
Method Article
Monitorización de la dinámica de interacción proteína-ARN in vivo a alta resolución temporal utilizando χCRAC
En este artículo
Erratum Notice
Resumen
La reticulación cinética y el análisis del ADNc es un método que permite la investigación de la dinámica de las interacciones proteína-ARN en células vivas a alta resolución temporal. Aquí se describe el protocolo en detalle, incluido el crecimiento de células de levadura, la reticulación UV, la recolección, la purificación de proteínas y los pasos de preparación de la biblioteca de secuenciación de próxima generación.
Resumen
La interacción entre las proteínas de unión al ARN (RBP) y sus sustratos de ARN exhibe fluidez y complejidad. Dentro de su vida útil, un solo ARN puede estar unido por muchas RBP diferentes que regularán su producción, estabilidad, actividad y degradación. Como tal, se ha hecho mucho para comprender la dinámica que existe entre estos dos tipos de moléculas. Un avance particularmente importante se produjo con la aparición de 'cross-l inking and immunoprecipitation' (CLIP). Esta técnica permitió una investigación rigurosa sobre qué ARN están unidos por un RBP particular. En resumen, la proteína de interés es UV reticulada a sus sustratos de ARN in vivo, purificada bajo condiciones altamente estrictas, y luego los ARN reticulados covalentemente a la proteína se convierten en bibliotecas de ADNc y se secuencian. Desde su concepción, se han desarrollado muchas técnicas derivadas para hacer que CLIP sea susceptible a campos particulares de estudio. Sin embargo, la reticulación mediante luz ultravioleta es notoriamente ineficiente. Esto da como resultado tiempos de exposición prolongados que hacen imposible el estudio temporal de las interacciones RBP-ARN. Para superar este problema, recientemente diseñamos y construimos dispositivos de irradiación UV y recolección de células muy mejorados. Usando estas nuevas herramientas, desarrollamos un protocolo para análisis resueltos en el tiempo de las interacciones RBP-ARN en células vivas a alta resolución temporal: enlace cinético CRoss-linking y Analysis of cDNAs (χCRAC). Recientemente utilizamos esta técnica para estudiar el papel de las RBP de levadura en la adaptación al estrés nutricional. Este manuscrito proporciona una descripción detallada del método χCRAC y presenta resultados recientes obtenidos con el RBP Nrd1.
Introducción
Los ARN a menudo dependen de las RBP para ejercer su función, lo que ha llevado a un gran interés en comprender la dinámica entre estas moléculas. Se han identificado muchas prácticas comerciales restrictivas en una amplia variedad de organismos. Sin embargo, siempre ha sido notoriamente difícil estudiar las interacciones RBP-ARN in vivo. Un gran avance en el estudio de tales interacciones se produjo con la aparición de CLIP1. Este método utiliza irradiación ultravioleta (UV, 254 nm) para inducir enlaces covalentes entre las RBP y sus ARN directamente unidos (es decir, reticulación a distancia cero). Posteriormente, la RBP de interés se inmunopurifica bajo condiciones estrictas para garantizar que solo se identifiquen los ARN reticulados covalentemente a las proteínas. Los ARN unidos se digieren parcialmente con RNasas y posteriormente se convierten en bibliotecas de ADNc para su secuenciación. El alto rigor de purificación es importante ya que aumenta en gran medida la especificidad de la recuperación de proteínas y ARN, que también se mejora aún más a través de la purificación SDS-PAGE del complejo ribonucleoproteína reticulada (RNP). CLIP y métodos relacionados también proporcionan información sobre la resolución de nucleótidos en el sitio de unión a proteínas, porque durante la preparación de la biblioteca de secuenciación, los aminoácidos que se entrecruzan al ARN con frecuencia terminan la transcriptasa inversa o hacen que la enzima introduzca mutaciones en este sitio 1,2,3.
Desde su introducción, el protocolo CLIP original ha producido una notable variedad de metodologías derivadas. Un avance particularmente importante se produjo con el desarrollo de HITS-CLIP (o CLIP-seq), que combina la secuenciación de alto rendimiento con el enfoque CLIP3. Desde entonces, esto ha sido adoptado por todas las metodologías basadas en CLIP. iCLIP introdujo mejoras en las técnicas de recorte y ligadura de adaptadores mediadas por RNasa que facilitan un mapeo más preciso de los sitios de unión de RBP4. PAR-CLIP combinó el etiquetado de 4tio-uridina/uracilo con la reticulación a 365 nm, lo que permitió mapear sitios de reticulación mediante el análisis de sustituciones T-C5. CRAC, urea-iCLIP, dCLIP y uvCLAP introdujeron condiciones de desnaturalización y pasos de purificación de afinidad dual que reducen aún más la unión de fondo a la resina de afinidad y aumentan aún más la especificidad de la captura de proteínas 2,6,7,8,9. Además, CRAC, uvCLAP y dCLIP introdujeron el etiquetado del RBP de interés con una etiqueta de afinidad, superando así la necesidad de generar anticuerpos específicos.
También se han realizado varias optimizaciones para acelerar la metodología CLIP. El protocolo CLIP original utilizaba radiomarcaje de los ARN capturados para visualizar los complejos RBP-ARN después de SDS-PAGE. Sin embargo, el uso de la radiactividad puede ser problemático para los laboratorios que no están preparados para este tipo de trabajo. irCLIP incorpora un adaptador acoplado a fluoróforos que facilita la visualización a través de imágenes infrarrojas10 y sCLIP utiliza biotinilación de ARN capturados para visualizarlos a través de HRP11 conjugado con estreptavidina. Además, eCLIP renuncia por completo al etiquetado de ARN; En cambio, la proteína se extirpa basándose únicamente en su tamaño conocido12. La purificación basada en estreptavidina también se ha utilizado para acelerar el proceso de preparación de la biblioteca en FAST-iCLIP, donde un adaptador 3' biotinilado se liga a los ARN y se utiliza para permitir la purificación después de la transcripción inversa y la circularización13. Las mejoras adicionales al protocolo iCLIP también aumentaron considerablemente la complejidad de las bibliotecas4.
Finalmente, CLIP ha sido modificado para permitir la captura de RBPs de diferentes subcompartimentos celulares 14,15, para visualizar RNAs recién transcritos usando inducción pulsada de ribonucleósidos fotoactivables 5,16,17, para capturar RNAs metilados18,19,20, para examinar las interacciones ARN-ARN 21,22, y para mapear 3' extremos 23,24.
A pesar de las grandes contribuciones de las técnicas basadas en CLIP para ayudar a nuestra comprensión de las interacciones entre las RBP y los ARN, se ha visto limitado por la ineficiencia de la reticulación UV. Aunque las células de cultivo cultivadas en una monocapa son generalmente relativamente fáciles de reticular, esto es significativamente más desafiante en tejidos o células en solución. Los tejidos pueden requerir múltiples rondas de exposición a los rayos UV para penetrar en las capas celulares requeridas, mientras que las células microbianas a menudo se cultivan en medios ricos que contienen compuestos aromáticos que absorben los rayos UV25. De hecho, se han utilizado tiempos de irradiación UV de hasta 30 min para generar suficiente reticulación entre las RBP y sus ARN unidos para tales muestras26,27,28. Esta exposición prolongada a los rayos UV induce respuestas de estrés dentro de la célula, como el daño del ADN inducido por los rayos UV, que puede contaminar los datos finales en algunas aplicaciones.
La mayoría de los estudios CLIP se han centrado en generar "instantáneas" únicas de interacciones específicas proteína-ARN en una célula. Sin embargo, las interacciones proteína-ARN son inherentemente dinámicas, particularmente cuando las células están sujetas a cambios en su entorno. Esto puede incluir una reducción repentina en la disponibilidad de nutrientes esenciales o cambios rápidos en la temperatura. Como tal, para comprender realmente el papel de una RBP durante el estrés, es mejor realizar análisis resueltos en el tiempo porque pueden capturar el espectro completo de objetivos de RBP durante el estrés y determinar en qué etapa de la respuesta al estrés está activa la RBP elegida. En particular, los estudios en levaduras mostraron que los primeros minutos de adaptación son absolutamente cruciales para la supervivencia y las vidas medias de ARN en bacterias pueden variar de minutos a segundos 29,30,31,32,33. Por lo tanto, tales análisis resueltos en el tiempo idealmente deberían realizarse a alta resolución temporal. Sin embargo, los largos tiempos de reticulación hacen que el estudio de las respuestas adaptativas en etapa temprana sea particularmente desafiante.
Para superar estos problemas, recientemente desarrollamos un método mejorado que es capaz de entrecruzar y recolectar células en escalas de tiempo de un minuto. Nuestro método χCRAC permite la medición cuantitativa de cambios dinámicos en las interacciones RBP-ARN a una resolución previamente no presenciada. Crucial para este método fue el desarrollo de un nuevo dispositivo de irradiación UV32 que reduce el tiempo de reticulación requerido en levaduras y bacterias en solución alrededor de 10 veces, congelando efectivamente las interacciones RBP-ARN instantáneamente. Además, para cosechar rápidamente las células después de la irradiación UV, desarrollamos un dispositivo de filtración al vacío que puede cosechar levadura de crecimiento exponencial en un cultivo de 0,5 L en alrededor de 30 s32. Estas innovaciones tecnológicas permiten el estudio de la dinámica de RBP-RNA a escala de minutos. Además, también introdujimos varias optimizaciones al protocolo CRAC original2 para aumentar su practicidad.
Usando χCRAC, recientemente estudiamos el objetivo de una RBP nuclear de levadura, Nab3, en respuesta a la privación de glucosa. En Saccharomyces cerevisiae, Nab3 puede formar un complejo con Nrd1, un RBP, y la helicasa de ARN Sen1 para formar el complejo NNS. La unión de NNS a la ARN polimerasa y la transcripción naciente puede desencadenar la terminación transcripcional34. Este complejo está involucrado principalmente en la eliminación de transcripciones crípticas de ARN no codificante, pero también se ha demostrado que regula la expresión de genes codificadores de proteínas. El estudio mostró una focalización diferencial de Nab3 a transcripciones no codificantes y codificantes después de solo un minuto de estrés32. Demostramos que la terminación co-transcripcional por Nab3 da como resultado una expresión muy transitoria, similar a un pulso, de genes de retrotransposón, que habría sido difícil de detectar utilizando enfoques tradicionales basados en CLIP. Además, los cortos tiempos de irradiación UV en nuestro reticulante UV también aumentaron significativamente la recuperación de ARN no codificantes de corta duración32. Es probable que χCRAC sea una herramienta crucial para dilucidar no solo cómo las RBP dan forma a la respuesta al estrés en escalas de tiempo inmediatas, sino también sus roles cambiantes durante todo el ciclo de vida de una respuesta. Este manuscrito proporciona una descripción detallada de todos los pasos del protocolo χCRAC. Con fines ilustrativos, el método se utilizó para estudiar la proteína de levadura Nrd1, que está involucrada en la terminación transcripcional y la desintegración del ARN35,36, y su objetivo de ARN en respuesta a la privación de glucosa en una multitud de puntos temporales. Finalmente, también demostramos que nuestra unidad de irradiación UV puede entrelazar rápidamente RBP con ARN en células HeLa, lo que permite realizar también análisis de alta resolución resueltos en el tiempo en células adherentes.
Protocolo
| TN150 |
| 50 mM Tris pH 7.8 |
| 150 mM NaCl |
| 0,1% NP-40 |
| Inhibidor de la proteasa 1X |
| TN1000 |
| 50 mM Tris pH 7.8 |
| 1M NaCl |
| 0,1% NP-40 |
| NP-PNK |
| 50 mM Tris-HCl pH 7.8 |
| 10 mM MgCl2 |
| 0,1% NP-40 |
| 5 mM de beta-mercaptoetanol |
| 5 x PNK |
| 250 mM Tris-HCl pH 7.8 |
| 50 mM MgCl2 |
| 50 mM de beta-mercaptoetanol |
| BM I |
| 50 mM Tris-HCl pH 7.8 |
| 300 mM NaCl |
| 10 mM de imidazol |
| 6M guanidina-HCl |
| 0,1% NP-40 |
| 5 mM de beta-mercaptoetanol |
| BM II |
| 50 mM Tris-HCl pH 7.8 |
| 50 mM NaCl |
| 10 mM de imidazol |
| 0,1% NP-40 |
| 5 mM de beta-mercaptoetanol |
| Búfer de elución |
| 50 mM Tris pH 7.8 |
| 50 mM NaCl |
| 250 mM de imidazol |
| 0,1% NP-40 |
| 5 mM de beta-mercaptoetanol |
| Tampón proteasa K |
| Tris de 50 mM |
| 0,1% NP-40 |
| 5 mM β-mercaptoetanol |
| 1% SDS |
| 5 mM EDTA |
| 50 mM NaCl2 |
| Tampón de lisis de mamíferos |
| 50 mM Tris-HCl pH 8 |
| NaCl de 100 mM |
| 0.5% v/v Triton X-100 |
| 0,25% p/v Na-desoxicolato |
| 0,1% p/v SDS |
| 5 mM EDTA |
| 1 mM TDT (añadido fresco) |
| Inhibidor de la proteasa 1X |
Tabla 1: Los buffers requeridos para χCRAC y sus composiciones.
1. Reticulación UV y producción de lisado
- Microorganismos en solución
- Inocular 3,5 L del medio deseado con levadura de un cultivo nocturno hasta un OD inicial600 de 0,05. Cultivar a 30 °C con agitación continua a 180 rpm.
- Durante el crecimiento, prepare otros materiales necesarios.
- Prepare un recipiente de nitrógeno líquido.
- Preparar 3 L de medio inductor de estrés y calentar a 30 °C en un baño maría.
- Configure el aparato filtrante, encienda la reticulación (Figura 2A) y etiquete los tubos cónicos de 50 ml, uno para cada punto de tiempo.
- Una vez que las células alcancen el OD 600 deseado, vierta500 ml de células directamente en el reticulante e irradie UV con 250 mJ de 254 nm UV. Consulte la Figura 2A y la Figura 3A para obtener detalles sobre el uso del reticulador.
NOTA: La energía de irradiación UV debe optimizarse cuidadosamente para cada proteína de interés. Consulte la discusión para obtener más detalles. - Después de la reticulación, filtre las células utilizando uno de los dispositivos de filtración al vacío (Figura 2B, C). Enrolle la membrana con las células filtradas, coloque en el tubo cónico t = 0 (tiempo cero) 50 ml y congele rápidamente en nitrógeno líquido.
- Filtre las celdas restantes en seis filtros diferentes. Resuspender las células recogidas en los 3 L de medio inductor de estrés previamente calentado dejando caer las membranas en el medio y mezclando vigorosamente con una tira durante 50 s. Después de los 50 s, prepárese para tomar la muestra t = 1.
- Después de 1 minuto, entrecruza 500 ml de células y cosecha a través de filtración como en los pasos 1.1.3–1.1.4. Repita después de 2, 4, 8, 14 y 20 minutos, o diferentes puntos de tiempo según sea necesario.
- Conservar los tubos cónicos que contienen las células a -80 °C. Ajuste la solución salina tamponada con fosfato (PBS) a 4 °C durante la noche.
- Al día siguiente, tome cada tubo cónico que contenga una muestra reticulada y resuspenda las células en 25 ml de PBS frío agitando vigorosamente.
- Transfiera las suspensiones celulares a nuevos tubos cónicos y gire a 4.600 x g, 5 min a 4 °C.
- Vierta el PBS, gire rápidamente de nuevo para recoger el PBS residual y luego decanta el líquido restante con una pipeta.
- Calcule el peso del pellet en el tubo comparándolo con un tubo vacío.
- Agregue dos volúmenes de pellets de TN150 helado, 60 μL de DNasa 1 y 10 μL de inhibidor de RNasa. Incubar en hielo durante 30 min.
- Por ejemplo, para 400 mg de células, agregue 800 μL de TN150 helado.
- La adición de la DNasa no es esencial para la mayoría de las proteínas solubles, pero es muy importante cuando se estudian proteínas unidas a la cromatina, como la ARN polimerasa. Además, reduce la viscosidad de los lisados bacterianos. Es muy importante utilizar exactamente dos volúmenes de pellets del tampón de lisis, o la eficiencia de lisis puede disminuir.
- Agregue tres volúmenes de pellets (en ml) de perlas de zirconia a la suspensión celular. Para la levadura, use perlas de 0.5 mm de diámetro y para bacterias use 0.1 mm.
- Por ejemplo, para 400 mg de células, mida 1,2 ml de perlas de zirconia en un tubo de 1,5 ml y agréguelas a las células resuspendidas en tampón de lisis.
- Vórtice las suspensiones celulares durante 1 min, luego colóquelas en hielo durante 1 min. Repita para un total de 5x.
- Agregue dos volúmenes de pellets de tampón TN150 y vórtice vigorosamente para mezclar.
- Centrifugar la suspensión en el tubo cónico a 4.600 g durante 20 min a 4 °C en una centrífuga de sobremesa.
- Después de la centrifugación, tome una muestra de 50 μL del sobrenadante para el futuro análisis de Western blot para examinar la expresión de la proteína de toda la célula.
- Transfiera los sobrenadantes a tubos de 1,5 ml y haga girar el lisado durante 20 minutos a 20.000 x g a 4 °C, en una microfuga.
- Alternativamente, si usa tubos de 5 ml, centrifugar a 13,000 x g durante 20 min.
- Después de la centrifugación, tomar una muestra de 50 μL del sobrenadante para el futuro análisis de Western blot para examinar la expresión soluble de la proteína.
- Proceda a la captura de RBP (sección 2).
- Células adherentes cultivadas
- Siembre suficientes células adherentes en una placa de Petri 24 h antes de la reticulación UV para que puedan alcanzar el 80% de confluencia al día siguiente. Cultivar durante la noche en el medio deseado en una incubadora de cultivo celular a 37 °C, 5%CO2.
NOTA: Si se utilizan placas de Petri de cuarzo, es beneficioso promover la adhesión celular a través del tratamiento del material de cultivo con poli-D-lisina (70,000-140,000 wt) y suero fetal de ternera (FCS) 2.5 h antes de la siembra. Agregue suficiente poli-D-lisina para cubrir toda la superficie de crecimiento e incube a temperatura ambiente (RT) durante 5 min. A continuación, la placa de Petri de cuarzo debe enjuagarse bien con agua y secarse en la incubadora de cultivo celular durante 2 h o hasta que esté completamente seca. Luego, agregue suficiente FCS para cubrir completamente la superficie de crecimiento y colóquelo en la incubadora durante al menos 30 minutos. El FCS debe eliminarse completamente antes de sembrar células. - Una vez que las células hayan alcanzado el 80% de confluencia, retire el medio y lave con 15 ml de PBS helado. A continuación, retire completamente todo el líquido restante y proceda inmediatamente al siguiente paso.
- Transfiera la placa de Petri a la bandeja para las células adherentes (Figura 3B) e irradie UV con 300 mJ de 254 nm UV. Consulte la Figura 2A y la Figura 3B para obtener detalles sobre el uso de la reticulación.
NOTA: La energía de irradiación UV debe optimizarse cuidadosamente para cada proteína de interés. Consulte la discusión para obtener más detalles. - Inmediatamente después de la reticulación, coloque la placa de Petri en hielo y agregue 10 ml de PBS helado. Recolectar células raspando y transfiéralas a un tubo cónico de 15 ml. Pellet por centrifugación a 300 x g durante 5 min a 4 °C.
- Retire el PBS y vuelva a suspender el pellet celular en 1 ml de PBS helado y transfiéralo a un tubo de microcentrífuga de 1,5 ml. Células de pellet de nuevo por centrifugación durante 5 min a 300 x g a 4 °C.
- Retire el PBS y congele los gránulos de células en hielo seco. Conservar los gránulos celulares a -80 °C hasta que sea necesario.
- Repita los pasos 1.2.3 a 1.2.6 para cada punto de tiempo.
- Resuspender los gránulos celulares en 1 ml de tampón de lisis y transferir a un tubo cónico de 15 ml. Luego, agregue 1 ml de tampón de lisis para un total de 2 ml.
- Añadir 5 μL de inhibidor de la RNasa de mamíferos.
- Sonicate 5x durante 10 s sobre hielo a 10 amperios. Espere 30 s entre rondas de sonicación.
- Calcule la concentración de proteína de cada muestra y normalice a la concentración más baja.
- Transfiera 1.98 ml de lisado a un tubo de 2 ml.
- Añadir 10 μL de DNasa I e incubar a 37 °C durante 5 min con agitación a 1.200 rpm.
- Centrifugar el lisado a 16.000 x g durante 20 min a 4 °C.
- Después de la centrifugación, tomar una muestra de 50 μL del sobrenadante para el futuro análisis de Western blot para examinar la expresión soluble de la proteína.
- Proceda a la captura de RBP (sección 2).
- Siembre suficientes células adherentes en una placa de Petri 24 h antes de la reticulación UV para que puedan alcanzar el 80% de confluencia al día siguiente. Cultivar durante la noche en el medio deseado en una incubadora de cultivo celular a 37 °C, 5%CO2.
2. Captura RBP
- Lave las perlas magnéticas anti-FLAG (75 μL de suspensión por muestra) o IgG agarosa (500 μL de suspensión por muestra) 3x con 5 ml de TN150. Resuspender en un volumen final de 700 μL de TN150 y añadir 100 μL de perlas lavadas a siete tubos cónicos de 15 ml.
- Almacenar en hielo hasta que sea necesario.
- Una vez aclarados los lisados, añadir el sobrenadante al tubo que contiene las perlas anti-FLAG/IgG.
- Nutar a 4 °C durante 2 h.
NOTA: Algunos protocolos describen incubaciones nocturnas con las perlas, pero esto no se recomienda, porque los largos tiempos de incubación pueden reducir drásticamente la recuperación de ARN reticulados.
3. Lavado de las perlas y el corte TEV de las etiquetas
- Coseche las perlas y retire el lisado.
- Tomar una muestra de 50 μL del sobrenadante para futuros análisis de Western blot para examinar la proteína no capturada.
- Vuelva a suspender las perlas en TN1000 helado y transfiéralas a un tubo de 1,5 ml. Lavar durante 10 min, 4 °C, con nutación. Repita para un total de tres lavados.
- Si usa perlas de agarosa IgG, lávelas con 5 ml de TN1000. Si utiliza perlas anti-FLAG, use 2 ml.
- A continuación, lave las perlas 3x con TN150, con el mismo volumen que el anterior.
- Después del tercer lavado, resuspender las perlas en 600 μL de TN150.
- Agregue 30 U de proteasa GST-TEV casera a la suspensión de perlas y gire durante 2 h a RT.
NOTA: La proteasa GST-TEV recombinante ahora también está disponible comercialmente, pero no se ha probado con este protocolo.- Durante la digestión, prepárese para los siguientes pasos configurando columnas de tres tubos de 1,5 ml para cada muestra (es decir, para siete muestras, tenga tres filas de siete columnas).
- A la última fila de tubos, añadir 0,4 g de clorhidrato de guanidio, 27 μL de cloruro de sodio 5M y 3 μL de imidazol 2,5 M (pH = 8). Tenga en cuenta que el pH del imidazol debe ser 8. Esto es fundamental para mantener la integridad del ARN.
- Además, lave el volumen requerido de perlas de níquel en WB I 3x. Utilice 100 μL de purín por muestra. Después del lavado final, vuelva a suspender las perlas en el mismo volumen original de WB I y guárdelas en hielo.
- Una vez completada la digestión del TEV, recoger el sobrenadante utilizando un bastidor magnético para perlas anti-FLAG o centrifugación para perlas IgG, y transferir a la primera fila de los tubos previamente configurados.
- Tomar una muestra de 50 μL del eluido TEV para el análisis de Western blot.
- Ajuste una incubadora termoblock a 37 °C. A la segunda fila de tubos, añadir 1 μL de cóctel de RNasa (dilución 1:50).
- Tome 550 μL de eluido TEV de la primera fila de tubos y agréguelos a la segunda fila (que contiene el cóctel RNasa). Pipetear vigorosamente para asegurar la mezcla.
- Después de completar esto para la primera muestra, coloque inmediatamente el tubo en el termobloque y encienda un temporizador. Pase a las muestras siguientes, de modo que cada una esté escalonada.
- Incubar durante exactamente 5 min. Una vez completado, retire la primera muestra del termobloque y transfiera la solución a la tercera fila de tubos (que contiene el polvo de clorhidrato de guanidio).
NOTA: Una incubación de 5 minutos con una dilución 1:50 del cóctel de RNasa suele ser adecuada para la mayoría de las proteínas, pero este paso deberá optimizarse cuidadosamente con diferentes tiempos de incubación o concentraciones para cada proteína para asegurarse de que los ARN reticulados sean del tamaño correcto (30-100 nt). - Inmediatamente vórtice durante un par de segundos a toda velocidad para disolver el polvo de guanidio y luego pasar a la siguiente muestra.
- Después de que todas las muestras se hayan transferido al polvo de guanidio, vuelva a vorátice para asegurarse de que todo el polvo esté completamente disuelto.
- Añadir 100 μL de perlas de níquel lavadas y rotar durante la noche a 4 °C. Esta incubación se puede acortar a 2 h.
4. Tratamiento de la fosfatasa alcalina en el cordón
- Ajuste un termobloqueo a 37 °C.
- Coloque una columna de centrifugado de purificación en un tubo de 2 ml, uno para cada muestra. Transfiera las perlas de níquel a las columnas y permita que el sobrenadante drene. Después, asegúrese de que todas las perlas de níquel se hayan retirado del tubo de 1,5 ml enjuagando con WB I y aplicándolas a la columna.
- Coloque 2 mL de tubos, seis por muestra (uno para recoger cada lavado). Mantenga el exterior de las columnas seco para mantener el flujo. Lave las perlas 3 veces con 500 μL de WB I y luego 3 veces con 500 μL de NP-PNK.
- Cierre la tapa de la columna de centrifugado y gire brevemente las perlas para eliminar el exceso de amortiguador.
- Coloque el tapón en la columna, coloque las columnas en tubos de 1,5 ml y agregue 60 μL de la mezcla de reacción que se ve en la Tabla 2.
| Componente | 1x | 7,5 veces |
| 5 x búfer PNK | 12 | 90 |
| Fosfatasa alcalina | 4 | 30 |
| Inhibidor de la RNasa | 2 | 15 |
| H2O | 42 | 315 |
| Volumen final | 60 μL | 450 μL |
Tabla 2: Mezcla de reacción de fosfatasa alcalina.
- Incubar las perlas durante 1 h a 37 °C.
- Lave las perlas 1x con 500 μL de WB I para inactivar la fosfatasa alcalina y luego 3x con 500 μL de tampón NP-PNK. Asegúrese de enjuagar bien el interior de la columna con el tampón NP-PNK para eliminar cualquier rastro de guanidio.
5. Ligadura en perlas del enlazador App-PE al extremo 3' del ARN
- Girar el tampón restante y añadir 60 μL de la mezcla especificada en la Tabla 3 (véase la Tabla 4 para la secuencia App-PE) a las columnas. Incubar la reacción durante 6 h a 25 °C.
| Componente | 1x | 7,5 veces |
| 5 x búfer PNK | 12 | 90 |
| Adaptador de App-PE (100 μM) | 0.6 | 4.5 |
| T4 ARN ligasa 2 truncada K227Q | 3 | 22.5 |
| Inhibidor de la RNasa | 1.5 | 11.25 |
| 50% PEG 8000 | 12 | 90 |
| H2O | 30.9 | 231.75 |
| Volumen final | 60 μL | 450 μL |
Tabla 3: Mezcla de reacción de ligadura del enlazador App-PE.
| Nombre del oligonucleótido | Secuencia (5'-3') | |||
| L5Aa | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrUrArArGrCrN-OH | |||
| L5Ab | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrArUrUrArGrCrN-OH | |||
| L5Ac | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrGrGrGrCrArGrCrCrN-OH | |||
| L5Ad | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrCrGrCrUrUrArGrCrCrN-OH | |||
| L5Ba | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrArGrArGrCrN-OH | |||
| L5Bb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrGrUrGrArGrCrN-OH | |||
| L5Bc | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrCrArCrUrGrCrCrN-OH | |||
| L5Bd | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrUrCrUrCrUrArGrCrN-OH | |||
| L5Ca | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrCrUrArGrCrN-OH | |||
| L5Cb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrCrUrNrNrNrUrGrGrArGrCrN-OH | |||
| L5CC | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrArCrUrCrArGrCrN-OH | |||
| L5Cd | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrGrArCrUrUrArGrCrN-OH | |||
| L5Da | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrCrGrUrGrArUrN-OH | |||
| L5Db | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrGrCrArCrUrArN-OH | |||
| L5Dc | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrUrArGrUrGrGrCrN-OH | |||
| L5Dd | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrArUrCrArCrGrN-OH | |||
| L5Ea | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrCrArCrUrGrUrN-OH | |||
| L5Eb | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrGrUrGrArCrArN-OH | |||
| L5Ec | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrNrUrGrUrCrArCrN-OH | |||
| L5Ed | invddT-ACACrGrArCrGrGrCrUrCrUrCrGrArUrCrUrNrNrNrArCrArGrUrGrGrN-OH | |||
| App_PE | App-NAGATCGGAAGAGCACACGTCTG-ddC | |||
Tabla 4: Las secuencias de los adaptadores de ADN y ARN necesarios para la ligadura en los extremos 5' y 3' de los ARN capturados. Estos se purificaron a través de HPLC sin RNasa.
- Lave las perlas 1x con 500 μL de WB I y 3x con 500 μL de tampón NP-PNK. Coloque la columna en un tubo nuevo y gire el tampón restante.
6. Fosforilación en el cordón de los extremos 5' del ARN
- Añadir 80 μL de la mezcla especificada en el cuadro 5 a las columnas. Incubar la reacción durante 40 min a 37 °C.
NOTA: Las muestras ahora serán altamente radiactivas. Por lo tanto, todo el trabajo posterior debe realizarse detrás de una pantalla protectora y los desechos deben eliminarse de acuerdo con las normas locales de salud y seguridad.
| Componente | 1x | 7,5 veces |
| 5 x búfer PNK | 16 | 120 |
| 32P-ɣATP (10 μCi/μL) | 3 | 22.5 |
| T4 PNK | 3 | 22.5 |
| H2O | 58 | 435 |
| Volumen final | 80 μL | 600 μL |
Tabla 5: Mezcla de reacción de fosforilación.
- Añadir 1 μL de ATP de 100 mM y dejar que la reacción proceda durante otros 20 min. Esto asegurará que casi todos los extremos 5' tengan fosfatos para facilitar la ligadura del enlazador 5'.
- Instale 2 mL de tubos, cinco por muestra.
- Lave las perlas 1x con 500 μL de WB I y 3x con 500 μL de tampón NP-PNK. Tenga en cuenta que estas eluciones serán muy radiactivas y, por lo tanto, deben eliminarse adecuadamente.
- Mueva la columna al tubo final y gire el búfer restante.
7. Ligadura en el cordón del enlazador de 5'
NOTA: Los enlazadores 5' contienen un código de barras de ARN que se utiliza para la identificación de cada muestra después de la secuenciación. Por lo tanto, es absolutamente crucial tener en cuenta qué enlazador se utiliza para qué muestra.
- Añadir 78 μL de la mezcla descrita en la tabla 6 a las columnas. Añadir 2 μL de adaptador de 5' (100 μM; ver Tabla 4) a cada tubo e incubar durante la noche a 18 °C.
| Componente | 1x | 7,5 veces |
| 5 x búfer PNK | 16 | 120 |
| ATP (10 mM) | 8 | 60 |
| Inhibidor de la RNasa | 2 | 15 |
| ARN ligasa T4 | 4 | 30 |
| H2O | 48 | 360 |
| Volumen final | 78 μL | 585 μL |
Tabla 6: Mezcla de reacción de ligadura de enlazador 5'.
- Al día siguiente, lavar las perlas 1x con 500 μL de WB I y 3x con 500 μL de WB II y transferir las columnas a un nuevo tubo de 2 mL.
8. Elución, SDS-PAGE y extracción de ARN
- Ajuste la centrífuga a 4 °C. Prepare dos filas de tubos de 1,5 ml por muestra para la elución.
- Gira el volumen vacío de las columnas con cuentas de níquel con un giro rápido. Coloque las columnas en la primera fila de tubos de elución y agregue 200 μL de tampón de elución. Espere 2 minutos, luego fuerce el búfer a través de la columna con un giro rápido.
- Mueva las columnas a la segunda fila de tubos y repita el paso 8.2. Cada muestra ahora tendrá 400 μL de eluido en total, divididos en dos tubos de 1,5 ml.
- Tome todos los eluidos y transfiéralos juntos a un tubo de 5 ml. Añadir 2 μL de 20 mg/ml de glucógeno. Por lo tanto, si se utilizan siete muestras, ahora habrá 2,8 ml de eluido agrupado en el tubo de 5 ml.
- Añadir 100 μL de ácido tricloroacético (ATC) por muestra [por ejemplo, 700 μL de ATC para 7 muestras (2,8 ml de eluido agrupado)] al tubo de 5 ml, y vórtice bien durante 30 s.
- Incubar en hielo durante 20 min.
- Centrifugadora durante 30 min a 17.000 x g, 4 °C, en una centrífuga de sobremesa.
- Retire con cuidado el sobrenadante del tubo cónico, comprobando la pipeta con un contador Geiger para asegurarse de que el pellet no se haya retirado accidentalmente. Si es así, devuelva el sobrenadante al tubo y centrifugar durante otros 10 minutos.
NOTA: El sobrenadante aún podría ser altamente radiactivo. Asegúrese de usar el blindaje adecuado. - Resuspender completamente el pellet en 2 ml de acetona helada.
- Centrifugadora durante 15 min a 17.000 x g, 4 °C.
- Retire la mayor cantidad posible de acetona con una pipeta P1000. Luego, gire brevemente el tubo para recoger pequeñas gotas de acetona y luego retire con una pipeta P10. Secar durante 2 min en una campana extractora.
NOTA: El sobrenadante de acetona todavía puede ser radiactivo. Asegúrese de usar el blindaje adecuado. - Resuspender la muestra en 30 μL de tampón de carga de proteína 1x. Para asegurarse de que el pellet se resuspende correctamente, compruebe que la gran mayoría de la radiactividad está ahora presente en el tampón de carga y no en el tubo de 1,5 ml retirando la solución en una pipeta P200 y midiendo la actividad que queda en el tubo de 1,5 ml utilizando un contador Geiger.
- Calentar la muestra durante 10 min a 65 °C. Cargar en un gel prefabricado Bis-Tris de 1 mm, 4-12% y ejecutar durante 1,5 h a 125 V en tampón MOPS.
- Después de que el gel haya terminado de funcionar, abra el casete de gel. El gel debe retenerse en la placa inferior. Deseche la parte superior.
- Envuelva el gel en una película adhesiva y luego asegúrelo con cinta adhesiva en el interior de un casete hermético. Asegúrese de que el casete tenga una pantalla amplificadora para mejorar la señal.
- Exponer una película autorradiográfica al gel y conservar el casete a -80 °C durante la exposición. El tiempo de exposición variará entre proteínas con diferentes eficiencias de reticulación.
- Al colocar la película, debe haber una forma de realinearla al casete para cortar la banda de interés en el paso posterior. Para garantizar esto, use una regla fluorescente y también asegúrese de que el gel esté en una esquina del casete, que luego está cubierto por la película también colocada en la esquina más extrema.
NOTA: Como regla general, los eluidos en el búfer de carga que dan una lectura de al menos ~ 250 cps cuando se muestran a un contador Geiger dan suficiente señal para una exposición de 3 h. De lo contrario, se realiza una exposición nocturna.
- Al colocar la película, debe haber una forma de realinearla al casete para cortar la banda de interés en el paso posterior. Para garantizar esto, use una regla fluorescente y también asegúrese de que el gel esté en una esquina del casete, que luego está cubierto por la película también colocada en la esquina más extrema.
- Desarrolla la película. Corte la película adhesiva que cubre el gel, pero no mueva el gel. De lo contrario, la imagen se desplazará del gel.
NOTA: Es probable que el gel sea altamente radiactivo. Asegúrese de usar el blindaje adecuado al cortar la rodaja de gel. - Coloque la película sobre el gel y elimine la banda de interés. Coloque la rodaja de gel en un tubo de 2 ml.
- Triture la rodaja de gel con una punta de pipeta P1000 y agregue 600 μL de tampón K de proteinasa K más 200 μg de proteinasa K (este protocolo utiliza 10 μL de una solución de proteinasa K de 20 mg / ml). Incubar durante 2 h a 55 °C con agitación vigorosa.
- Luego, corte el extremo de una punta P1000 con un bisturí limpio y transfiera las piezas sobrenadantes y de gel a una columna de centrifugado colocada en un tubo de 2 ml.
- Gire la columna durante 1 minuto a 17.000 x g en RT. Recoja el flujo que contiene los ARN radiactivos aislados.
- Realizar una extracción de fenol:cloroformo.
- Añadir 50 μL de acetato de sodio 3 M, pH = 5,2, y 500 μL de fenol:cloroformo y vórtice bien. Girar durante 5 minutos a 17.000 x g. Retire la capa superior acuosa y colóquela en un nuevo tubo de 1,5 ml.
- Añadir 500 μL de cloroformo y vórtice vigorosamente durante 10–15 s. Girar durante 5 min a 17.000 x g en RT. Retire la capa acuosa y colóquela en un nuevo tubo de 1,5 ml.
- Agregue 1 μL de 20 mg / ml de glucógeno y 1 ml de etanol helado, 96%. Precipitar durante 30 min a -80 °C o durante la noche a -20 °C.
- Centrifugadora durante 30 min a 4 °C, 17.000 x g. Retirar el sobrenadante, añadir 500 μL de etanol al 70% y centrifugar durante 5 min, 4 °C a 17.000 x g. Retire todo el etanol, realice un giro rápido para recoger residuos y elimine el exceso con una pipeta P10.
- Seque el pellet durante ~ 3 minutos en una campana extractora. Resuspender en 20 μL de agua tratada con DEPC.
- Almacene el ARN a -80 °C durante la noche o proceda inmediatamente al paso de transcripción inversa.
9. Transcripción inversa
- Añadir 2 μL de 10 μM RT oligo (PE_reverse; ver Tabla 7) y 4 μL de 5 mM dNTPs a los 20 μL de ARN.
| Nombre del oligonucleótido | Secuencia (5'-3') | |||
| P5 hacia adelante | AATGATACGGCGACCGAGATCTACTACCTTTCCCTACACGACGCTCTCCGATCT | |||
| BC1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC7 | CAAGCAGAAGACGGCATACGAGATCAGATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC8 | CAAGCAGAAGACGGCATACGAGATTAGCTTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC9 | CAAGCAGAAGACGGCATACGAGATCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC10 | CAAGCAGAAGACGGCATACGAGATATCACGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| PE_reverse | CAGACGTGTGCTCTTCCGATCT | |||
Tabla 7: Los cebadores de PCR (incluidas las secuencias de códigos de barras) y el cebador de transcripción inversa.
- Transfiera a un termobloque precalentado a 85 °C durante 3 min, luego enfríe sobre hielo durante 5 min. Recoja el contenido del tubo mediante una breve centrifugación y luego agregue 8 μL de tampón de transcriptasa inversa 5x, 2 μL de DTT de 100 mM y 2 μL de inhibidor de RNasa.
- Incubar la mezcla a 50 °C durante 3 min y, a continuación, añadir 2 μL de transcriptasa inversa e incubar durante 1 h a 50 °C.
- Inactivar la transcriptasa inversa por incubación a 65 °C durante 15 min.
- Transfiera los tubos a un termobloque precalentado a 37 °C y deje actuar durante 3 minutos para que se aclimaten.
- Añadir 2 μL de RNasa H e incubar durante 30 min a 37 °C.
- Aísle el ADNc usando perlas SPRI.
- Añadir dos volúmenes de 84 μL de cuentas. Incubar durante 15 min. Coloque las cuentas en una rejilla magnética y déjelas durante 1 minuto para cosechar las cuentas.
- Retire y deseche el sobrenadante y agregue 200 μL de etanol al 70%. No retire las perlas de la rejilla magnética. Incubar las perlas con el etanol durante 30 s.
- Retire el etanol y repita el paso de lavado. Retire todo el etanol residual con una punta P10.
- Coloque las perlas en una campana extractora durante 2 minutos para secarlas. Retire las perlas de la rejilla, vuelva a suspenderlas en 12 μL de agua y luego vuelva a colocar las perlas en la rejilla. Eliminar 11 μL de sobrenadante.
- Congelar el ADNc a -20 °C o proceder inmediatamente a la etapa de PCR.
10. Reacción de qPCR
- Antes de la PCR final para la amplificación de los ADNc, se realiza una reacción en cadena de la polimerasa cuantitativa (qPCR) para identificar el número óptimo de ciclos para amplificar los ADNc para evitar la sobreamplificación de la biblioteca.
- Configure una reacción de qPCR en hielo de acuerdo con la Tabla 8. Consulte la Tabla 7 para todos los cebadores.
| Componente | 1x |
| 2x reacción qPCR mastermix | 5 |
| Imprimación P5 de 0,1 μM (hacia adelante) | 0.8 |
| Imprimación BC de 0,1 μM (inversa) | 0.8 |
| ADNc (o agua como control negativo) | 1 |
| H2O | 2.4 |
| Volumen final | 10 μL |
Tabla 8: Mezcla de reacción qPCR.
- Para una cuantificación adecuada de los ciclos necesarios para la amplificación, utilice tres réplicas técnicas para el ADNc y tres controles negativos (es decir, agua).
- Selle las placas con una película ópticamente transparente y ejecute la qPCR de acuerdo con las instrucciones del fabricante del kit.
- Analizar las muestras a través de un método de cuantificación absoluta para identificar el número de ciclos (n) en los que se alcanza la rodilla de crecimiento exponencial (ver Figura 4C para un ejemplo). Este número de ciclos se utiliza para la amplificación final del resto del ADNc.
11. Reacción de PCR y extracción en gel
- Configure la reacción de PCR en hielo de acuerdo con la Tabla 9. Consulte la Tabla 7 para todos los cebadores.
NOTA: Sólo se utilizan 5 μL de la biblioteca de ADNc.
| Componente | 1x |
| 10 x tampón de polimerasa de lectura de pruebas | 5 |
| Cebador P5 de 10 μM (hacia adelante) | 1 |
| Cebador BC de 10 μM (reverso) | 1 |
| 5 mM dNTPs | 2.5 |
| Revisión de la enzima polimerasa | 1 |
| ADNc | 5 |
| H2O | 34.5 |
| Volumen final | 50 μL |
Tabla 9: Mezcla de reacción PCR.
- Ejecute la PCR de la siguiente manera: 95 °C durante 2 min; n ciclos de 98 °C durante 20 s, 52 °C durante 30 s y 72 °C durante 1 min; y 72 °C durante 5 min. El número (n) de ciclos para amplificar la biblioteca χCRAC viene determinado por la qPCR descrita en la sección 10.
- Añadir 1 μL de exonucleasa I e incubar a 37 °C durante 60 min.
- Limpie el ADNc amplificado utilizando perlas SPRI como se describió anteriormente utilizando dos volúmenes de perlas (es decir, 100 μL). Eluto en 11 μL.
- Agregue 3 μL de tinte de carga 6x y ejecute un gel prefabricado de 6% TBE a 100 V durante 1 h en 1x tampón TBE. Utilice una escalera apropiada para la cuantificación de fragmentos cortos de ADN.
- Una vez terminado, retire el gel del casete y colóquelo en un recipiente hermético adecuado con suficiente 1x TBE para cubrir el gel (por ejemplo, ~ 50 ml). Añadir una cantidad adecuada de colorante seguro SYBR (por ejemplo, para 50 ml, utilizar 5 μL de un colorante 10.000x)
- Deje que el gel se manche a través de una mezcla suave durante 15 minutos en RT. Drene el TBE que contiene SYBR y reemplácelo con 1x TBE limpio. Lave el gel durante 10 minutos agitando suavemente en RT.
- Drene el 1x TBE y coloque el gel en una carpeta transparente. Corte la carpeta a un tamaño adecuado.
- Imagen del gel a través de un medio apropiado, como un generador de imágenes de fósforo. Extirpar fragmentos de ADN entre ~175 pb y ~400 pb. Coloque la rodaja de gel en un tubo de 1,5 ml.
- Triturar bien la rodaja de gel con una punta P1000 y añadir 400 μL deH2O. Incubar a 37 °C agitando durante 1 h en un termobloque.
- Congelar la muestra en hielo seco durante 10 min, luego volver a colocar en el termobloque a 37 °C agitando durante 1 h.
- Cree una unidad de filtro tomando una columna de filtro e insertando dos filtros de microfibra de vidrio en su interior. Coloque la unidad en un tubo de 1,5 ml.
- Corte el extremo de una punta P1000 con un bisturí limpio y tome la suspensión de gel TBE aplastada, luego dispense en la unidad de filtro creada en el paso 11.12. Girar a 17.000 x g durante 30 s.
- Agregue 1 μL de glucógeno al sobrenadante, junto con 40 μL de acetato de sodio, pH = 5.2, y 1 ml de etanol al 96%. Incubar a -80 °C durante 30 min.
- Centrifugadora durante 30 min a 17.000 x g, 4 °C. Desechar el sobrenadante y lavar con 500 μL de etanol al 70%.
- Girar durante 5 minutos, retirar el etanol por completo y luego secar el pellet en una campana extractora durante 3 minutos.
- Resuspender en 10 μL deH2Oy medir la concentración de ADN.
Resultados
Para demostrar la eficacia del método χCRAC, se realizó un experimento de curso temporal con cepas de levadura que expresan una proteína Nrd1 marcada con HTP. En la figura 1 se proporciona una representación esquemática detallada que describe cómo funciona el método. Al igual que Nab3, Nrd1 está involucrado en la desintegración del ARN nuclear de una variedad de transcripciones de ARN37. Trabajos previos del laboratorio de Corden sugirieron que la unión de Nrd1 a sus objetivos de ARN cambia significativamente cuando las células son sometidas a falta de glucosa28,38. Como tal, las células que crecen exponencialmente en un medio que contiene glucosa (SD-TRP) se cambiaron al mismo medio sin glucosa (S-TRP) durante un curso de tiempo para monitorear los cambios dinámicos en las interacciones Nrd1-ARN. Las muestras se tomaron y se reticularon en la cámara de enlace Vari-X (Figura 3A) antes del turno y luego después de 1, 2, 4, 8, 14 y 20 min. El medio utilizado para el crecimiento celular fue deliberadamente deficiente en triptófano para reducir la absorción UV por este aminoácido aromático. Tenga en cuenta que es mejor utilizar un medio sintético que esté esterilizado por filtro, ya que el autoclave del medio puede conducir a la caramelización de los azúcares. Esto reduce la eficiencia de reticulación.
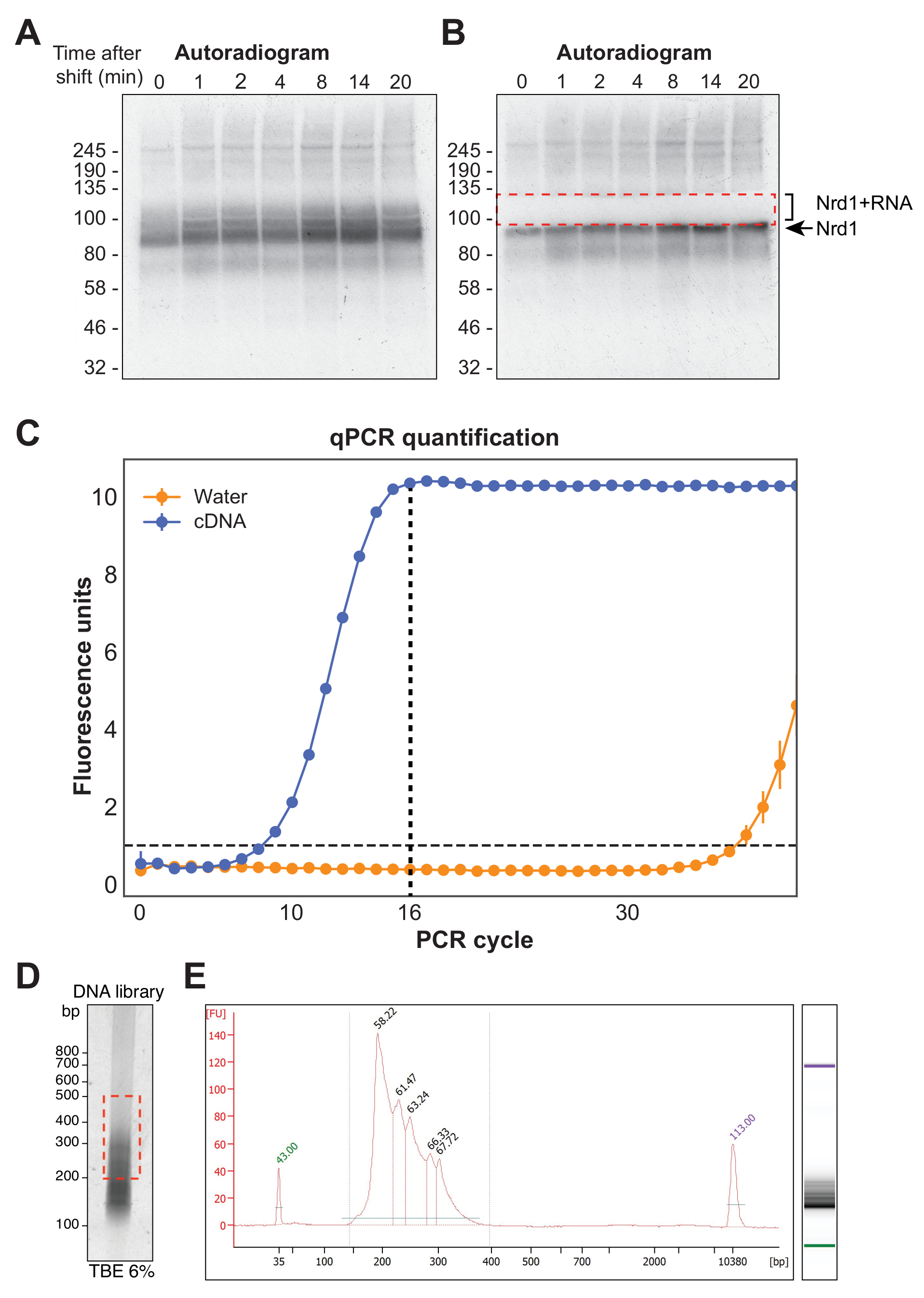
La figura 4A muestra una autorradiografía representativa de un experimento χCRAC. Tenga en cuenta que en este ejemplo, las muestras no se agruparon. En cambio, cada uno se ejecutó individualmente en el gel. Esto se recomienda para las pruebas experimentales iniciales para demostrar que la proteína se cruza eficazmente con el ARN en todos los puntos de tiempo probados. Se observó una señal particularmente intensa en el peso molecular esperado de la RBP, que representa la proteína unida a ARN radiomarcados muy cortos que no son susceptibles de secuenciación. Por lo tanto, se aisló la señal de mancha por encima de esta banda, que es la proteína reticulada a fragmentos de ARN más largos. El fragmento fue cortado justo por encima de la banda de proteína más alrededor de 30 kDa. La figura 4B muestra un autorradiograma después de la escisión, con la proteína reticulada a ARN cortos que quedan en el gel y la señal previamente manchada ahora extirpada.
Después de la transcripción inversa, la biblioteca de ADNc debe amplificarse mediante PCR. Sin embargo, se debe evitar la sobreamplificación de la biblioteca, ya que esto puede introducir sesgos hacia secuencias amplificadas preferentemente por la polimerasa y generar artefactos de PCR. Las bibliotecas sobreamplificadas también contienen un gran número de secuencias duplicadas que desperdician lecturas en el secuenciador. Para calcular el número ideal de ciclos de PCR para la amplificación de la biblioteca final, se amplificó una alícuota del ADNc mediante qPCR utilizando los oligonucleótidos P5 y BC. El primer ciclo en el que la biblioteca alcanzó la fluorescencia máxima se eligió como el recuento del ciclo de PCR. La Figura 4C da un ejemplo de una qPCR de una biblioteca típica de ADNc, que produjo un recuento de ciclos máximos de 16. Este valor se utilizó para la PCR final de χCRAC. Para procesar los datos secuenciados, utilizamos un software previamente desarrollado en nuestro laboratorio (pyCRAC) y el pipeline correspondiente para el análisis de los datos cinéticos de CRAC (Nues et al., 2017; https://git.ecdf.ed.ac.uk/sgrannem/pycrac, https://bitbucket.org/sgrann/kinetic_crac_pipeline/src/default/). Estas herramientas de software de código abierto permiten la demultiplexación y el recorte de los datos, la eliminación de duplicados de PCR, la identificación de picos estadísticamente significativos, las lecturas de clúster en secuencias contiguas y la identificación de motivos de unión39. Más detalles sobre cómo funcionan estas herramientas se encuentran en sus respectivas páginas web.
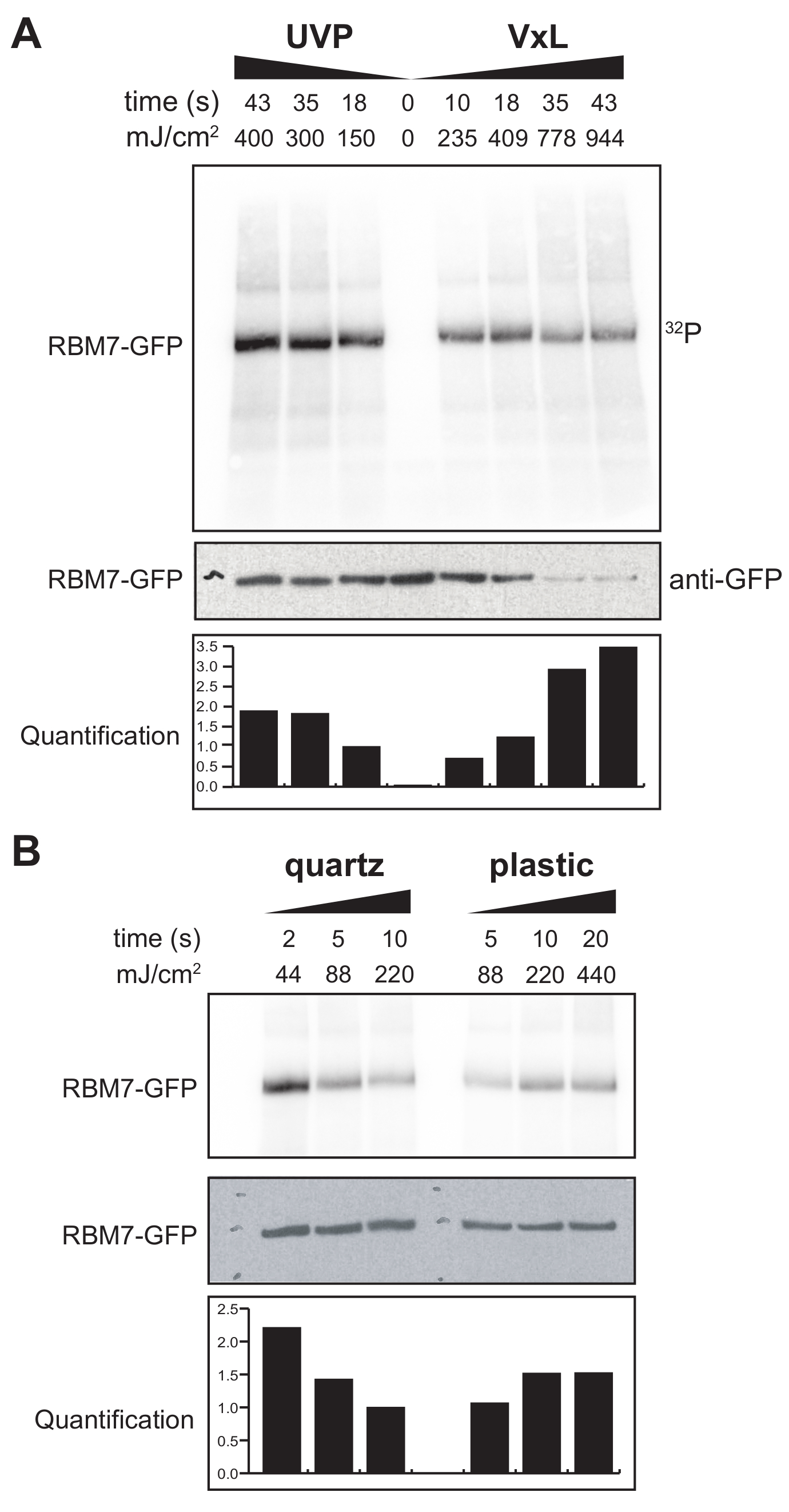
También comenzamos a desarrollar un protocolo χCRAC para células de mamíferos. La mayoría de las líneas celulares de mamíferos se cultivan como una monocapa y la bandeja en nuestro reticulante con la bolsa permeable a los rayos UV no es adecuada para experimentos con células adherentes. Para superar este problema, desarrollamos una etapa en la que los usuarios pueden irradiar UV 1-2 placas de Petri (150 mm de diámetro y 25 mm de profundidad) con células adherentes (Figura 3B). Como primera prueba, la eficiencia del reticulante para células de mamíferos se midió a través de la reticulación y la captura de GFP-RBM7 marcados de manera estable utilizando anticuerpos anti-GFP y una purificación tradicional basada en CLIP. Como se muestra en la Figura 5A, el reticulante fue capaz de recuperar complejos proteína-ARN de células de mamíferos cultivadas como una monocapa utilizando irradiación UV de 254 nm con eficiencias comparables a un dispositivo de irradiación UV ampliamente utilizado. Sin embargo, los artículos de plástico de cultivo celular estándar que normalmente se usan para experimentos de reticulación UV son impenetrables a 254 nm UV. Por lo tanto, en nuestro reticulante las células sólo recibirían irradiación del banco superior de lámparas UV. Para superar esto, desarrollamos una placa de Petri de cuarzo permeable a los rayos UV para el crecimiento celular y la reticulación. El uso del material de cultivo de cuarzo mostró una recuperación robusta de complejos proteína-ARN con tan solo 2 s de irradiación UV (Figura 5B). Cuando se combinan con métodos de captura de RBP para células de mamíferos como las tecnologías CLIP, estos cortos tiempos de reticulación son susceptibles con cursos de tiempo para recuperar perfiles de unión al ARN espaciotemporal de RBP en respuesta a tensiones genotóxicas o agotamientos rápidos de factores proteicos, o en paralelo con la sincronización transcripcional o del ciclo celular.
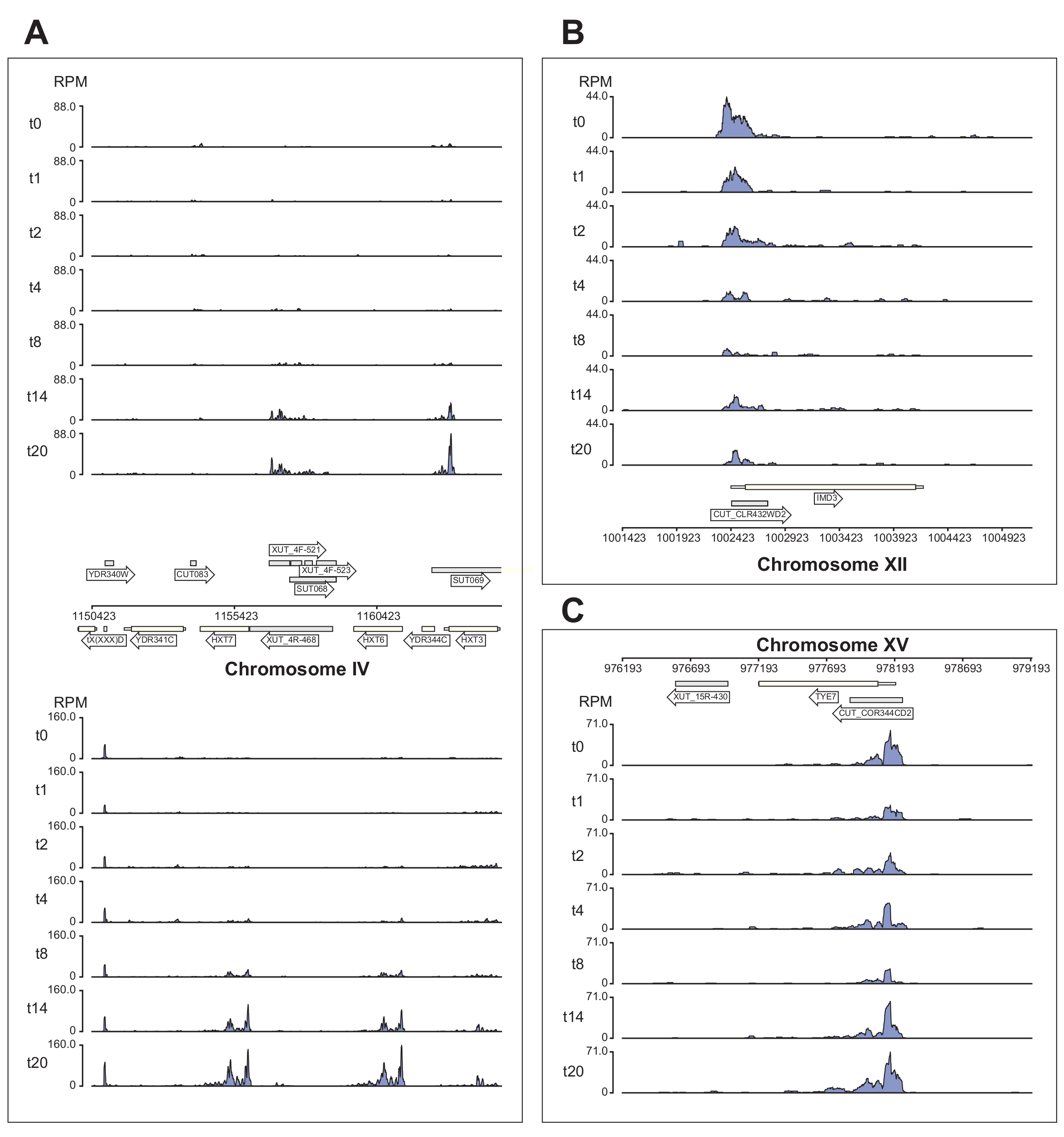
La Figura 6 muestra varios ejemplos de los datos Nrd1 procesados por la canalización χCRAC. Esta figura se preparó utilizando los archivos bedgraph generados por la canalización y el paquete python GenomeBrowser (https://pypi.org/project/GenomeBrowser/1.6.3/), que diseñamos para simplificar la creación de imágenes de los datos del navegador del genoma con calidad de publicación. Los rectángulos grises representan regiones genómicas que expresaron ARN no codificantes, como la transcripción inestable críptica (CUT), las transcripciones estables no caracterizadas (SUT)40 y las transcripciones inestables sensibles a Xrn1 (XUT)41. Los datos de la Figura 6 muestran que Nrd1 se une a muchas de estas transcripciones de ARN no codificantes, lo que coincide con la idea de que esta proteína está implicada en la degradación de esta clase de transcripciones42. La Figura 6A muestra una región de ~15 kb en el cromosoma IV. Aquí hubo un aumento significativo en la unión de Nrd1 a las transcripciones que codifican los transportadores de glucosa de alta afinidad HXT6 y HXT7, los cuales están regulados al alza durante la inanición de glucosa. Es probable que la terminación de la transcripción por el complejo NNS pueda influir en la cinética de inducción de estos genes durante la falta de glucosa. La Figura 6B muestra un ejemplo de enlace cruzado Nrd1 a la transcripción Imd3, que se sabe que está regulada por Nab343. En este caso, los datos demostraron una reducción significativa en la unión a la falta de glucosa. Trabajos previos mostraron una disminución de la unión de Nab3 a la transcripción de Tye7 durante la falta de glucosa44. De acuerdo con esta observación, los datos de χCRAC sugieren que la unión de Nrd1 disminuyó durante la inanición de glucosa y la reticulación de Nrd1 a Tye7 estaba en su punto más bajo después de 8 min de estrés (Figura 4C). Sin embargo, parece que este efecto fue solo transitorio, porque después de 14 minutos de inanición de glucosa, la unión a Nrd1 volvió a los niveles iniciales.

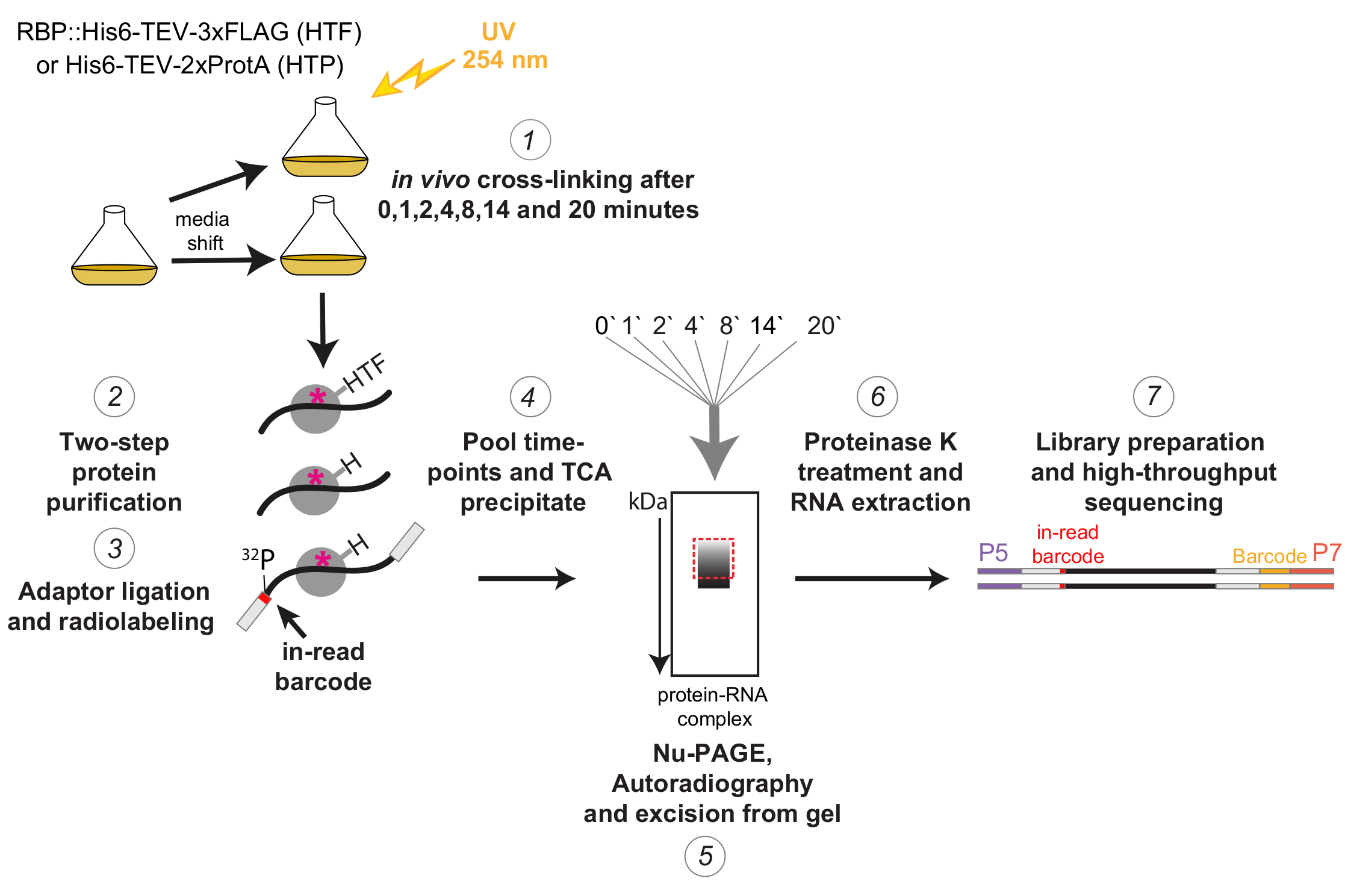
Figura 1: Representación esquemática del protocolo χCRAC. Las cepas etiquetadas se cultivaron hasta la densidad deseada. RBP indica proteína de unión al ARN. Posteriormente, se tomó una muestra de referencia y se entrecruzó con luz UV de 254 nm. Las células restantes se cosecharon por filtración y luego se cambiaron rápidamente al medio inductor de estrés. Para el experimento χCRAC descrito aquí, se tomaron muestras y se entrecruzaron 1, 2, 4, 8, 14 y 20 minutos después del turno (1). El RBP de interés se purificó utilizando una purificación de afinidad de dos pasos altamente estricta (2). A continuación, los ARN reticulados capturados se digirieron parcialmente con RNasas, se marcaron radiactivamente en el extremo 5' y los adaptadores se ligaron a ellos (3). Los adaptadores de 5' contenían secuencias de códigos de barras únicas "en lectura" para que las muestras individuales pudieran separarse bioinformáticamente después de la secuenciación. Los complejos RBP-RNA se eluyeron, se agruparon y precipitaron juntos (4), se resolvieron mediante SDS-PAGE y se visualizaron mediante autorradiografía (5). Posteriormente, una sola rebanada de gel que contenía la señal radiactiva justo encima de la banda principal, ilustrada con una caja roja discontinua en la imagen de autorradiografía, se cortó del gel (5). Las rodajas de gel se trataron con proteasa K y posteriormente se extrajo el ARN (6), se convirtió en ADNc y se amplificó mediante PCR (7). El paso PCR introdujo códigos de barras adicionales (bloque amarillo introducido por P7 oligo) para que muchas bibliotecas pudieran multiplexarse en un solo carril. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Reticulación y filtración al vacío. (A) El reticulante. La suspensión celular se vierte en un embudo ubicado en la parte superior derecha de la máquina (consulte también la Figura 3A para un primer plano) y se mantiene en una bolsa transparente a los rayos UV ubicada en la bandeja central. Esta bolsa está flanqueada por dos persianas que permanecen cerradas hasta que el usuario indica a la máquina que inicie el paso de irradiación. Las células se irradian con luz UV de las bandejas tanto arriba como abajo. La máquina viene suministrada con lámparas UV de 254 y 365 nm, siendo esta última aplicable para experimentos PAR-CLIP. La máquina se opera a través de un panel de pantalla táctil ubicado en la parte superior derecha que permite controlar la dosis de UV o el tiempo de exposición. (B) Después de la reticulación, las celdas se drenan desde el lado izquierdo de la máquina. Las suspensiones celulares se recuperan al vacío y se drenan en un matraz de vidrio donde posteriormente se pueden verter en un dispositivo de filtración al vacío para su recolección. (C) Dispositivos de filtración al vacío. Estos se abren y cierran a través de un clip y se inserta un filtro entre ellos. Se utilizaron cuatro dispositivos de filtración en paralelo durante series temporales muy cortas para no perder tiempo como resultado del cambio de filtros. (D) Después de la filtración, el sobrenadante del medio se drenó en matraces para su posterior eliminación. Se instalaron válvulas debajo de los dispositivos de filtración al vacío para mantener el vacío en el sistema cuando se retira el filtro. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Reticulación de células suspendidas vs. adherentes. (A) El reticulante con la cámara de reticulación Vari-X para celdas de suspensión. El cultivo celular se vierte en la entrada de la muestra (embudo) ubicada en la parte superior derecha de la bandeja. (B) Bandeja que puede contener placas de Petri de plástico o cuarzo para reticular células adherentes o pequeños volúmenes de células de suspensión. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Preparación de la biblioteca. (A) Ejemplo de un autorradiograma de un experimento Nrd1-HTP χCRAC. La señal fuerte y concentrada representa la proteína reticulada a ARN muy cortos, mientras que el frotis anterior representa la proteína reticulada a ARN de longitud suficiente para la secuenciación. (B) El frotis fue extirpado como se muestra en un autoradiograma tomado después de la escisión en gel. (C) Una qPCR representativa de una biblioteca de ADNc χCRAC. En este ejemplo, la amplificación máxima del ADNc se alcanzó a los 16 ciclos. Por lo tanto, se utilizaron 16 ciclos para la amplificación final. La barra de error representa la desviación estándar de tres réplicas técnicas de qPCR. (D) Ejemplo de una imagen de fósforo de una biblioteca de ADNc en un gel TBE al 6%. (E) análisis de longitud y calidad del ADNc a partir de una electroforesis capilar basada en chips. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Prueba de alta RNasa Experimento iCLIP para probar la reticulación en células de mamíferos. Se muestran autoradiogramas de experimentos GFP-RBM7 iCLIP que probaron la eficiencia de la recuperación de RNP a través de varias energías de reticulación. Las inmunoprecipitaciones se realizaron utilizando anticuerpos anti-GFP acoplados a perlas magnéticas en células reticuladas que expresaron de manera estable GFP-RBM7. Los inmunoprecipitados se incubaron con altas concentraciones de RNasa I para recortar los ARN asociados a longitudes cortas y uniformes. Las RNP fueron visualizadas por el etiquetado 32Py SDS-PAGE y migran como una banda definida, cerca de la migración de la proteína no reticulada. La cuantificación indica los resultados de los análisis densitométricos de la señal de ARN RBM7 radiomarcada normalizada a la señal Western blot anti-GFP. (A) Curso temporal de reticulación del reticulante UVP comúnmente utilizado frente a nuestro reticulante (Vari-X-linker; VxL). (B) Entrecruzamiento del curso temporal de nuestro reticulante en artículos de cultivo de cuarzo (izquierda) y plástico (derecha). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Ejemplos de gráficos del navegador del genoma que muestran el poder de χCRAC para mostrar la unión temporal diferencial de Nrd1 a sus objetivos. Cada cuadro muestra gráficos para regiones genómicas individuales. Las flechas indican en qué hebra están codificados los genes (flecha que apunta a la izquierda = menos hebra; flecha que apunta a la derecha = hebra más). Los puntos de tiempo (min) se indican mediante t0, t1, t2, etc. en los ejes y de cada subgráfico. Se muestran números romanos que indican los cromosomas y las coordenadas. (A) Tras la privación de glucosa, Nrd1 se une a dos transportadores de glucosa de alta afinidad, HXT6 y HXT7, que están regulados al alza en esta condición. (B) Se observa que Nrd1 se une a Imd3, un objetivo ya validado de Nab344, con una intensidad reductora después de la falta de glucosa. (C) La unión Nrd1 de Tye7 exhibe una naturaleza dinámica y transitoria, disminuyendo después de la falta de glucosa a un mínimo después de 8 min de estrés. Sin embargo, la unión posteriormente vuelve a los niveles basales después de 14 min. Las lecturas se normalizaron a "lecturas por millón" (RPM; eje y). Los recuadros grises indican regiones que codifican ARN no codificantes. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El método χCRAC, combinado con los nuevos dispositivos de reticulación y recolección celular, tiene un gran potencial porque es aplicable a una amplia gama de organismos modelo y, por lo tanto, debería ser de interés general para el campo del ARN. Hay muchas áreas en las que se puede utilizar χCRAC. Por ejemplo, el método podría usarse para medir el ensamblaje jerárquico de proteínas en grandes complejos macromoleculares, como el espliceosoma y el ribosoma, que a menudo implica interacciones dinámicas entre proteínas y moléculas de ARN. Ahora también lo usamos rutinariamente para monitorear las interacciones entre los factores de descomposición del ARN y sus sustratos cuando las células están sujetas a diversos tipos de estrés. Esto nos permite determinar en qué etapa de la respuesta adaptativa estos factores son más activos, a qué sustratos se unen y qué tan dinámicas son estas interacciones. Estos datos deberían permitir a los investigadores determinar la contribución relativa de cada factor en la adaptación a los cambios ambientales.
χCRAC utiliza etiquetas de purificación de afinidad dual (HTF o HTP) para purificar la proteína en condiciones altamente estrictas y desnaturalizantes. Esto asegura que el ARN copurificado esté altamente enriquecido para los ARN que fueron reticulados covalentemente a la proteína de interés. Sin embargo, confiar en las etiquetas de afinidad tiene desventajas. Por ejemplo, la etiqueta podría interferir con la función de la proteína, lo que podría dar una lectura distorsionada de su interactoma de unión al ARN. Además, para algunos organismos modelo puede que no siempre sea posible utilizar etiquetas porque las herramientas genéticas para integrar fragmentos de ADN en el genoma o para transformar plásmidos de expresión aún no están disponibles. Sin embargo, es sencillo alterar algunas partes del protocolo χCRAC para hacerlo compatible con los protocolos basados en CLIP que dependen de anticuerpos para la purificación de la RBP. De hecho, este estudio demostró que es posible combinar purificaciones basadas en iCLIP con nuestro reticulante. Ahora estamos en el proceso de desarrollar protocolos CLIP para estudiar la asociación temporal de proteínas de unión al ARN humano con transcripciones de ARN nacientes.
Cuando se realiza χCRAC en una nueva proteína, la exposición a los rayos UV debe optimizarse para inducir la máxima reticulación. Esto es importante porque las altas exposiciones a los rayos UV pueden reducir la recuperación del ARN durante la etapa de purificación. Las células que expresaban la RBP recombinante fueron expuestas a varias dosis UV, 100 mJ/cm 2, 250 mJ/cm 2, 500 mJ/cm 2 y 1 J/cm 2. Los RNP fueron capturados y los RNAs fueron fragmentados y radiomarcados. Posteriormente, las RNP fueron resueltas por SDS-PAGE y se tomó un autoradiograma para deducir qué exposición daba la señal más intensa (es decir, la reticulación máxima).
Una vez optimizadas las condiciones experimentales, se recomiendan varios experimentos de control al realizar χCRAC. Primero, se puede usar una muestra irradiada con UV y sin etiquetar para monitorear la unión de fondo a las perlas de purificación. En segundo lugar, cuando se aplica χCRAC durante un experimento de turno, una segunda serie temporal en la que las células se desplazan de nuevo al medio original permite investigar si la filtración de las células en sí induce cambios en los niveles de ARN o en las interacciones proteína-ARN.
Como se mencionó en la Introducción, numerosos artículos publicados recientemente sugieren una serie de optimizaciones al protocolo CLIP. Esto incluye el uso de adaptadores marcados con fluorescencia para detectar el complejo proteína-ARN a través del escaneo infrarrojo10, así como optimizaciones para varios pasos de purificación de ácidos nucleicos y selección de tamaño que han demostrado aumentar la complejidad de las bibliotecas resultantes12,45. Actualmente estamos implementando algunas de estas mejoras para refinar aún más el protocolo χCRAC. El protocolo presentado aquí ya contiene una serie de mejoras a los protocolos CRAC y χCRAC originales que aumentan la complejidad de los datos. Por ejemplo, anteriormente, después de resolver los complejos radiactivos de proteína-ARN reticulados en geles SDS-PAGE, se transferían a una membrana de nitrocelulosa y el ARN reticulado se aislaba de la mancha. Sin embargo, la transferencia del RNP y la posterior extracción de ARN pueden ser muy ineficientes, particularmente cuando se trata de grandes RBP como las subunidades de ARN polimerasa. Esto puede resultar en una reducción significativa en la recuperación del ARN reticulado. En el protocolo actual, el ARN reticulado se extrae directamente de las rodajas de gel SDS-PAGE, como se ilustra en la Figura 1. Esto aumentó la recuperación de ARN reticulados. Además, después de la amplificación por PCR de los ADNc, el producto se resolvió originalmente en geles de agarosa al 3%, a baja temperatura de fusión, y luego se extrajeron productos de PCR de 175-300 pb del gel. Sin embargo, estos geles pueden sobrecargarse fácilmente, lo que resulta en una separación muy pobre del ADN. La sustitución de los geles de agarosa por geles TBE prefabricados dio como resultado una separación de tamaño más consistente y una mejor recuperación de los productos de PCR.
Divulgaciones
A. Langford y W. Worboys están afiliados a UVO3, una compañía comercial. No tuvieron ningún papel en el diseño del estudio, la recopilación e interpretación de datos, o la decisión de presentar el trabajo para su publicación.
Agradecimientos
Este trabajo fue apoyado por subvenciones del Wellcome Trust (091549 a SG y 109093 / Z / 15 / A a SM), la subvención básica del Centro Wellcome Trust para Biología Celular (092076) y la Beca de Investigación Senior No Clínica del Consejo de Investigación Médica (MR / R008205 / 1 a SG), la Organización Europea de Biología Molecular bajo una beca postdoctoral a largo plazo (ALTF 1070-2017 a R.A.C), y el Fondo de Investigación Independiente de Dinamarca (T.H.J).
Materiales
| Name | Company | Catalog Number | Comments |
| 1,4-dithioreitol | Merck | 10708984001 | Buffer component in mammalian cell lysis |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | General reaction tube |
| 2 mL tubes | Eppendorf | 0030 123.344 | For holding columns and collection of waste |
| 32P-yATP | Perkin Elmer | NEG502Z-250 | For radiolabelling the 5' end of the RNA |
| 4-12% Bis-Tris gel | Invitrogen | NP0321BOX | SDS-PAGE gel |
| 4X loading buffer | Novex | NP0008 | Protein loading dye concentrate |
| 50 bp ladder | New England Biolabs | N3236 | Reference ladder for excising region of interest from the amplified cDNA library |
| 50% PEG | NEB | B100045 | For the L5 linker ligation |
| 6% TBE gel | Invitrogen | EC6265BOX | For separation and purification of the cDNA library |
| Acetone | ACROS Organics | 423245000 | Washing of TCA-precipitated proteins |
| anti-FLAG beads | Sigma Aldrich | M8823-1ML | For purifcation of FLAG-tagged RBPs |
| ATP (100 mM) | Thermo Fisher Scientific | R0441 | For ligation of the L5 linker onto the 5' end of captured RNAs |
| Beta-mercaptoethanol | Sigma Aldrich | M3148-100ML | Buffer component |
| Biomax MS intensifying screen | Sigma Aldrich | Z363162-1EA | For intensifying the autoradiogram signal |
| Chloroform | Thermo Fisher Scientific | 1010219 | For phenol-chloroform extraction following RNA purification |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 11873580001 | For inhibition of cellular proteases after lysis |
| Complete supplement mixture -TRP | Formedium | DCS0149 | For preparation of synthetic defined medium |
| Costar Spin-X 0.22 µm filters | Sigma Aldrich | CLS8160 | For isolating the excised cDNAs following gel extraction |
| DNase RQ1 | Promega | M6101 | For DNA digest following cell lysis |
| dNTPs (10 mM) | Sigma Aldrich | 4638956001 | For reverse transcription and PCR |
| Ethanol | Thermo Fisher Scientific | 10041814 | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Ethylenediaminetetraacetic acid | Invitrogen | AM9261 | For protease K buffer |
| Exonuclease I | New England Biolabs | M0293 | For degradation of primers following PCR |
| Glass microfiber filters | Whatman | 1823-010 | For isolating the excised cDNAs following gel extraction |
| Glucose | Formedium | GLU03 | For preparation of glucose-containing, synthetic defined medium |
| Glycogen (20 mg/mL) | Roche | 10901393001 | Precipitation of proteins, RNA and DNA |
| GST-TEV | Homemade | Construct and purification protocol is available upon request | |
| Guanidium hydrochloroide | Thermo Fisher Scientific | 10071503 | Required for pulldown denaturing conditions and washing buffer |
| IgG beads | GE Healthcare | 17-0969-01 | For purification of protein A-tagged RBPs |
| Imidazole | Sigma Aldrich | I2399-100G | For elution of captured proteins from Nickel beads |
| Isoamyl alcohol | Thermo Fisher Scientific | A393-500 | For phenol-chloroform extraction following RNA purification |
| Luna Universal One-Step RT-qPCR | NEB | E3005S | For qPCR of the cDNA in order to calculate required number of PCR cycles |
| Magnesium chloride | Fluka Analytical | 63020-1L | For PNK buffer |
| Membrane filters | Millipore | AAWP09000 for yeast or HAWP09000 for bacteria | For vacuum filtration of cells |
| Micro bio-spin columns | Biorad | 732-6204 | For collecting eluate after gel extraction |
| Ni-NTA beads | Qiagen | 30210 | For secondary protein capture |
| NP-40 | Sigma Aldrich | I8896-100ML | Buffer component |
| Pfu polymerase | Promega | M7741 | For amplification of the cDNA library |
| Phenol | Sigma Aldrich | P4682-400ML | For phenol-chloroform extraction following RNA purification |
| Pierce spin columns | Thermo Fisher Scientific | 69725 | For on-column enzymatic reactions |
| Protease K | Roche | 3115887001 | For degradation of the RBP following gel extraction |
| Quartz Petri dish | UVO3 | N/A | For cross-linking of adherent cells. Available from https://www.vari-x-link.com for 400 GBP |
| Radiography films | Amersham | 28906843 | For autoradiography visualisation |
| RNAClean XP beads | Beckmann | A63987 | SPRI beads for clean up of RNAs and cDNAs |
| RNase H | New England Biolabs | M0297 | For degradation of RNAs following reverse transcription |
| RNase-It | Agilent | 400720 | For RNA digestion |
| rRNasin | Promega | N2511 | For inhibition of any contaminating RNases during enzymatic reaction |
| Sodium acetate | Sigma Aldrich | S2889-1KG | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Sodium chloride | Thermo Fisher Scientific | 7647-14-5 | Buffer component |
| Sodium deoxycholate | Sigma Aldrich | D6750-100G | Buffer component in mammalian cell lysis |
| Sodium dodecylsulfate | Sigma Aldrich | L3771-1KG | For protease K buffer |
| SUPERase-In | Invitrogen | AM2694 | For inhibition of cellular RNases after lysis |
| SuperScript IV | Thermo Fisher Scientific | 18090010 | For reverse transcription |
| T4 PNK | New England Biolabs | M0201 | For radiolabelling the 5' end of the RNA |
| T4 RNA ligase 1 | New England Biolabs | M0204 | For ligation of the L5 adaptor onto the RNA 5' end |
| T4 RNase ligase 2, truncated K222Q | NEB | M0351S | For ligation of the App_PE linker onto the 3' end of captured RNAs |
| TBE buffer (10X) | Invitrogen | 15581-028 | For running TBE gels |
| TEV protease | Homemade | For eluting captured proteins following FLAG capture | |
| Thermosensitive alkaline phosphatase | Promega | M9910 | For 5' and 3' dephosphorylation of RNAs |
| Trichloroacetic acid (100%) | Sigma Aldrich | T0699-100ML | For precipitation of RBP-RNA complexes |
| Tris hydrochloride | Invitrogen | 15504-020 | Buffer component |
| Triton X-100 | Sigma Aldrich | T8787-100ML | Buffer component in mammalian cell lysis |
| Vari Filter | UVO3 | N/A | Device for vacuum harvesting cells. Available from https://www.vari-x-link.com for 100 GBP |
| Vari-X-Linker | UVO3 | N/A | Cross-linker for cross-linking cells. Available from https://www.vari-x-link.com for 16,000 GBP |
| Yeast nitrogen base | Formedium | CYN0410 | For preparation of synthetic defined medium |
| Zirconia beads | Thistle | 11079105Z for yeast or 11079101Z for bacteria | For cell lysis via bead beating |
Referencias
- Ule, J., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 302 (5648), 1212-1215 (2003).
- Granneman, S., Kudla, G., Petfalski, E., Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 106 (24), 9613-9618 (2009).
- Licatalosi, D. D., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 456 (7221), 464-469 (2008).
- König, J., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature Structural & Molecular Biology. 17 (7), 909-915 (2010).
- Hafner, M., et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Aktaş, T., et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 544 (7648), 115-119 (2017).
- Huppertz, I., et al. iCLIP: Protein–RNA interactions at nucleotide resolution. Methods. 65 (3), 274-287 (2014).
- Li, X., et al. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Research. 41 (6), 3805-3818 (2013).
- Rosenberg, M., et al. Denaturing CLIP, dCLIP, Pipeline Identifies Discrete RNA Footprints on Chromatin-Associated Proteins and Reveals that CBX7 Targets 3′ UTRs to Regulate mRNA Expression. Cell Systems. 5 (4), 368-385 (2017).
- Zarnegar, B. J., et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nature Methods. 13 (6), 489-492 (2016).
- Kargapolova, Y., Levin, M., Lackner, K., Danckwardt, S. sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Research. 45 (10), 6074-6086 (2017).
- Van Nostrand, E. L., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods. 13 (6), 508-514 (2016).
- Flynn, R. A., et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA. 21 (1), 135-143 (2015).
- Brugiolo, M., Botti, V., Liu, N., Müller-McNicoll, M., Neugebauer, K. M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Research. 45 (18), 10452-10465 (2017).
- Sanford, J. R., et al. Identification of Nuclear and Cytoplasmic mRNA Targets for the Shuttling Protein SF2/ASF. PLOS ONE. 3 (10), e3369 (2008).
- Garzia, A., Meyer, C., Morozov, P., Sajek, M., Tuschl, T. Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods. 118-119, 24-40 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22 (10), 2031-2042 (2012).
- Chen, K., et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
- Ke, S., et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes & Development. 29 (19), 2037-2053 (2015).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Kudla, G., Granneman, S., Hahn, D., Beggs, J. D., Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proceedings of the National Academy of Sciences. 108 (24), 10010-10015 (2011).
- Sugimoto, Y., et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 519 (7544), 491-494 (2015).
- Hwang, H. W., et al. cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron. 95 (6), 1334-1349 (2017).
- Hwang, H. W., et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Reports. 15 (2), 423-435 (2016).
- Lee, F. C. Y., Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Molecular Cell. 69 (3), 354-369 (2018).
- Beckmann, B. M. RNA interactome capture in yeast. Methods. 118-119, 82-92 (2017).
- Granneman, S., Petfalski, E., Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. The EMBO Journal. 30 (19), 4006-4019 (2011).
- Schaughency, P., Merran, J., Corden, J. L. Genome-Wide Mapping of Yeast RNA Polymerase II Termination. PLOS Genetics. 10 (10), e1004632 (2014).
- Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Kresnowati, M. T. A. P., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology. 2, 49 (2006).
- Marguerat, S., Lawler, K., Brazma, A., Bähler, J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biology. 11 (6), 702-714 (2014).
- van Nues, R., et al. Kinetic CRAC uncovers a role for Nab3 in determining gene expression profiles during stress. Nature Communications. 8 (1), 12 (2017).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Tudek, A., Candelli, T., Libri, D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité?. Biochimie. 117, 28-36 (2015).
- Lingaraju, M., et al. The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nature Communications. 10 (1), 1-11 (2019).
- Lubas, M., et al. Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell. 43 (4), 624-637 (2011).
- Conrad, N. K., et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 154 (2), 557-571 (2000).
- Darby, M. M., Serebreni, L., Pan, X., Boeke, J. D., Corden, J. L. The Saccharomyces cerevisiae Nrd1-Nab3 Transcription Termination Pathway Acts in Opposition to Ras Signaling and Mediates Response to Nutrient Depletion. Molecular and Cellular Biology. 32 (10), 1762-1775 (2012).
- Webb, S., Hector, R. D., Kudla, G., Granneman, S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 15 (1), R8 (2014).
- Jensen, T. H., Jacquier, A., Libri, D. Dealing with Pervasive Transcription. Molecular Cell. 52 (4), 473-484 (2013).
- van Dijk, E. L., et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 475 (7354), 114-117 (2011).
- Thiebaut, M., et al. Futile Cycle of Transcription Initiation and Termination Modulates the Response to Nucleotide Shortage in S. cerevisiae. Molecular Cell. 31 (5), 671-682 (2008).
- Merran, J., Corden, J. L. Yeast RNA-Binding Protein Nab3 Regulates Genes Involved in Nitrogen Metabolism. Molecular and Cellular Biology. 37 (18), e00154-e00117 (2017).
- Bresson, S., Tuck, A., Staneva, D., Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Molecular Cell. 65 (5), 787-800 (2017).
- Buchbender, A., et al. Improved library preparation with the new iCLIP2 protocol. Methods. , (2019).
Erratum
Formal Correction: Erratum: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC
Posted by JoVE Editors on 2/17/2023. Citeable Link.
An erratum was issued for: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC. The Authors section was updated from:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Synthetic and Systems Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
to:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Mehak Chauhan1
Niki Christopoulou1
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Engineering Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados