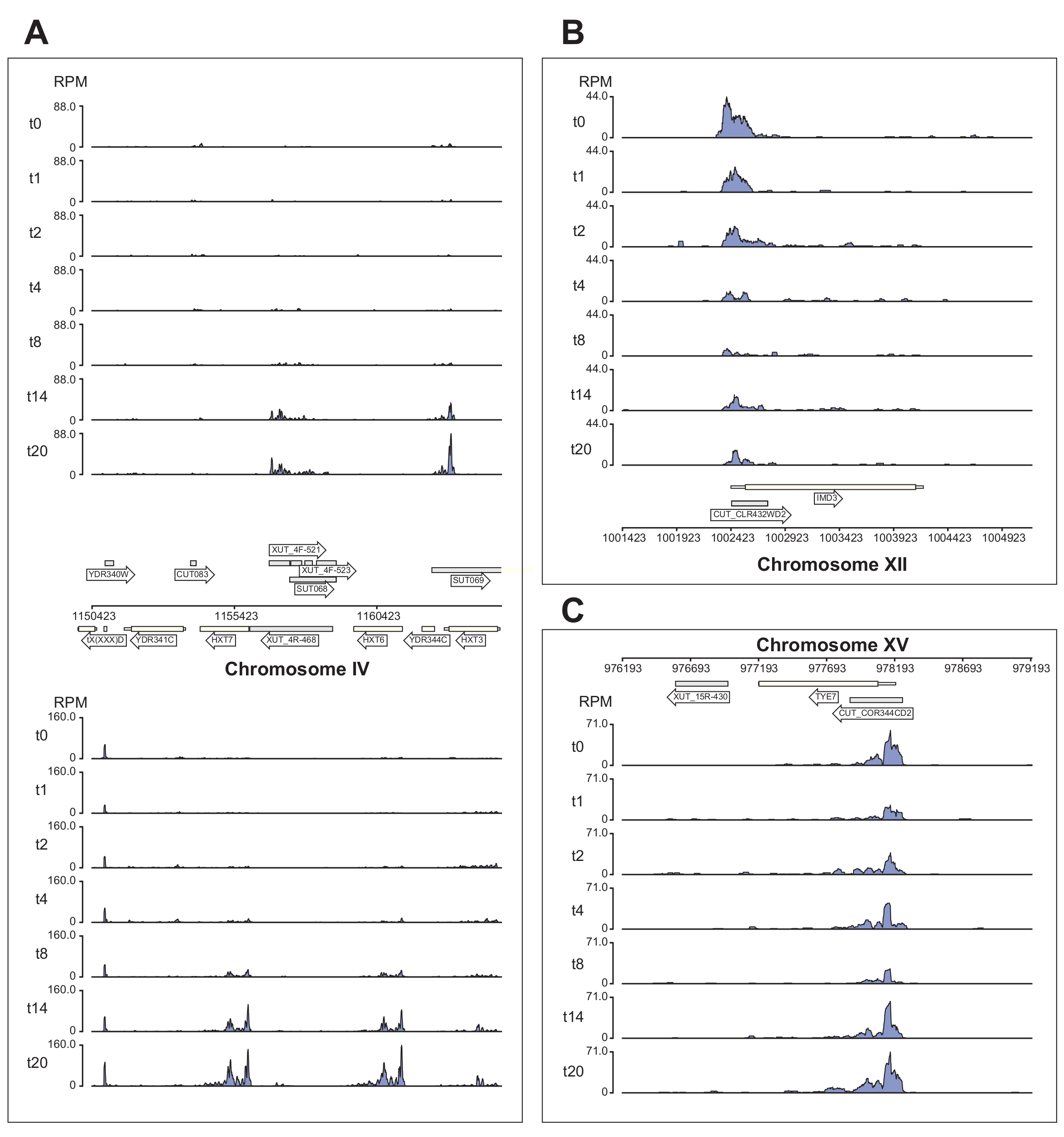
図6 は、χCRACパイプラインによって処理されるNrd1データのいくつかの例を示しています。この図は、パイプラインによって生成されたベッドグラフファイルと、データの公開品質のゲノムブラウザー画像の作成を簡素化するために設計されたpython GenomeBrowserパッケージ(https://pypi.org/project/GenomeBrowser/1.6.3/)を使用して作成されました。灰色の長方形は、不可解な不安定転写物(CUT)、安定な特徴付けられない転写物(SUT)40、Xrn1感受性不安定転写物(XUT)41など、ノンコーディングRNAを発現するゲノム領域を表します。 図6 のデータは、Nrd1がこれらの非コードRNA転写物の多くに結合することを示しており、このタンパク質がこのクラスの転写産物の分解に関与しているという考えと一致している42。 図6A は、IV染色体上の~15 kb領域を示しています。ここでは、グルコース飢餓中にアップレギュレーションされる高親和性グルコーストランスポーターHXT6およびHXT7をコードする転写物へのNrd1の結合の有意な増加があった。NNS複合体による転写終結は、グルコース飢餓中のこれらの遺伝子の誘導動態に影響を与える可能性があります。 図6B は、Nab343によって調節されることが知られているImd3転写産物へのNrd1架橋の例を示す。この場合、データはグルコース飢餓時の結合の有意な減少を示した。以前の研究では、グルコース飢餓中にNab3のTye7転写物への結合が減少したことが示されました44。この観察結果と一致して、χCRACデータは、Nrd1の結合がグルコース飢餓中に減少し、Nrd1のTye7への架橋が8分間のストレス後に最も低くなることを示唆しています(図4C)。しかし、グルコース飢餓の14分後、Nrd1結合が開始レベルに戻ったため、この効果は一過性にすぎなかったようです。

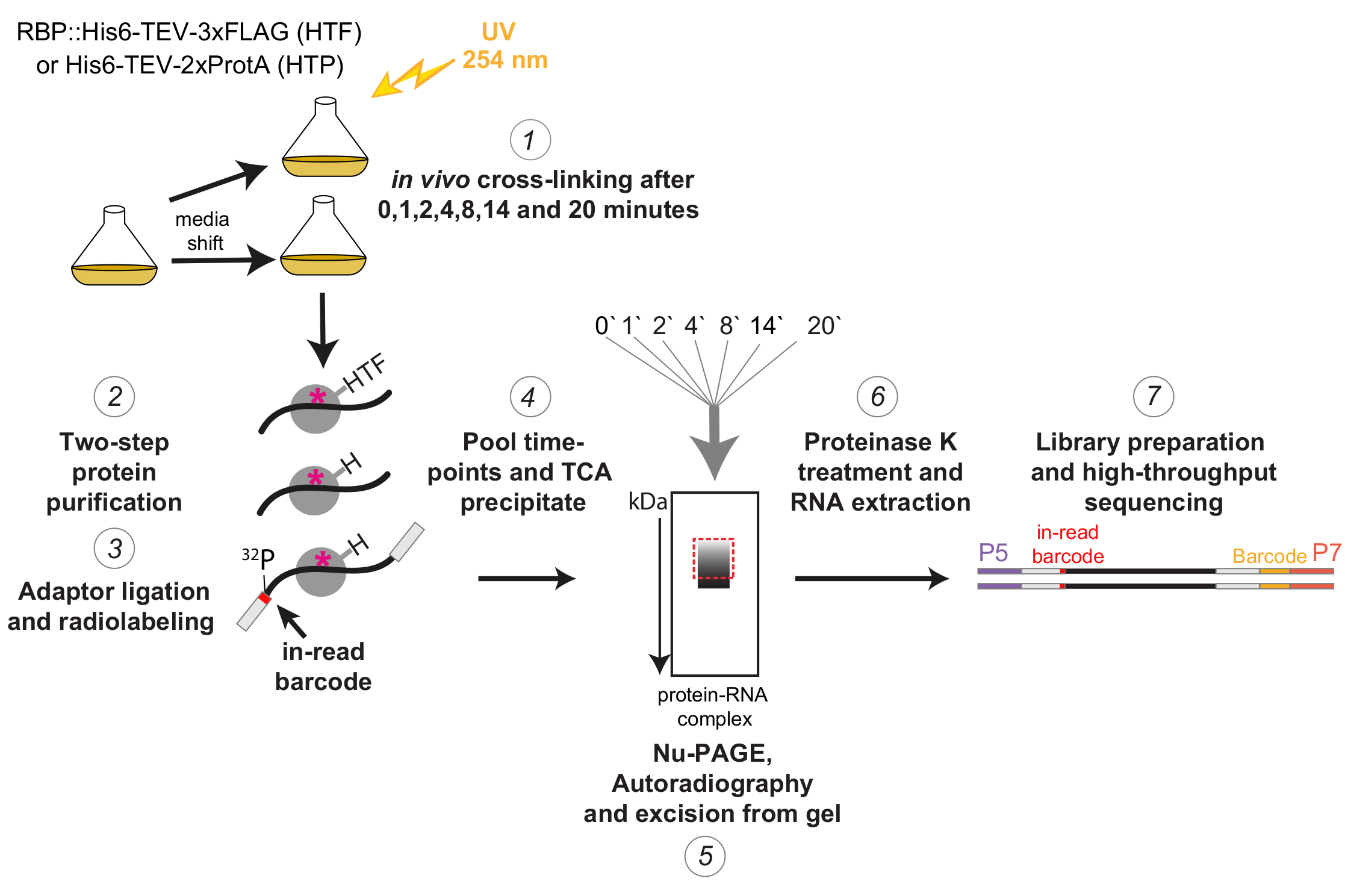
図1: χCRACプロトコルの概略図。タグ付き株は、所望の密度になるまで増殖させた。RBPはRNA結合タンパク質を示す。その後、参照サンプルを採取し、254nmのUV光で架橋した。残りの細胞は濾過によって回収され、次いで急速にストレス誘発培地に移行した。ここで説明するχCRAC実験では、シフトの1、2、4、8、14、および20分後にサンプルを採取し、架橋しました(1)。次に、目的のRBPを、非常に厳密な2段階のアフィニティー精製(2)を用いて精製した。次に、捕捉された架橋RNAをRNaseで部分的に消化し、5'末端で放射性標識し、アダプターをそれらの上にライゲーションしました(3)。5'アダプターには、シーケンシング後に個々のサンプルをバイオインフォマティクス的に分離できるように、固有の「読み取り中」バーコードシーケンスが含まれていました。次に、RBP-RNA複合体を溶出、プール、および沈殿させ(4)、SDS-PAGEによって分解し、オートラジオグラフィーによって視覚化しました(5)。続いて、オートラジオグラフィ画像に破線の赤いボックスで例示した、メインバンドのすぐ上の放射性シグナルを含む単一のゲルスライスを、ゲルから切り取った(5)。ゲルスライスをプロテアーゼKで処理し、続いてRNAを抽出し(6)、cDNAに変換し、PCRで増幅しました(7)。PCRステップでは、追加のバーコード(P7オリゴによって導入された黄色のブロック)が導入され、多くのライブラリを1つのレーンに多重化できるようになりました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:架橋と真空ろ過。 (a)架橋剤。細胞懸濁液を機械の右上にある漏斗に注ぎ(クローズアップについては 図3A も参照)、中央トレイにあるUV透過性バッグに保持します。このバッグの側面には2つのシャッターがあり、ユーザーが照射ステップを開始するように機械に指示するまで閉じたままになります。細胞は、上下の両方のトレイからUV光を照射される。このマシンには、254nmと365nmのUVランプが付属しており、後者はPAR-CLIP実験に適用できます。機械は右上にあるタッチスクリーンパネルを介して操作され、UV投与量または露光時間を制御できます。(B)架橋に続いて、セルは機械の左側から排出されます。細胞懸濁液は真空で回収され、ガラスフラスコに排出され、その後、回収のために真空ろ過装置に注がれます。(C)真空ろ過装置。これらはクリップを介して開閉され、フィルターがその間に挿入されます。4つのろ過装置を非常に短い時系列で並行して使用し、フィルターを交換した結果として時間を無駄にしないようにしました。(D)濾過後、培地上清をフラスコに排出し、その後の処分を行う。フィルターを取り外したときにシステム内の真空を維持するために、真空ろ過装置の下にバルブが設置されました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:架橋浮遊細胞と接着細胞。 (a)浮遊細胞用のVari-Xリンカーチャンバーを備えた架橋剤。細胞培養物をトレイの右上にあるサンプルインレット(漏斗)に注ぎます。(B)接着細胞または少量の浮遊細胞を架橋するためのプラスチックまたは石英ペトリ皿を保持できるトレイ。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:ライブラリの準備。 (A)Nrd1-HTPχCRAC実験からのオートラジオグラムの例。強くて集中したシグナルは、非常に短いRNAに架橋されたタンパク質を表し、上記の塗抹標本は、シーケンシングに十分な長さのRNAに架橋されたタンパク質を表します。(B)塗抹標本は、ゲル切除後に撮影したオートラジオグラムに示すように切除した。(C)χCRAC cDNAライブラリからの代表的なqPCR。この例では、cDNAの最大増幅は16サイクルで到達した。したがって、16サイクルが最終増幅に使用された。エラーバーは、3回のテクニカルqPCR反復の標準偏差を表します。(D)6%TBEゲル上のcDNAライブラリからのリン画像の例。(e)チップベースのキャピラリー電気泳動からのcDNA長および品質解析。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5: 哺乳類細胞における架橋をテストするための高RNaseテストiCLIP実験。 これは、さまざまな架橋エネルギーにわたるRNP回収の効率をテストしたGFP-RBM7 iCLIP実験からのオートラジオグラムです。免疫沈降は、GFP-RBM7を安定に発現する架橋細胞上の磁気ビーズに結合した抗GFP抗体を用いて行った。免疫沈降物を高濃度のRNase Iとともにインキュベートして、関連するRNAを短く均一な長さにトリミングしました。RNPは 、32P標識およびSDS-PAGEによって視覚化され、非架橋タンパク質の移動に近い、定義されたバンドとして移動しました。定量は、抗GFPウェスタンブロットシグナルに正規化された放射性標識RBM7-RNAシグナルの濃度測定分析の結果を示す。(A)一般的に使用されるUVP架橋剤と当社の架橋剤(Vari-X-linker;(B)石英(左)およびプラスチック(右)培養器上の架橋剤の架橋経時変化。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図6:Nrd1の標的への差次的で時間的な結合を示すχCRACのパワーを示すゲノムブラウザプロットの例。 各ボックスには、個々のゲノム領域のプロットが表示されます。矢印は、遺伝子がコードされている鎖を示します(左向き矢印=マイナス鎖、右向き矢印=プラス鎖)。タイムポイント(分)は、各サブプロットのy軸上のt0、t1、t2などで示されます。染色体と座標を示すローマ数字が表示されます。(A)グルコース欠乏時に、Nrd1は2つの高親和性グルコーストランスポーター、HXT6およびHXT7に結合し、これらは両方ともこの状態でアップレギュレーションされます。(B)Nrd1は、Nab344のすでに検証済みの標的であるImd3に結合し、グルコース飢餓後に強度が低下することが観察されています。(C)Tye7のNrd1結合は動的かつ一過性の性質を示し、グルコース飢餓後に8分間のストレス後に最小に減少する。しかしながら、結合はその後14分後に基礎レベルに戻る。読み取りは "100 万回あたりの読み取り数" (RPM; y 軸) に正規化されました。灰色のボックスは、ノンコーディングRNAをコードする領域を示す。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
χCRAC法は、新しい架橋および細胞採取装置と組み合わせることで、幅広いモデル生物に適用可能であり、RNA分野に一般的な関心が寄せられるため、大きな可能性を秘めています。χCRACを活用できる分野はたくさんあります。例えば、この方法は、タンパク質とRNA分子間の動的相互作用を伴うことが多いスプライソソームやリボソームなどの大きな高分子複合体へのタンパク質の階層的集合を測定するために使用できます。また、細胞が多様なストレスを受けた際のRNA崩壊因子とその基質との相互作用をモニタリングするためにも日常的に使用しています。これにより、適応応答のどの段階でこれらの因子が最も活発であるか、どの基質に結合するか、およびこれらの相互作用がどれほど動的であるかを判断できます。このようなデータにより、研究者は環境変化への適応における各要因の相対的な貢献を決定できるはずです。
χCRACは、デュアルアフィニティー精製タグ(HTFまたはHTP)を使用して、非常に厳格で変性した条件下でタンパク質を精製します。これにより、共精製されたRNAは、目的のタンパク質に共有結合で架橋されたRNAに対して高度に濃縮されます。ただし、アフィニティタグに依存することには欠点があります。例えば、タグはタンパク質機能を妨害する可能性があり、RNA結合相互作用の歪んだ読み出しを与える可能性があります。さらに、一部のモデル生物では、DNA断片をゲノムに組み込んだり、発現プラスミドを形質転換したりするための遺伝的ツールがまだ利用できないため、タグを常に利用できるとは限りません。ただし、χCRACプロトコルの一部を変更して、RBPの精製に抗体に依存するCLIPベースのプロトコルと互換性を持たせるのは簡単です。実際、この研究は、iCLIPベースの精製を当社の架橋剤と組み合わせることが可能であることを示しました。現在、ヒトRNA結合タンパク質と新生RNA転写産物との時間的関連を研究するためのCLIPプロトコルを開発中です。
新しいタンパク質に対してχCRACを実行する場合、最大の架橋を誘導するためにUV曝露を最適化する必要があります。高いUV曝露は精製ステップ中のRNAの回収率を低下させる可能性があるため、これは重要です。組換えRBPを発現する細胞を、100 mJ/cm2、250 mJ/cm2、500 mJ/cm2、および1 J/cm2の様々なUV用量に曝露した。次に、RNPを捕捉し、RNAを断片化して放射性標識しました。その後、RNPはSDS-PAGEによって解決され、どの曝露が最も強い信号(すなわち、最大架橋)を与えたかを推測するためにオートラジオグラムが撮影されました。
実験条件が最適化されると、χCRACを実行する際にいくつかの対照実験が推奨されます。まず、UV照射されたタグなしサンプルを使用して、精製ビーズへのバックグラウンド結合を監視できます。第二に、シフト実験中にχCRACを適用する場合、細胞を元の培地に戻す第2の時系列により、細胞自体のろ過がRNAレベルまたはタンパク質-RNA相互作用の変化を誘発するかどうかを調べることができます。
はじめにで述べたように、最近発表された多くの論文は、CLIPプロトコルに対するいくつかの最適化を示唆しています。これには、赤外線スキャン10を介してタンパク質−RNA複合体を検出するための蛍光標識アダプターの使用、ならびに得られるライブラリー12,45の複雑さを増大させることが示される様々な核酸精製およびサイズ選択ステップの最適化が含まれる。現在、χCRACプロトコルをさらに改良するために、これらの改善のいくつかを実装しています。ここで紹介するプロトコルには、元のCRACおよびχCRACプロトコルに対する多くの改善がすでに含まれており、データの複雑さが増しています。例えば、SDS-PAGEゲル上の架橋放射性タンパク質-RNA複合体を分離した後、それらをニトロセルロース膜に移し、架橋RNAをブロットから単離した。しかしながら、RNPの転写およびその後のRNA抽出は、特にRNAポリメラーゼサブユニットのような大きなRBPを扱う場合、非常に非効率的であり得る。これにより、架橋RNAの回収率が大幅に低下する可能性があります。現在のプロトコルでは、図1に示すように、架橋RNAはSDS-PAGEゲルスライスから直接抽出されます。これにより、架橋RNAの回収率が増加しました。さらに、cDNAのPCR増幅後、生成物は最初に3%の低融点アガロースゲルで分離され、次に175〜300 bp PCR産物がゲルから抽出されました。しかしながら、これらのゲルは容易に過負荷になる可能性があり、その結果、DNAの分離が非常に不十分になる。アガロースゲルをプレキャストTBEゲルに置き換えることで、より一貫したサイズ分離とPCR産物の回収率が向上しました。
開示事項
A. LangfordとW. Worboysは、営利企業であるUVO3と提携しています。彼らは、研究デザイン、データ収集と解釈、または出版のために研究を提出する決定において何の役割も果たしていませんでした。
謝辞
この研究は、ウェルカムトラスト(SGに091549、S.M.に109093 / Z / 15 / A)、ウェルカムトラスト細胞生物学センターコアグラント(092076)、医学研究評議会非臨床上級研究フェローシップ(MR / R008205 / 1からSG)、長期ポスドクフェローシップ(ALTF 1070-2017からR.A.C.)、からの助成金によってサポートされました。 独立研究基金デンマーク(T.H.J)。
資料
| Name | Company | Catalog Number | Comments |
| 1,4-dithioreitol | Merck | 10708984001 | Buffer component in mammalian cell lysis |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | General reaction tube |
| 2 mL tubes | Eppendorf | 0030 123.344 | For holding columns and collection of waste |
| 32P-yATP | Perkin Elmer | NEG502Z-250 | For radiolabelling the 5' end of the RNA |
| 4-12% Bis-Tris gel | Invitrogen | NP0321BOX | SDS-PAGE gel |
| 4X loading buffer | Novex | NP0008 | Protein loading dye concentrate |
| 50 bp ladder | New England Biolabs | N3236 | Reference ladder for excising region of interest from the amplified cDNA library |
| 50% PEG | NEB | B100045 | For the L5 linker ligation |
| 6% TBE gel | Invitrogen | EC6265BOX | For separation and purification of the cDNA library |
| Acetone | ACROS Organics | 423245000 | Washing of TCA-precipitated proteins |
| anti-FLAG beads | Sigma Aldrich | M8823-1ML | For purifcation of FLAG-tagged RBPs |
| ATP (100 mM) | Thermo Fisher Scientific | R0441 | For ligation of the L5 linker onto the 5' end of captured RNAs |
| Beta-mercaptoethanol | Sigma Aldrich | M3148-100ML | Buffer component |
| Biomax MS intensifying screen | Sigma Aldrich | Z363162-1EA | For intensifying the autoradiogram signal |
| Chloroform | Thermo Fisher Scientific | 1010219 | For phenol-chloroform extraction following RNA purification |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 11873580001 | For inhibition of cellular proteases after lysis |
| Complete supplement mixture -TRP | Formedium | DCS0149 | For preparation of synthetic defined medium |
| Costar Spin-X 0.22 µm filters | Sigma Aldrich | CLS8160 | For isolating the excised cDNAs following gel extraction |
| DNase RQ1 | Promega | M6101 | For DNA digest following cell lysis |
| dNTPs (10 mM) | Sigma Aldrich | 4638956001 | For reverse transcription and PCR |
| Ethanol | Thermo Fisher Scientific | 10041814 | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Ethylenediaminetetraacetic acid | Invitrogen | AM9261 | For protease K buffer |
| Exonuclease I | New England Biolabs | M0293 | For degradation of primers following PCR |
| Glass microfiber filters | Whatman | 1823-010 | For isolating the excised cDNAs following gel extraction |
| Glucose | Formedium | GLU03 | For preparation of glucose-containing, synthetic defined medium |
| Glycogen (20 mg/mL) | Roche | 10901393001 | Precipitation of proteins, RNA and DNA |
| GST-TEV | Homemade | Construct and purification protocol is available upon request | |
| Guanidium hydrochloroide | Thermo Fisher Scientific | 10071503 | Required for pulldown denaturing conditions and washing buffer |
| IgG beads | GE Healthcare | 17-0969-01 | For purification of protein A-tagged RBPs |
| Imidazole | Sigma Aldrich | I2399-100G | For elution of captured proteins from Nickel beads |
| Isoamyl alcohol | Thermo Fisher Scientific | A393-500 | For phenol-chloroform extraction following RNA purification |
| Luna Universal One-Step RT-qPCR | NEB | E3005S | For qPCR of the cDNA in order to calculate required number of PCR cycles |
| Magnesium chloride | Fluka Analytical | 63020-1L | For PNK buffer |
| Membrane filters | Millipore | AAWP09000 for yeast or HAWP09000 for bacteria | For vacuum filtration of cells |
| Micro bio-spin columns | Biorad | 732-6204 | For collecting eluate after gel extraction |
| Ni-NTA beads | Qiagen | 30210 | For secondary protein capture |
| NP-40 | Sigma Aldrich | I8896-100ML | Buffer component |
| Pfu polymerase | Promega | M7741 | For amplification of the cDNA library |
| Phenol | Sigma Aldrich | P4682-400ML | For phenol-chloroform extraction following RNA purification |
| Pierce spin columns | Thermo Fisher Scientific | 69725 | For on-column enzymatic reactions |
| Protease K | Roche | 3115887001 | For degradation of the RBP following gel extraction |
| Quartz Petri dish | UVO3 | N/A | For cross-linking of adherent cells. Available from https://www.vari-x-link.com for 400 GBP |
| Radiography films | Amersham | 28906843 | For autoradiography visualisation |
| RNAClean XP beads | Beckmann | A63987 | SPRI beads for clean up of RNAs and cDNAs |
| RNase H | New England Biolabs | M0297 | For degradation of RNAs following reverse transcription |
| RNase-It | Agilent | 400720 | For RNA digestion |
| rRNasin | Promega | N2511 | For inhibition of any contaminating RNases during enzymatic reaction |
| Sodium acetate | Sigma Aldrich | S2889-1KG | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Sodium chloride | Thermo Fisher Scientific | 7647-14-5 | Buffer component |
| Sodium deoxycholate | Sigma Aldrich | D6750-100G | Buffer component in mammalian cell lysis |
| Sodium dodecylsulfate | Sigma Aldrich | L3771-1KG | For protease K buffer |
| SUPERase-In | Invitrogen | AM2694 | For inhibition of cellular RNases after lysis |
| SuperScript IV | Thermo Fisher Scientific | 18090010 | For reverse transcription |
| T4 PNK | New England Biolabs | M0201 | For radiolabelling the 5' end of the RNA |
| T4 RNA ligase 1 | New England Biolabs | M0204 | For ligation of the L5 adaptor onto the RNA 5' end |
| T4 RNase ligase 2, truncated K222Q | NEB | M0351S | For ligation of the App_PE linker onto the 3' end of captured RNAs |
| TBE buffer (10X) | Invitrogen | 15581-028 | For running TBE gels |
| TEV protease | Homemade | For eluting captured proteins following FLAG capture | |
| Thermosensitive alkaline phosphatase | Promega | M9910 | For 5' and 3' dephosphorylation of RNAs |
| Trichloroacetic acid (100%) | Sigma Aldrich | T0699-100ML | For precipitation of RBP-RNA complexes |
| Tris hydrochloride | Invitrogen | 15504-020 | Buffer component |
| Triton X-100 | Sigma Aldrich | T8787-100ML | Buffer component in mammalian cell lysis |
| Vari Filter | UVO3 | N/A | Device for vacuum harvesting cells. Available from https://www.vari-x-link.com for 100 GBP |
| Vari-X-Linker | UVO3 | N/A | Cross-linker for cross-linking cells. Available from https://www.vari-x-link.com for 16,000 GBP |
| Yeast nitrogen base | Formedium | CYN0410 | For preparation of synthetic defined medium |
| Zirconia beads | Thistle | 11079105Z for yeast or 11079101Z for bacteria | For cell lysis via bead beating |
参考文献
- Ule, J., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 302 (5648), 1212-1215 (2003).
- Granneman, S., Kudla, G., Petfalski, E., Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 106 (24), 9613-9618 (2009).
- Licatalosi, D. D., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 456 (7221), 464-469 (2008).
- König, J., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature Structural & Molecular Biology. 17 (7), 909-915 (2010).
- Hafner, M., et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Aktaş, T., et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 544 (7648), 115-119 (2017).
- Huppertz, I., et al. iCLIP: Protein–RNA interactions at nucleotide resolution. Methods. 65 (3), 274-287 (2014).
- Li, X., et al. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Research. 41 (6), 3805-3818 (2013).
- Rosenberg, M., et al. Denaturing CLIP, dCLIP, Pipeline Identifies Discrete RNA Footprints on Chromatin-Associated Proteins and Reveals that CBX7 Targets 3′ UTRs to Regulate mRNA Expression. Cell Systems. 5 (4), 368-385 (2017).
- Zarnegar, B. J., et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nature Methods. 13 (6), 489-492 (2016).
- Kargapolova, Y., Levin, M., Lackner, K., Danckwardt, S. sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Research. 45 (10), 6074-6086 (2017).
- Van Nostrand, E. L., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods. 13 (6), 508-514 (2016).
- Flynn, R. A., et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA. 21 (1), 135-143 (2015).
- Brugiolo, M., Botti, V., Liu, N., Müller-McNicoll, M., Neugebauer, K. M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Research. 45 (18), 10452-10465 (2017).
- Sanford, J. R., et al. Identification of Nuclear and Cytoplasmic mRNA Targets for the Shuttling Protein SF2/ASF. PLOS ONE. 3 (10), e3369 (2008).
- Garzia, A., Meyer, C., Morozov, P., Sajek, M., Tuschl, T. Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods. 118-119, 24-40 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22 (10), 2031-2042 (2012).
- Chen, K., et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
- Ke, S., et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes & Development. 29 (19), 2037-2053 (2015).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Kudla, G., Granneman, S., Hahn, D., Beggs, J. D., Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proceedings of the National Academy of Sciences. 108 (24), 10010-10015 (2011).
- Sugimoto, Y., et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 519 (7544), 491-494 (2015).
- Hwang, H. W., et al. cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron. 95 (6), 1334-1349 (2017).
- Hwang, H. W., et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Reports. 15 (2), 423-435 (2016).
- Lee, F. C. Y., Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Molecular Cell. 69 (3), 354-369 (2018).
- Beckmann, B. M. RNA interactome capture in yeast. Methods. 118-119, 82-92 (2017).
- Granneman, S., Petfalski, E., Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. The EMBO Journal. 30 (19), 4006-4019 (2011).
- Schaughency, P., Merran, J., Corden, J. L. Genome-Wide Mapping of Yeast RNA Polymerase II Termination. PLOS Genetics. 10 (10), e1004632 (2014).
- Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Kresnowati, M. T. A. P., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology. 2, 49 (2006).
- Marguerat, S., Lawler, K., Brazma, A., Bähler, J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biology. 11 (6), 702-714 (2014).
- van Nues, R., et al. Kinetic CRAC uncovers a role for Nab3 in determining gene expression profiles during stress. Nature Communications. 8 (1), 12 (2017).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Tudek, A., Candelli, T., Libri, D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité?. Biochimie. 117, 28-36 (2015).
- Lingaraju, M., et al. The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nature Communications. 10 (1), 1-11 (2019).
- Lubas, M., et al. Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell. 43 (4), 624-637 (2011).
- Conrad, N. K., et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 154 (2), 557-571 (2000).
- Darby, M. M., Serebreni, L., Pan, X., Boeke, J. D., Corden, J. L. The Saccharomyces cerevisiae Nrd1-Nab3 Transcription Termination Pathway Acts in Opposition to Ras Signaling and Mediates Response to Nutrient Depletion. Molecular and Cellular Biology. 32 (10), 1762-1775 (2012).
- Webb, S., Hector, R. D., Kudla, G., Granneman, S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 15 (1), R8 (2014).
- Jensen, T. H., Jacquier, A., Libri, D. Dealing with Pervasive Transcription. Molecular Cell. 52 (4), 473-484 (2013).
- van Dijk, E. L., et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 475 (7354), 114-117 (2011).
- Thiebaut, M., et al. Futile Cycle of Transcription Initiation and Termination Modulates the Response to Nucleotide Shortage in S. cerevisiae. Molecular Cell. 31 (5), 671-682 (2008).
- Merran, J., Corden, J. L. Yeast RNA-Binding Protein Nab3 Regulates Genes Involved in Nitrogen Metabolism. Molecular and Cellular Biology. 37 (18), e00154-e00117 (2017).
- Bresson, S., Tuck, A., Staneva, D., Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Molecular Cell. 65 (5), 787-800 (2017).
- Buchbender, A., et al. Improved library preparation with the new iCLIP2 protocol. Methods. , (2019).
Erratum
Formal Correction: Erratum: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC
Posted by JoVE Editors on 2/17/2023. Citeable Link.
An erratum was issued for: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC. The Authors section was updated from:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Synthetic and Systems Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
to:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Mehak Chauhan1
Niki Christopoulou1
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Engineering Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved