
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
ニワトリ頭蓋神経堤細胞培養物の作製と形態解析
要約
この汎用性の高いプロトコルは、ニワトリ胚からの脳神経襞の切除による遊走性神経堤細胞(NCC)の単離について説明しています。プレーティングとインキュベーションを行うと、遊走性NCCが神経襞の外植片から出現し、単純化された2D環境での細胞形態と遊走の評価を可能にします。
要約
脊椎動物の発生中、神経堤細胞(NCC)は広範囲に移動し、頭蓋顔面骨格や末梢神経系などの構造に寄与するさまざまな細胞型に分化します。NCCの移動を3D胚の文脈で理解することは重要ですが、2D培養で遊走細胞を単離することで、可視化と機能特性評価が容易になり、胚研究を補完します。本プロトコルは、初代NCC培養物を生成するためにニワトリ脳神経襞を単離する方法を示す。遊走性NCCは、フィブロネクチンでコーティングされた基質に播種された神経襞の外植片から出現します。これにより、分散した付着性NCC集団が得られ、染色および定量的形態学的分析によって評価できます。この単純化された文化アプローチは適応性が高く、他の手法と組み合わせることができます。例えば、NCCの遊走および移動行動は、タイムラプスイメージングによって評価するか、または遺伝子発現の阻害剤または実験的操作(例えば、DNA、モルホリノ、またはCRISPRエレクトロポレーション)を含むことによって機能的に照会することができる。その汎用性のために、この方法は頭蓋NCC発達を調査するための強力なシステムを提供します。
概要
神経堤細胞(NCC)は、脊椎動物の胚の一過性細胞集団です。NCCは神経板の境界で特定され、背側神経管1から移行するために上皮間葉転換(EMT)を受けます。EMT後、NCCは胚全体に広範囲に分散し、最終的には頭蓋顔面骨格、心臓の流出路、末梢神経系の大部分など、さまざまな構造を分化させて寄与します2。細胞極性、細胞骨格、および接着特性の変化は、遊走性細胞集団から遊走性細胞集団へのこのシフトの根底にあります3。NCC EMTと遊走を研究することで、細胞の運動性の基本的なメカニズムについての洞察が得られ、先天性欠損症と癌転移を予防および治療するための取り組みに情報を提供します。
in vivo分析は、胚の文脈でNCC発生プロセスを理解するために不可欠ですが、in vitroメソッドは、追加の実験手段を容易にする視覚的および物理的なアクセシビリティを提供します。単純化された2D環境では、NCC形態、細胞骨格構造、および移動距離を評価できます。さらに、運動性NCCの移動行動に対する遺伝的または可溶性因子の摂動の影響を分析することができます4、5、6、7、8、9、10。さらに、単離された遊走性または移動性NCCを収集、プールし、プロテオミクス、トランスクリプトミクス、およびエピゲノムプロファイリングを通じてNCCの発生調節を研究するためのハイスループット方法論に使用することができます7,11。さまざまな発生モデル生物から頭蓋NCCを調製する方法が利用可能である12,13,14が利用可能ですが、この記事では、ニワトリ胚から頭蓋NCCを培養することを最初に学んだ人のためのアプローチの仕組みを示します。
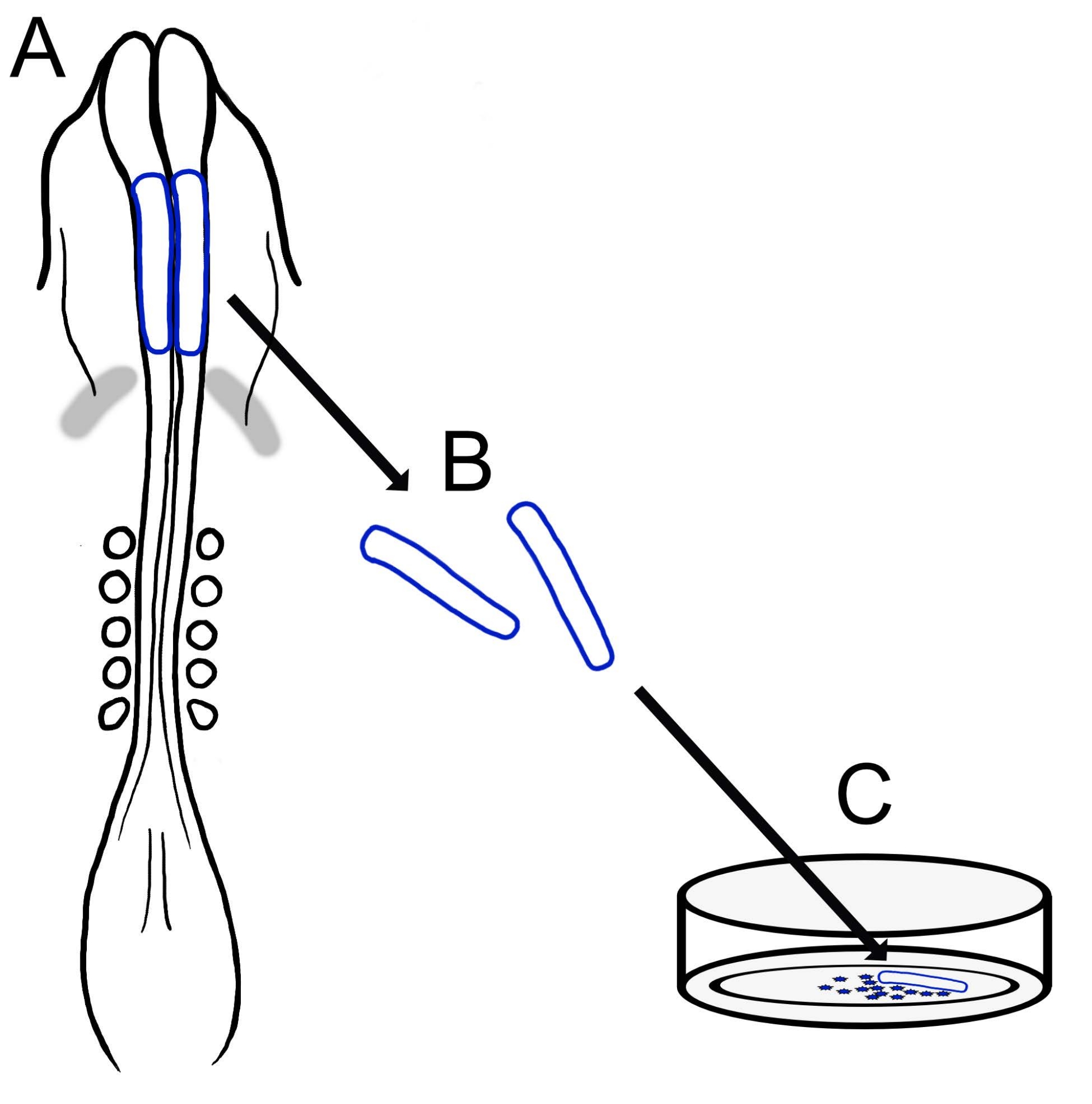
現在のプロトコルは、ニワトリ頭蓋NCC培養物を調製するための汎用性の高い技術を記載しています(図1)。NCCは外植された神経襞から培養基質に容易に移動するため、ニワトリNCCは胚組織から自然に分離し、初代培養物が容易に生成されます。中脳NCCが脳神経襞から一斉に移動すると(体幹15の細胞ごとの層間剥離とは対照的に)、これらの培養物は主に遊走性脳神経堤細胞で構成され、最初の神経襞切除は遊走性NCCの収集方法を提供します。ニワトリ脳神経襞を解剖して培養するための基本的な方法を詳述し、この方法のさまざまな用途とバリエーションの提案を提供します。

図1:ニワトリ脳神経襞培養プロトコルの概略図。 (A,B)脳神経襞(青色で輪郭が描かれている)は、5つの体節を持つニワトリ胚から切除されます(Aの背側図で示されています)。灰色の帯、心臓の三日月。(C)フィブロネクチンにプレーティングすると、遊走性神経堤細胞が神経襞から出現し、基質上に分散する。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
プロトコル
ホワイトレグホーン、ゴールデンセックスリンク、ロードアイランドレッドなど、あらゆる種類のガル スガルス 品種を使用できます。本研究で使用した鶏卵は様々な品種であり、地元の農場や孵化場を含む複数の供給源から入手した。
1.溶液と材料の調製
- 123.3 mM NaCl、1.53 mM CaCl 2、4.96 mM KCl、0.809 mM Na 2 HPO 4、および0.147 mM KH2PO4を混合してリンゲル溶液を調製します(材料表を参照)。pHを7.4に調整し、100mLボトルにろ過滅菌します。清潔で乾燥した場所に保管してください。ボトルを石鹸で洗わないでください(組織培養ガラス製品として扱います)。
- FNを滅菌リンゲル溶液に溶解して1 mg/mLのフィブロネクチン(FN)ストック溶液( 材料の表を参照)を調製し、100 μLアリコートで-80°Cで保存します(少なくとも4年間安定)。
- L15培地に0.8%L-グルタミン、0.08%ペニシリン/ストレプトマイシン、10%FBS、および10%ニワトリ胚抽出物を添加して、完全培養培地を調製します12。3 mLアリコートを作成し、-20°Cまたは-80°Cで保存します。
注:培養物をCO2インキュベーターでインキュベートする場合は、DMEM/F12 を空気用に緩衝されたL15に置き換える必要があります。 - 1x PBSで4%パラホルムアルデヒド溶液を調製します。pHを7.4に調整します。10 mLアリコートを作成し、-20°Cで保存します。
注意: パラホルムアルデヒドはドラフトで取り扱う必要があります。 - ろ紙のサポートフレーム(長軸に2つまたは3つのパンチ穴が重なっている約1 cm x 1.5 cmの長方形の用紙)を準備します。
- 標準の3穴パンチを使用して大きなろ紙の端に沿って穴を開け、打ち抜いたエッジをストリップにカット/トリミングしてから、穴の間のストリップを長方形~長さ1.5cmにカットします。使用前にオートクレーブろ紙を貼ってください(オプション)。
- 細かい鉗子、鈍い鉗子、解剖はさみ、ばねはさみ、鋭利なタングステン針などの解剖ツールを収集します16。
2.胚のインキュベーション
- 上記の供給源のいずれかから受精卵を入手してください。
- 受精卵を38°C(100°F)の加湿インキュベーター内で直立位置(尖った端を下/鈍い面を上にして卵の垂直の長軸)に置きます。
- 4〜7個の体節が形成されるまでインキュベートし(ステージ8 + -9)17、これには約35時間かかります。
注:潜伏時間は、株、鶏の年齢、季節などによって大きく異なる可能性があるため、実験的に決定する必要があります。プログラム可能なアウトレットタイマーは、夜間にインキュベーションを開始して目的のタイミングを得るのに役立ちます。 - インキュベーターから取り出した後、卵に70%エタノールを徹底的にスプレーし、それらを乾燥させて殻を消毒します。
注:胚はすぐに収集できますが、卵を室温まで冷やすと、卵黄の脆弱性と収集中の胚の損失が減少します。
3.培養皿の準備
- ダウンストリームアプリケーションに適したニューラルフォールド培養皿( 材料表を参照)を選択します。
- 固定および染色される培養物には、マルチウェル組織培養皿に入れたガラスカバーガラスを使用します。
注意: ウェルサイズに一致するカバーガラスは、流体の混合による移動が少なくなり、外植片がずれにくくなり、皿の底ではなくカバーガラスに付着する可能性が高くなります。 - 倒立顕微鏡によるライブイメージング用に、ガラス底のシングルチャンバーまたは分割チャンバーディッシュ( 材料表を参照)を選択します。
- 正立顕微鏡または実体顕微鏡でのライブイメージングには、6ウェル、12ウェル、または24ウェルの光学的に適切な組織培養プレートを使用してください。
- ハイスループット分析のために遊走性神経堤細胞の大量収集のためのプラスチック組織培養皿を選択してください。
- 固定および染色される培養物には、マルチウェル組織培養皿に入れたガラスカバーガラスを使用します。
- 10 U / mLのペニシリンと10 μg / mLのストレプトマイシンを滅菌リンゲル溶液の100 mLボトルに追加します(たとえば、100 μLの10,000 U / mLペニシリンと10,000 μg / mLストレプトマイシンを使用して、リンゲルのP / Sを作成します)。1週間以内にリンガーのP / Sを使用してください。
- 1 mg/mLのFNのアリコートを氷上で解凍します。リンガーP / Sで10〜100μg / mLのFNの濃度に希釈します。
- 井戸または皿の底を覆うのに十分なFN溶液をピペットする。例えば、24ウェルプレートではウェルあたり100 μL、35 mmディッシュでは500 μLです。
- 蓋を元に戻し、加湿インキュベーター(または蒸留水に浸したペーパータオルが入った蓋付きトレイ)で皿またはプレートをFN溶液とともに38°C(100°F)で少なくとも1時間インキュベートし、神経襞を解剖します。
4.ニワトリ胚の分離
- 卵の向きを維持するように注意してください(孵卵中に胚は卵黄の上に浮かびます)。はさみまたは鈍い鉗子を使用して、卵の長さの約1 / 4〜1 / 3の小さな穴を殻に開けます。
- はさみまたは鈍い鉗子の先端を小さな穴に慎重に挿入し、卵黄を乱さないようにし、卵の周りの卵殻を切り裂き、卵殻の上部を取り除きます(図2A)。
- 胚を分離のために配置します。
注:胚がシェルカップに理想的に配置されていない場合は、鈍い鉗子を使用して卵黄を慎重に回転させ、胚が上になるようにします。または、卵黄を手袋をはめたカップ状の手に注ぎ、卵黄を壊さないように注意し、アルブミンを排出させます(図2B)。次に、卵黄を手から手へと動かすことによって胚を配置することができます。 - 閉じた鈍い鉗子の平らな端をそっと使用して胚を準備し、胚を覆う卵黄の表面に残っている余分なアルブミンを拭き取ります(図2C)。余分なアルブミンは、繊細なタスクワイパーで穏やかな圧力を使用して除去することもできます。
注意: アルブミンが取り除かれると、卵黄の表面は滑らかではなくテクスチャーで見えるはずです。十分なアルブミンを拭き取らないと、セクション4、ステップ4のろ紙支持体の接着が阻害されます。 - 鉗子を使用して、胚をフレームのウィンドウに入れた状態で、胚の上にろ紙サポートフレームを置きます。ろ紙をそっと押し下げて、卵黄に付着させます。
- ろ紙フレームの外側を解剖ハサミで切り取ります(図2D)。鉗子またははさみの先端を使用してフレームの端をつかみ、胚を卵黄からそっと持ち上げます(図2E)。斜めに持ち上げると、卵黄から胚をきれいに取り除くのに役立ちます。
- 紙枠を下にして(胚腹側を上にして)胚をリンガーP / Sで満たされた60mmまたは100mmのペトリ皿に入れます(図2F)。RNAまたはタンパク質感受性の下流アプリケーションのために収集する場合は、胚の皿を氷の上に置いてください。
注:胚を腹側を上にして配置しないと、胚が紙枠から剥がれるリスクがあります。複数の胚を収集し、神経襞解剖ステップに移行する前に最大1時間リンガーのP / Sに保存することができます。
5.神経襞の解剖
- リンゲルのP / S溶液を含むきれいな皿に胚を移し、濾紙フレームを鉗子で持ち、ゆっくりと前後に振り回して、胚の視界を覆い隠す卵黄を取り除きます。呼び出し音のP / Sを交換するか、曇った場合は新鮮な皿に移します。
- 解剖顕微鏡下で胚の背側/フレーム側を上にして配置します。胚を紙枠に残してぴんと張って所定の位置に保持し、鉗子を使用して卵子膜を取り除き、神経ひだを露出させます。
注:胚が紙枠から落ちた場合は、皿に平らに配置するか、解剖のためにシルガードコーティングされた皿18 に固定することができます。 - スプリングハサミまたは鋭利なタングステン針を使用して、中脳神経ひだを慎重に切除します。拡大する視神経小胞への組織尾と、菱形の狭窄が現れ始めたばかりの後脳への吻側を含めます(心臓三日月も有用な指標です、 図3B、C)。神経襞の最背側を切除し、神経管と非神経外胚葉の汚染を最小限に抑えるように注意してください(図3C)。
- P20ピペッターまたは卵黄リンガーP/Sですすいだ滅菌ガラス製パスツールピペットを使用して、神経襞をリンガーP/Sを含む清潔な皿に移します(これはプラスチックまたはガラスをブロックして組織がくっつくのを防ぎます)。追加の折り目を解剖しながら、集めた折り目を氷の上に保管します。
6.神経襞のメッキ
- 完全培養培地のアリコートを解凍します(セクション1、ステップ3)。100 U/mLのペニシリンと100 μg/mLのストレプトマイシンを加え、フィルター滅菌します。準備した培地を37〜38°Cに保ちながら、他の手順を実行します。
- 培養皿をインキュベーターから取り出します(セクション3、ステップ5)。ピペッターまたはパスツールピペットを使用して、カバーガラス、皿、またはウェルからFN溶液を取り除きます。FNコーティング基板をリンゲルP/Sですすいだ後、適切な量の完全培養培地をディッシュまたはウェルに加えます(24ウェルプレートのウェルの場合は500 μL、35 mmディッシュまたは6ウェルプレートの場合は2 mL)。
注:容量を小さくすると、高価な試薬(たとえば、24ウェルプレートの場合は200 μL)を保存できますが、ディッシュが蒸発しないように十分に加湿されていることを確認してください。 - p20またはp200ピペッターを使用して、まずピペットチップを卵黄リンガーのP / Sですすぎ、プラスチックをブロックし、組織が付着するのを防ぎます。次に、孤立した神経襞をFNコーティングされたカバーガラスに移し、リンガーのP / Sをできるだけ少なくするように注意します。折り目をFNコーティングされたカバーガラスの中央に向かって配置します。
注:1つまたはいくつかの神経襞は、19 mmウェルの12 mmカバーガラスにメッキでき、35 mmプレートには最大50個までメッキできます。 - 外植片を10〜15分間落ち着かせた後、播種した神経襞で培養皿をゆっくりと注意深く運んで、38°C(100°F)の加湿チャンバーに入れます。これにより、ウェル内の神経襞の移動が最小限に抑えられ、分散したままでカバーガラスに付着します。
注:L15(DMEMではない)を使用する場合、外植片培養皿は、湿らせたペーパータオルでカバー付きのトレイを使用して卵インキュベーターでインキュベートすることもできます。 - 加湿インキュベーター内で神経襞培養物を能動的遊走の期間中インキュベートする(合計約16〜20時間、 図4)。
7. 形態素解析のための培養移動性NCCの固定と染色
- パスツールピペットで培地を取り出し、フィルター滅菌した1x PBSでウェルをすすぎます。
- 室温で4%パラホルムアルデヒドを15分間加えます。
注:固定時間は実験的に決定する必要がある場合があります。パラホルムアルデヒドを培地に直接10分間(50:50)添加し、除去し、希釈していない4%パラホルムアルデヒドと10分間交換して、より繊細な細胞構造を保持することもできます。 - パラホルムアルデヒドを除去し、1x PBSで3回すすぎます。
- 形態学的評価のために、固定されたNCCを適切な染料で染色します。一例として、オレゴングリーンコンジュゲートファロイジンによる染色がここで詳述される。
注:ファロイジン19でアクチン細胞骨格を視覚化すると、比較的均一な染色強度で遊走性NCCの構造的複雑さが明らかになります。ただし、ファロイジンはすべての細胞領域を強調表示するわけではありません。原形質膜を標識する色素(例えば、小麦胚芽凝集素またはDiI)または細胞質も使用することができるが、明るく染色する細胞体に対する微細な突起のイメージングを複雑にする。さらに、免疫蛍光法は、神経堤細胞をHNK-1で標識するため、または目的のタンパク質の細胞内局在を決定するために同時に行うことができる7,20。- 最後のPBSリンスを取り除き、カバーガラスを覆うためにPBS + 0.5%Triton X-100(PBST)+ 5%血清(FBSまたは抗体との共染色に適した別の動物由来)を加え、室温のプラットフォームシェーカーで10分間インキュベートします。
- Pipet 200 nMのオレゴングリーンコンジュゲートファロイジン(PBST + 5%血清で希釈)を、各カバーガラスの滑らかな表面(フレキシブルパラフィンシーリングフィルムなど)上に配置します。カバーガラスが12 mmの場合は、容量30 μLのピペットで固定します。
- 一対の鉗子を使用して、PBST + 5%血清からカバーガラスを取り除き、細胞の向きが上を向いていることを確認します。カバーガラスの端を繊細なタスクワイパーに軽く触れて、余分な液体を吸い取ります。
- 各カバーガラスセル側を下にして、希釈したファロイジンの滴の上にそっと置きます。室温で30分間インキュベートします。染色中はカバーガラスを暗所に保管してください(アルミホイルで覆われた皿で覆うか、引き出しに入れます)。
- インキュベーション中に、培養皿のウェルにPBSTを加えます(24ウェルプレートの各ウェルに750 μLのPBST)。
- インキュベーション期間が経過したら、染色液からカバーガラスを持ち上げて培養皿に戻し、カバーガラスを裏返して細胞側が上になるようにします。カバーガラスがPBSTで覆われていることを確認し、カバーガラスを覆い、暗闇の中で10分間プラットフォームシェーカーに置きます。PBSTを取り外し、この手順を2回繰り返して、合計3回の10分間の洗浄を行います。
- 封入剤( 材料の表を参照)を顕微鏡スライドに1滴(カバーガラスごとに)置きます。25 μLの容量は、12 mmのカバーガラスに適しています。
- PBSTで最後に洗浄した後、カバーガラスの端を繊細なタスクワイパーに触れて余分な液体を取り除き、カバーガラスのセル側を追跡します。
- カバースリップセルを下にして、カバースリップをインパウンティングメディアに斜めにゆっくりと下げて、気泡が発生しないようにします。イメージングの前にメディアをセットします。
8. 培養渡り性NCCの形態学的評価
- 染色された細胞を画像化し、.tiffファイルとしてエクスポートします。
注:40倍の対物レンズはイメージング培養に適していますが、10倍(移動性NCCの全フィールドをキャプチャするため)から100倍(単一のNCC)の範囲の対物レンズを使用して、形態学的評価用の画像を収集することができます。本研究の画像は、倒立マルチモーダルイメージングプラットフォームを使用してキャプチャされました( 材料の表を参照)。 - 画像を画像解析ソフトウェア(ImageJ21、 材料表を参照)にアップロードします。[ 画像] > [複製] をクリックして、分析に使用する各画像の 2 番目のコピーを作成し、ほとんどの画像処理を元に戻すことができないため、元の画像は未編集のままにします。
- 画像をクリックして >明るさ/コントラスト>調整 し、スライドバーを使用して画像の明るさまたはコントラストを調整します。
- 以下の手順に従って、画像をグレースケールに変換します。
- 画像がRGBでエクスポートされた場合、マージされたチャネルとしてRGBでアップロードされます。画像を分離するには、[ 画像>カラー]>[チャンネルの分割 ]をクリックして、シングルチャンネルのグレースケール画像を取得します。
- 画像がすでに分離されている場合は、 画像>「8ビット 」と入力して>ファイルをグレースケールに変換します。
- [画像]をクリックして>[しきい値>調整]をクリックして、ピクセルがセル(前景、黒)または背景(白)として識別されるバイナリ画像に変換します。スライドバーを使用するか、[自動]をクリックして、背景からセルを明確に定義するしきい値設定を選択します。
- 細胞が重なり合っているか、または染色が連続していない場合は、流域または穴埋め機能22を使用してさらに処理する必要があるかもしれません。これにより、セルが互いに分離されるか、分析するセル内のギャップが埋められます。[ バイナリ>プロセス]>[バイナリの作成] をクリックして、最初に画像を8ビットバイナリ形式に変換します。次に 、[集水域>>バイナリ処理 ]または [穴を埋め]をクリックします。
注意: このソフトウェアは、信号を分離または入力する必要がある場所に最適であり、必要に応じてプラグインを使用してさらに調整できます。
- 細胞が重なり合っているか、または染色が連続していない場合は、流域または穴埋め機能22を使用してさらに処理する必要があるかもしれません。これにより、セルが互いに分離されるか、分析するセル内のギャップが埋められます。[ バイナリ>プロセス]>[バイナリの作成] をクリックして、最初に画像を8ビットバイナリ形式に変換します。次に 、[集水域>>バイナリ処理 ]または [穴を埋め]をクリックします。
- キャプチャする測定値を選択します。[ 分析>測定値の設定 ]をクリックし、[形状記述子]チェックボックスをオンにして、真円度(円形)、アスペクト比(AR)、真円度(円形)、および固さを分析します。
注:必要な分析のタイプに応じて、面積や周長などの他の測定値も含まれる場合があります。 - [ 解析]>[パーティクルの解析]をクリックし、[パーティクルの解析]メニューからパラメータを設定します。
- [サイズ] で、Size パラメーターが 0-Infinity (たとえば、 図 5 で 500-Infinity) に設定されている場合、バックグラウンド信号またはより小さなセル デブリも含まれるため、対象のセルまたはパーティクルの大きさをピクセル単位で大まかに推定します。
注: [サイズ]を狭い範囲に調整すると、解析に必要な粒子よりも小さい粒子や大きい粒子を除外できます。 - [円形度]で、0〜1.0のままにして、画像内のすべてのセル形状を測定します。
- [表示]で、ドロップダウンメニューから [ベアアウトライン ]を選択して、[パーティクルの分析]機能で選択したセルのアウトラインを調べます。アウトラインをスキャンして、重なり合うセルが区別されるようにしますが、セルが不必要に分割されていないことを確認します。また、「結果の表示」、「要約」、「マネージャーに追加」のメニューボックスにチェックを入れます。
- [サイズ] で、Size パラメーターが 0-Infinity (たとえば、 図 5 で 500-Infinity) に設定されている場合、バックグラウンド信号またはより小さなセル デブリも含まれるため、対象のセルまたはパーティクルの大きさをピクセル単位で大まかに推定します。
- すべてのパラメータを設定したら、[ OK]をクリックします。
注:4つの別々のウィンドウがポップアップし、(1)画像で識別されたセルの裸の輪郭、(2)画像でカウントされたセル、(3)各セルの測定結果、および(4)カウントされたセルの総数とその平均測定値の概要が表示されます。- 破片がカウントされた場合、重複するセルが1つとしてカウントされた場合、単一のセルが倍数としてカウントされた場合、または多くのセルがカウントから除外された場合、元の未編集の画像に戻り、別の複製を作成し、「しきい値」またはその他のパラメーターを調整します。
結果
本プロトコルの概要を 図1に示す。孵化した卵を開き、表面に胚を置いた卵黄を手袋をはめた手のひらにそっと注ぐことによって単離しました(図2A、B)。アルブミンを取り除いた後(図2C)、胚を囲む卵黄膜にろ紙フレームを適用して、卵黄膜が切断されるとこぼれ始める卵黄から胚を切断して持ち上げやすくしまし...
ディスカッション
ここで説明する技術は、ニワトリの神経襞を分離し、それらをメッキして移動性頭蓋NCCの培養物を作成する適応可能な方法を提供します。これらの培養物は、ニワトリNCCの移動および形態の容易な分析のための単純化された2D条件を提供し、卵子イメージング法におけるより技術的に困難なものを補完することができる24、25、26
開示事項
著者には利益相反はありません。
謝辞
コリーヌ・A・フェアチャイルドとケイティ・L・バーミリオンが、ニワトリ脳神経襞培養プロトコルの開発に参加してくださったことに感謝します。
資料
| Name | Company | Catalog Number | Comments |
| AxioObserver equipped with an LSM710 confocal scan head controlled by ZEN 3.0 SR software | Zeiss | Used alpha Plan-Apochromat 100x/1.46 Oil DIC M27 objective | |
| CaCl2 | Sigma-Aldrich | C3306 | |
| Chamber dishes (glass bottom, single or divided) | MatTek; Cell Vis | P35G-1.5-14-C (MatTek) X000NOJQGX (Cellvis) X000NOK1OJ (Cellvis) | Single chamber 35 mm or 4 chamber 35 mm |
| Cover glass | Carolina Biological Supply Company | 633029, 633031, 633033, 633035, 633037 | circles, 0.13–0.17 mm thickness, available in 12-25 mm diameter |
| DMEM/F12 | ThermoFisher Scientific | 11320033 | Alternative for L15 media |
| Egg incubator | Sportsman | 1502 | |
| FBS | Life Technologies | 10437-028 | |
| Fibronectin | Fisher Scientific | CB-40008A | |
| Filter paper | Whatman | grade 3MM chromatography | |
| Forceps (blunt) | Fisher Scientific; Thomas Scientific | 08-890 (Fisher);1141W97 (Thomas) | |
| Forceps (fine) | Fine Science Tools | 11252-20 | Dumont #5 |
| Image J | https://fiji.sc/ | Free image analysis software | |
| KCl | Sigma-Aldrich | P3911 | |
| KH2PO4 | Sigma-Aldrich | P0662 | |
| L15 media | Invitrogen | 11415064 | |
| L-glutamine | Invitrogen | 25030 | |
| Mounting Media (Vectashield or ProLong Gold) | Vector Laboratories; Thermofisher Scientific | H-1700 (Vectashield); P36930 (ProLong Gold) | |
| Na2HPO4 | Sigma-Aldrich | S9638 | |
| NaCl | Sigma-Aldrich | S9888 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| Penicillin/streptomycin | Life Technologies | 15140-148 | 10,000 Units/mL Penicillin; 10,000 mg/mL Streptomycin |
| Petri Dishes | VWR (or similar) | 60 mm, 100 mm | |
| Phalloidin | Sigma-Aldrich | P1951 | multiple flurophores available |
| Pin holder | Fine Science Tools | 26016-12 | For tungsten needle (alternative for spring scissors) |
| Scissors (dissection) | Fine Science Tools | 14061-10 | |
| Spring Scissors | Fine Science Tools | 15000-08 | 2.5 mm cutting edge (alternative for tungsten needle) |
| Sylgard | Krayden | Sylgard 184 | |
| Syringe Filters | Sigma-Aldrich | SLGVM33RS | Millex-GV Syringe Filter Unit, 0.22 µm, PVDF, 33 mm, gamma sterilized |
| Tissue culture dishes | Sarstedt | 83-3900 | 35 mm culture dishes for bulk neural fold cultures |
| Triton X-100 | Sigma-Aldrich | X100 | |
| Tungsten wire | Variety of sources | 0.01" diameter for tungsten needle (alternative for spring scissors) |
参考文献
- Pla, P., Monsoro-Burq, A. H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Developmental Biology. 444, 36-46 (2018).
- Tang, W., Bronner, M. E. Neural crest lineage analysis: From past to future trajectory. Development. 147 (20), (2021).
- Piacentino, M. L., Li, Y., Bronner, M. E. Epithelial-to-mesenchymal transition and different migration strategies as viewed from the neural crest. Current Opinion in Cell Biology. 66, 43-50 (2020).
- McLennan, R., et al. Neural crest cells bulldoze through the microenvironment using Aquaporin 1 to stabilize filopodia. Development. 147 (1), 185231 (2020).
- Carmona-Fontaine, C., et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Developmental Cell. 21 (6), 1026-1037 (2011).
- Giovannone, D., et al. Slits affect the timely migration of neural crest cells via robo receptor. Developmental Dynamics. 241 (8), 1274-1288 (2012).
- Vermillion, K. L., Lidberg, K. A., Gammill, L. S. Cytoplasmic protein methylation is essential for neural crest migration. Journal of Cell Biology. 204 (1), 95-109 (2014).
- Yang, X., Li, J., Zeng, W., Li, C., Mao, B. Elongator Protein 3 (Elp3) stabilizes Snail1 and regulates neural crest migration in Xenopus. Scientific Reports. 6 (1), 1-9 (2016).
- Gonzalez Malagon, S. G., et al. Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nature Communications. 9 (1), 1-15 (2018).
- Bhattacharya, D., Azambuja, A. P., Simoes-Costa, M. Metabolic reprogramming promotes neural crest migration via yap/tead signaling. Developmental Cell. 53 (2), 199-211 (2020).
- Jacques-Fricke, B. T., et al. Profiling NSD3-dependent neural crest gene expression reveals known and novel candidate regulatory factors. Developmental Biology. 475, 118-130 (2021).
- Bronner-Fraser, M., García-Castro, M. Chapter 4 manipulations of neural crest cells or their migratory pathways. Methods in Cell Biology. 87, 75-96 (2008).
- Milet, C., Monsoro-Burq, A. H. Dissection of xenopus laevis neural crest for in vitro explant culture or in vivo transplantation. Journal of Visualized Experiments. (85), e51118 (2014).
- Malagon, S. G. G., et al. Dissection, culture and analysis of primary cranial neural crest cells from mouse for the study of neural crest cell delamination and migration. Journal of Visualized Experiments. (152), e60051 (2019).
- Theveneau, E., Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Developmental Biology. 366 (1), 34-54 (2012).
- Conrad, G. W., Bee, J. A., Roche, S. M., Teillet, M. A. Fabrication of microscalpels by electrolysis of tungsten wire in a meniscus. Journal of Neuroscience Methods. 50 (1), 123-127 (1993).
- Hamburger, V., Hamilton, H. L. A series of normal stages in the development of the chick embryo. Journal of Morphology. 88 (1), 49-92 (1951).
- Gammill, L. S., Jacques-Fricke, B., Roffers-Agarwal, J. Embryological and genetic manipulation of chick development. Methods in Molecular Biology. 1920, 75-97 (2019).
- Vandekerckhove, J., Deboben, A., Nassal, M., Wieland, T. The phalloidin binding site of F-actin. The EMBO Journal. 4 (11), 2815-2818 (1985).
- Bronner-Fraser, M. Analysis of the early stages of trunk neural crest migration in avian embryos using monoclonal antibody HNK-1. Developmental Biology. 115 (1), 44-55 (1986).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Soille, P., Vincent, L. Determining watersheds in digital pictures via flooding simulations. Visual Communications and Image Processing '90: Fifth in a Series. (1360), 240-250 (1990).
- Haupt, A., Minc, N. How cells sense their own shape - mechanisms to probe cell geometry and their implications in cellular organization and function. Journal of Cell Science. 131 (6), (2018).
- Ezin, M., Fraser, S. Chapter 11 time-lapse imaging of the early avian embryo. Methods in Cell Biology. 87, 211-236 (2008).
- Kulesa, P. M., Bailey, C. M., Cooper, C., Fraser, S. E. In ovo live imaging of avian embryos. Cold Spring Harbor Protocols. 5 (6), (2010).
- McKinney, M. C., Kulesa, P. M. Live imaging of the neural crest cell epithelial-to-mesenchymal transition in the chick embryo. Methods in Molecular Biology. 2179, 107-114 (2021).
- Gustafson, C. M., Roffers-Agarwal, J., Gammill, L. S. Chick cranial neural crest cells release extracellular vesicles that are critical for their migration. Journal of Cell Science. , (2022).
- Williams, R., Sauka-Spengler, T. Ex ovo electroporation of early chicken embryos. STAR Protocols. 2 (2), 100424 (2021).
- Moulton, J. D. Using morpholinos to control gene expression. Current Protocols in Nucleic Acid Chemistry. 68 (1), 4-30 (2017).
- Gandhi, S., et al. A single-plasmid approach for genome editing coupled with long-term lineage analysis in chick embryos. Development. 148 (7), (2021).
- Williams, R. M., et al. Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Developmental Cell. 51 (2), 255-267 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved