
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Preparazione e analisi morfologica di colture cellulari di cresta neurale cranica di pulcino
In questo articolo
Riepilogo
Questo protocollo versatile descrive l'isolamento delle cellule della cresta neurale premigratoria (NCC) attraverso l'escissione di pieghe neurali craniche da embrioni di pulcino. Dopo la placcatura e l'incubazione, gli NCC migratori emergono dagli espianti di piega neurale, consentendo la valutazione della morfologia cellulare e della migrazione in un ambiente 2D semplificato.
Abstract
Durante lo sviluppo dei vertebrati, le cellule della cresta neurale (NCC) migrano ampiamente e si differenziano in vari tipi di cellule che contribuiscono a strutture come lo scheletro craniofacciale e il sistema nervoso periferico. Mentre è fondamentale comprendere la migrazione NCC nel contesto di un embrione 3D, l'isolamento delle cellule migratorie in coltura 2D facilita la visualizzazione e la caratterizzazione funzionale, integrando gli studi embrionali. Il presente protocollo dimostra un metodo per isolare le pieghe neurali craniche dei pulcini per generare colture NCC primarie. Gli NCC migratori emergono da espianti di piega neurale placcati su un substrato rivestito di fibronectina. Ciò si traduce in popolazioni NCC disperse e aderenti che possono essere valutate mediante colorazione e analisi morfologiche quantitative. Questo approccio culturale semplificato è altamente adattabile e può essere combinato con altre tecniche. Ad esempio, l'emigrazione NCC e i comportamenti migratori possono essere valutati mediante imaging time-lapse o interrogati funzionalmente includendo inibitori o manipolazioni sperimentali dell'espressione genica (ad esempio, DNA, morfolino o elettroporazione CRISPR). Grazie alla sua versatilità, questo metodo fornisce un potente sistema per studiare lo sviluppo di NCC cranici.
Introduzione
Le cellule della cresta neurale (NCC) sono una popolazione di cellule transitorie negli embrioni vertebrati. Le NCC sono specificate ai bordi della piastra neurale e subiscono una transizione epiteliale-mesenchimale (EMT) per migrare dal tubo neurale dorsale1. Dopo l'EMT, le NCC si disperdono ampiamente in tutto l'embrione, differenziandosi e contribuendo a varie strutture, tra cui lo scheletro craniofacciale, il tratto di deflusso del cuore e la maggior parte del sistema nervoso periferico2. I cambiamenti nella polarità cellulare, nel citoscheletro e nelle proprietà di adesione sono alla base di questo passaggio da una popolazione cellulare preliminaria a una popolazione di cellule migratorie3. Lo studio dell'EMT e della migrazione NCC fornisce approfondimenti sui meccanismi fondamentali della motilità cellulare e informa gli sforzi per prevenire e curare i difetti alla nascita e le metastasi del cancro.
Mentre l'analisi in vivo è vitale per comprendere i processi di sviluppo NCC in un contesto embrionale, i metodi in vitro offrono accessibilità visiva e fisica che facilitano ulteriori vie sperimentali. In un ambiente 2D semplificato, è possibile valutare la morfologia NCC, le strutture citoscheletriche e la distanza migrata. Inoltre, gli effetti della perturbazione genetica o solubile dei fattori sui comportamenti migratori delle NCC mobili possono essere analizzati 4,5,6,7,8,9,10. Inoltre, NCC premigratory o migratorie isolate possono essere raccolte, raggruppate e utilizzate per metodologie ad alto rendimento per studiare la regolazione dello sviluppo delle NCC attraverso profili proteomici, trascrittomici ed epigenomici 7,11. Mentre sono disponibili metodi per la preparazione di NCC cranici da vari organismi modello di sviluppo12,13,14, questo articolo dimostra la meccanica dell'approccio per coloro che imparano per la prima volta a coltivare NCC cranici da embrioni di pulcino.
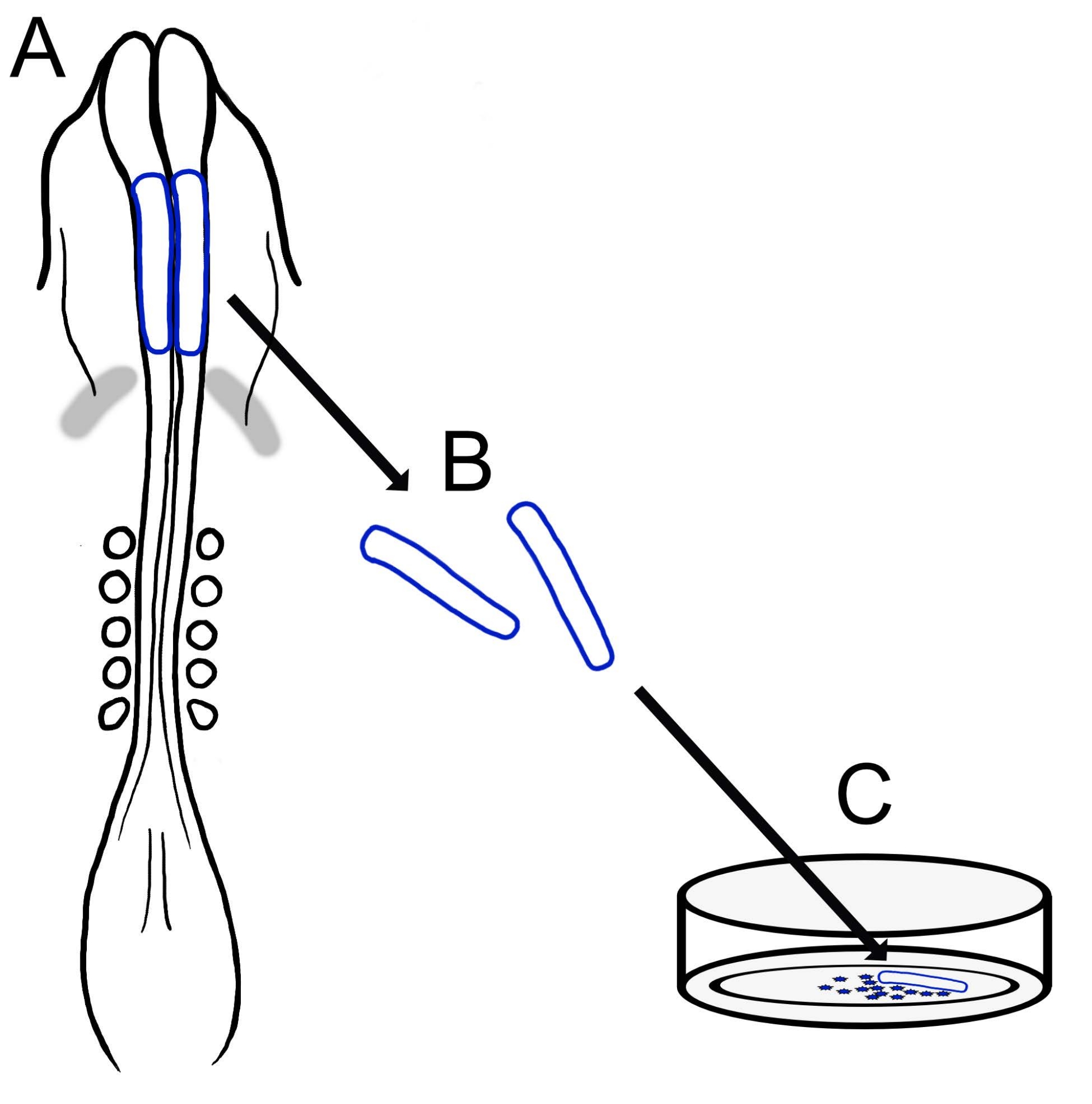
L'attuale protocollo descrive una tecnica versatile per la preparazione di colture di NCC craniche di pulcino (Figura 1). Poiché gli NCC migrano facilmente dalle pieghe neurali espiantate su un substrato di coltura, gli NCC di pulcino si separano naturalmente dal tessuto embrionale e le colture primarie sono facilmente generate. Poiché le NCC del mesencefalo migrano in massa dalle pieghe neurali craniche (in contrasto con la delaminazione prolungata, cellula per cellula nel tronco15), queste colture consistono principalmente di cellule migratorie della cresta neurale cranica, con l'escissione iniziale della piega neurale che fornisce un metodo di raccolta per le NCC premigatorie. Viene dettagliato un metodo di base per sezionare e coltivare le pieghe neurali craniche dei pulcini e vengono offerti suggerimenti per diverse applicazioni e variazioni di questo metodo.

Figura 1: Panoramica schematica del protocollo di coltura neurale cranica del pulcino. (A,B) Le pieghe neurali craniche (delineate in blu) sono asportate da un embrione di pulcino con cinque somiti (mostrati in vista dorsale in A). Bande grigie, mezzaluna cardiaca. (C) Quando placcate sulla fibronectina, le cellule della cresta neurale migratoria emergono dalle pieghe neurali e si disperdono sul substrato. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Protocollo
È possibile utilizzare qualsiasi varietà di razze Gallus gallus , tra cui White Leghorn, Golden Sex Link o Rhode Island Red. Le uova di gallina utilizzate nel presente studio erano di varie razze e ottenute da molteplici fonti, tra cui allevamenti e vivai locali.
1. Preparazione di soluzioni e materiali
- Preparare la soluzione di Ringer miscelando 123,3 mM NaCl, 1,53 mM CaCl 2, 4,96 mM KCl, 0,809 mM Na 2 HPO 4 e 0,147 mM KH2PO4 (vedere Tabella dei materiali). Regolare il pH a 7,4 e sterilizzare il filtro in flaconi da 100 ml. Conservare in luogo pulito e asciutto. Non lavare le bottiglie con sapone (trattare come vetreria per colture di tessuti).
- Preparare 1 mg/mL di soluzione madre di fibronectina (FN) (vedere Tabella dei materiali) sciogliendo FN in soluzione sterile di Ringer e conservare in 100 μL di aliquote a -80 °C (stabile per almeno 4 anni).
- Preparare terreni di coltura completi integrando terreni L15 con 0,8% L-glutammina, 0,08% penicillina / streptomicina, 10% FBS e 10% estratto di embrione di pulcino12. Creare 3 mL di aliquote e conservare a -20 °C o -80 °C.
NOTA: Se le colture sono incubate in un incubatore di CO2 , DMEM / F12 deve essere sostituito con L15, che è tamponato per l'aria. - Preparare una soluzione di paraformaldeide al 4% in 1x PBS. Regolare il pH a 7,4. Creare 10 ml di aliquote e conservarle a -20 °C.
ATTENZIONE: La paraformaldeide deve essere maneggiata in una cappa aspirante. - Preparare i telai di supporto della carta da filtro (circa 1 cm x 1,5 cm rettangoli di carta con due o tre fori perforati sovrapposti sull'asse lungo).
- Forare i fori lungo il bordo di un grande pezzo di carta da filtro usando un punzone standard a tre fori, tagliare / tagliare il bordo perforato in una striscia, quindi tagliare la striscia tra i fori in rettangoli ~ 1,5 cm di lunghezza. Carta da filtro per autoclave prima dell'uso (opzionale).
- Raccogli strumenti di dissezione, tra cui pinze sottili , pinze smussate, forbici da dissezione, forbici a molla e aghi di tungsteno affilati16.
2. Incubazione embrionale
- Ottenere uova fecondate da una qualsiasi delle fonti sopra menzionate.
- Posizionare le uova fecondate in posizione verticale (asse lungo dell'uovo verticale con l'estremità appuntita verso il basso / lato smussato verso l'alto) all'interno di un'incubatrice umidificata a 38 ° C (100 ° F).
- Incubare fino a formare da quattro a sette somiti (stadi 8+-9)17, che richiede circa 35 ore.
NOTA: Il tempo di incubazione può variare in modo significativo a seconda del ceppo, dell'età delle galline, della stagione, ecc. E deve essere determinato sperimentalmente. I timer di uscita programmabili sono utili per avviare l'incubazione durante la notte per ottenere i tempi desiderati. - Dopo aver rimosso dall'incubatore, spruzzare accuratamente le uova con etanolo al 70% e lasciarle asciugare per disinfettare i gusci.
NOTA: Gli embrioni possono essere raccolti immediatamente, ma lasciare raffreddare le uova a temperatura ambiente diminuisce la fragilità dei tuorli e la perdita di embrioni durante la raccolta.
3. Preparazione di piatti di cultura
- Selezionare i piatti di coltura a piega neurale (vedere la tabella dei materiali) appropriati per l'applicazione a valle.
- Per le colture che saranno fissate e macchiate, utilizzare vetrini di copertura collocati in piatti di coltura di tessuti multi-pozzetto.
NOTA: i coprivetrini che corrispondono alle dimensioni del pozzo si muoveranno meno con la miscelazione fluida, rendendo meno probabile che gli espianti si spostino e più che aderiscano al coprivetrino piuttosto che al fondo del piatto. - Selezionare una camera singola con fondo in vetro o piatti a camera divisa (vedere la tabella dei materiali) per l'imaging dal vivo con un microscopio invertito.
- Per l'imaging dal vivo su un microscopio verticale o stereo, utilizzare piastre di coltura tissutale otticamente appropriate a 6, 12 o 24 pozzetti.
- Selezionare piatti di coltura di tessuti plastici per la raccolta di massa di cellule della cresta neurale migratoria per analisi ad alto rendimento.
- Per le colture che saranno fissate e macchiate, utilizzare vetrini di copertura collocati in piatti di coltura di tessuti multi-pozzetto.
- Aggiungere 10 U/mL di penicillina e 10 μg/mL di streptomicina a un flacone da 100 mL di soluzione sterile di Ringer (ad esempio, 100 μL di 10.000 U/mL di penicillina e 10.000 μg/mL di streptomicina, per produrre P/S di Ringer). Utilizzare R / S di Ringer entro 1 settimana.
- Scongelare un'aliquota di 1 mg/mL di FN sul ghiaccio. Diluire con P/S di Ringer ad una concentrazione di 10-100 μg/mL di FN.
- Pipet abbastanza soluzione FN per coprire il fondo del pozzo o del piatto. Ad esempio, 100 μL per pozzetto in una piastra da 24 pozzetti, 500 μL per un piatto da 35 mm.
- Sostituire il coperchio e incubare piatti o piatti con una soluzione di FN in un'incubatrice umidificata (o in un vassoio coperto con asciugamani di carta distillati imbevuti d'acqua) a 38 °C (100 °F) per almeno 1 ora mentre si sezionano le pieghe neurali.
4. Isolamento degli embrioni di pulcino
- Fare attenzione a mantenere l'orientamento dell'uovo (l'embrione galleggia verso la parte superiore del tuorlo durante l'incubazione). Usa le forbici o una pinza smussata per praticare un piccolo foro nel guscio, circa 1/4-1/3 della lunghezza dell'uovo.
- Inserire con attenzione la punta delle forbici o delle pinze smussate nel piccolo foro, assicurandosi di non interrompere il tuorlo, e tagliare il guscio d'uovo attorno all'uovo, rimuovendo la parte superiore del guscio d'uovo (Figura 2A).
- Posizionare l'embrione per l'isolamento.
NOTA: Se l'embrione non si trova idealmente nella coppa del guscio, utilizzare una pinza smussata per girare con attenzione il tuorlo, in modo che l'embrione sia in cima. In alternativa, versare il tuorlo in una mano guantata a coppa, facendo attenzione a non rompere il tuorlo e lasciando che l'albumina defluisca (Figura 2B). Quindi, l'embrione può essere posizionato spostando il tuorlo di mano in mano. - Preparare l'embrione usando delicatamente il bordo piatto di una pinza smussata chiusa per rimuovere l'albumina in eccesso che rimane sulla superficie del tuorlo che copre l'embrione (Figura 2C). L'albumina in eccesso può anche essere rimossa usando una leggera pressione con un delicato tergicristallo.
NOTA: Una volta eliminata l'albumina, la superficie del tuorlo dovrebbe apparire strutturata anziché liscia. La mancata rimozione di albumina sufficiente inibirà l'adesione del supporto di carta da filtro nella sezione 4, fase 4. - Utilizzare una pinza per posizionare una cornice di supporto in carta da filtro sopra l'embrione, con l'embrione nella finestra del telaio. Premere delicatamente sulla carta da filtro per farla aderire al tuorlo.
- Tagliare intorno all'esterno del telaio della carta da filtro con le forbici per dissezione (Figura 2D). Utilizzare una pinza o punte a forbice per afferrare il bordo del telaio e sollevare delicatamente l'embrione dal tuorlo (Figura 2E). Il sollevamento ad angolo aiuterà a rimuovere l'embrione in modo pulito dal tuorlo.
- Posizionare l'embrione con il telaio di carta rivolto verso il basso (lato ventrale dell'embrione verso l'alto) in una capsula di Petri da 60 mm o 100 mm riempita con P/S di Ringer (Figura 2F). Conservare il piatto di embrioni sul ghiaccio se si raccoglie per un'applicazione a valle sensibile all'RNA o alle proteine.
NOTA: Il mancato posizionamento degli embrioni con il lato ventrale verso l'alto rischia di staccarli dalle loro cornici di carta. Più embrioni possono essere raccolti e conservati nel R / S per un massimo di 1 ora prima di passare alle fasi di dissezione della piega neurale.
5. Dissezione delle pieghe neurali
- Trasferire un embrione in un piatto pulito contenente la soluzione P/S di Ringer e sciacquare l'embrione tenendo il telaio di carta da filtro con una pinza e facendo scorrere delicatamente avanti e indietro per eliminare qualsiasi tuorlo che oscura la vista dell'embrione. Sostituire il R / S del Ringer o trasferirlo su un piatto fresco se diventa torbido.
- Posizionare l'embrione dorsale/telaio verso l'alto sotto un microscopio da dissezione. Lasciando l'embrione sul telaio di carta per mantenerlo teso e tenuto in posizione, rimuovere la membrana vitellina usando una pinza per esporre le pieghe neurali.
NOTA: Se l'embrione cade dal telaio di carta, può essere steso piatto nel piatto o appuntato su un piatto rivestito sylgard18 per la dissezione. - Usando forbici a molla o un ago di tungsteno affilato, asportare con cura le pieghe neurali del mesencefalo. Includere il tessuto caudale alle vescicole ottiche in espansione e rostrale al cervello posteriore, dove le costrizioni rombomeriche stanno appena iniziando a comparire (anche la mezzaluna cardiaca è un indicatore utile, Figura 3B,C). Fare attenzione ad asportare l'aspetto più dorsale della piega neurale con un tubo neurale contaminante minimo e ectoderma non neurale (Figura 3C).
- Trasferire le pieghe neurali in un piatto pulito contenente P/S di Ringer usando un pipettor P20 o una pipetta Pasteur sterile in vetro risciacquata con P/S di Ringer (questo blocca la plastica o il vetro per evitare che il tessuto si attacchi). Conservare le pieghe raccolte sul ghiaccio mentre si sezionano ulteriori pieghe.
6. Placcatura pieghe neurali
- Scongelare un'aliquota di terreni di coltura completi (sezione 1, fase 3). Aggiungere 100 U/mL di penicillina e 100 μg/ml di streptomicina e filtrare sterilizzare. Mantenere i mezzi preparati a 37-38 °C mentre si eseguono altri passaggi.
- Rimuovere i piatti di coltura dall'incubatore (sezione 3, passaggio 5). Utilizzando un pipettor o una pipetta Pasteur, rimuovere la soluzione FN da coperchi, stoviglie o pozzetti. Dopo aver risciacquato il substrato rivestito di FN con P/S di Ringer, aggiungere un volume appropriato di terreno di coltura completo alle piastre o ai pozzetti (500 μL per i pozzetti in una piastra da 24 pozzetti, 2 ml per un piatto da 35 mm o una piastra a sei pozzetti).
NOTA: volumi più piccoli possono preservare reagenti costosi (ad esempio, fino a 200 μL per una piastra a 24 pozzetti) ma garantire che il piatto sia sufficientemente umidificato per evitare l'evaporazione. - Utilizzando un pipettatore p20 o p200, in primo luogo, sciacquare la punta della pipetta con il tuorlo Ringer P / S per bloccare la plastica e impedire che il tessuto si attacchi. Quindi, trasferire le pieghe neurali isolate sui coprivetrini rivestiti di FN, avendo cura di trasferire il minor R / S possibile. Posizionare la piega o le pieghe verso il centro del coprislip rivestito in FN.
NOTA: Una o più pieghe neurali possono essere placcate su un coprivetrino da 12 mm in un pozzetto da 19 mm, fino a 50 su una piastra da 35 mm. - Dopo aver lasciato riposare gli espianti per 10-15 minuti, metterli in una camera umidificata a 38 ° C (100 ° F) trasportando lentamente e con attenzione i piatti di coltura con pieghe neurali placcate. Ciò ridurrà al minimo lo spostamento delle pieghe neurali nei pozzi, assicurando che rimangano disperse e aderiscano a eventuali scivolamenti di copertura.
NOTA: Quando si utilizza L15 (non DMEM), i piatti di coltura espiantati possono anche essere incubati in un'incubatrice per uova utilizzando un vassoio coperto con asciugamani di carta inumiditi. - Incubare colture di pieghe neurali nell'incubatore umidificato per tutta la durata della migrazione attiva (circa 16-20 ore totali, Figura 4).
7. Fissazione e colorazione di NCC migratori in coltura per analisi morfologiche
- Rimuovere il terreno di coltura con una pipetta Pasteur e sciacquare i pozzetti con 1x PBS sterilizzato con filtro.
- Aggiungere il 4% di paraformaldeide per 15 minuti a temperatura ambiente.
NOTA: potrebbe essere necessario determinare sperimentalmente il tempo di fissazione. È anche possibile aggiungere paraformaldeide direttamente ai supporti per 10 minuti (50:50), rimuovere e sostituire con paraformaldeide non diluita al 4% per 10 minuti per mantenere strutture cellulari più delicate. - Rimuovere la paraformaldeide e risciacquare tre volte con 1x PBS.
- Colorare NCC fissi con un colorante appropriato per la valutazione morfologica. Ad esempio, la colorazione con falloidina coniugata Oregon Green è dettagliata qui.
NOTA: La visualizzazione del citoscheletro di actina con falloidina19 rivela la complessità strutturale del NCC migratorio con intensità di colorazione relativamente uniforme; Tuttavia, la falloidina potrebbe non evidenziare tutte le aree cellulari. Possono essere utilizzati anche coloranti che etichettano la membrana plasmatica (ad esempio, agglutinina del germe di grano o DiI) o il citoplasma, ma complicano l'imaging di sporgenze fini rispetto al corpo cellulare che colora brillantemente. Inoltre, l'immunofluorescenza può essere eseguita contemporaneamente per marcare le cellule della cresta neurale con HNK-1 o per determinare la localizzazione subcellulare di una proteina di interesse 7,20.- Rimuovere il risciacquo finale PBS e aggiungere PBS + 0,5% Triton X-100 (PBST) + 5% siero (FBS o da un altro animale adatto per il costing con un anticorpo) per coprire i coprivetrini e incubare per 10 minuti su uno shaker a piattaforma a temperatura ambiente.
- Pipet 200 nM di falloidina coniugata Oregon Green (diluita in siero PBST + 5%) su una superficie liscia (come pellicola sigillante flessibile per paraffina) per ogni vetrino. Per un coprivetrino da 12 mm, pipettare un volume di 30 μL.
- Utilizzando un paio di pinze, rimuovere il coprislip dal siero PBST + 5%, assicurandosi di mantenere l'orientamento delle cellule rivolte verso l'alto. Toccare brevemente il bordo del coprislip su un tergicristallo delicato per assorbire il liquido in eccesso.
- Posizionare delicatamente ogni lato della cellula di copertura verso il basso sulla goccia di falloidina diluita. Incubare per 30 minuti a temperatura ambiente. Tenere i coperchi al buio durante la colorazione (coprire con un piatto coperto di alluminio o mettere in un cassetto).
- Durante l'incubazione, aggiungere PBST ai pozzetti del piatto di coltura (750 μL di PBST per ogni pozzetto di una piastra da 24 pozzetti).
- Dopo il periodo di incubazione, sollevare i coperchi dalla soluzione colorante e rimetterli nel piatto di coltura, capovolgendo il vetrino di copertura in modo che il lato della cellula sia verso l'alto. Assicurarsi che il coprivetrino sia coperto con PBST e posizionarlo su uno scuotitore a piattaforma per 10 minuti, mantenendo i coprivetrini coperti e al buio. Rimuovere PBST e ripetere questo passaggio due volte, per un totale di tre lavaggi di 10 minuti.
- Posizionare una goccia (per vetrino) di supporto di montaggio (vedere Tabella dei materiali) su un vetrino da microscopio. Il volume di 25 μL funziona bene per un coprivetrino da 12 mm.
- Dopo il lavaggio finale con PBST, toccare il bordo del coprivetrino su un delicato tergicristallo per rimuovere il liquido in eccesso, tenendo traccia del lato cellulare del vetrino.
- Montare la cella del coprislip con il lato rivolto verso il basso abbassando lentamente il coprislip ad angolo sul supporto di montaggio per evitare la creazione di bolle. Consentire l'impostazione del supporto prima dell'imaging.
8. Valutazione morfologica dei NCC migratori in coltura
- Immagine delle celle colorate ed esportazione come file .tiff.
NOTA: Un obiettivo 40x funziona bene per le colture di imaging, ma obiettivi che vanno da 10x (per catturare un intero campo di NCC migratori) a 100x (singoli NCC) possono essere utilizzati per raccogliere immagini per la valutazione morfologica. Le immagini nel presente studio sono state catturate utilizzando una piattaforma di imaging multimodale invertita (vedi Tabella dei materiali). - Caricare le immagini in un software di analisi delle immagini (ImageJ21, vedere Tabella dei materiali). Fare clic su Immagine > Duplica per creare una seconda copia di ogni immagine da utilizzare per l'analisi, lasciando così l'originale non modificato, poiché la maggior parte dell'elaborazione delle immagini non può essere invertita.
- Fare clic su Immagine > Regola > luminosità / contrasto e utilizzare le barre di scorrimento per regolare la luminosità o il contrasto delle immagini.
- Converti le immagini in scala di grigi seguendo i passaggi seguenti.
- Se le immagini vengono esportate in RGB, verranno caricate in RGB come canale unito. Per separare le immagini, fare clic su Colore > immagine > Canali divisi per acquisire immagini in scala di grigi a canale singolo.
- Se le immagini sono già separate, vai a Immagine > Digitare > 8 bit per convertire il file in scala di grigi.
- Fare clic su Immagine > Regola > soglia per convertirla in un'immagine binaria in cui i pixel sono identificati come celle (primo piano, nero) o sfondo (bianco). Utilizzare la barra di scorrimento e/o fare clic su Auto per selezionare l'impostazione Soglia che definisce chiaramente le celle dallo sfondo.
- Se le celle si sovrappongono o la colorazione non è continua, potrebbe essere necessario elaborare ulteriormente utilizzando le funzioni Spartiacque o Fori di riempimento22. Questo separerà le cellule l'una dall'altra o riempirà le lacune all'interno delle celle da analizzare. Fare clic su Elabora > binario > Crea binario per convertire prima l'immagine in un formato binario a 8 bit. Quindi fare clic su Elabora > binario > Spartiacque o Riempi fori.
NOTA: il software si adatta meglio ai punti in cui i segnali devono essere separati o compilati, che possono essere regolati ulteriormente utilizzando plug-in, se necessario.
- Se le celle si sovrappongono o la colorazione non è continua, potrebbe essere necessario elaborare ulteriormente utilizzando le funzioni Spartiacque o Fori di riempimento22. Questo separerà le cellule l'una dall'altra o riempirà le lacune all'interno delle celle da analizzare. Fare clic su Elabora > binario > Crea binario per convertire prima l'immagine in un formato binario a 8 bit. Quindi fare clic su Elabora > binario > Spartiacque o Riempi fori.
- Selezionare le misure da acquisire. Fare clic su Analizza > Imposta misure e selezionare le caselle Descrittori forma per l'analisi di circolarità (Circ.), Proporzioni (AR), Rotondità (Arrotondamento) e Solidità.
NOTA: possono essere incluse anche altre misure, come Area e Perimetro, a seconda del tipo di analisi necessaria. - Fare clic su Analizza > Analizza particelle e impostare i parametri dal menu "Analizza particelle".
- In "Dimensioni", stimare approssimativamente quanto sono grandi le celle o le particelle di interesse in pixel, poiché verranno inclusi anche il segnale di sfondo o i detriti di celle più piccoli se il parametro Size rimane impostato su 0-Infinity (ad esempio, impostato come 500-Infinity nella Figura 5).
NOTA: la regolazione delle dimensioni su un intervallo più ristretto può escludere particelle più piccole o più grandi di quelle necessarie per l'analisi. - In "Circolarità", lasciare 0-1,0 per misurare tutte le forme di cella nell'immagine.
- In "Mostra", selezionare Contorni nudi dal menu a discesa per ispezionare i contorni delle celle selezionati con la funzione "Analizza particelle". Eseguire la scansione dei contorni per assicurarsi che le celle sovrapposte siano distinte, ma le celle non vengano divise inutilmente. Inoltre, seleziona le caselle di menu per "Visualizza risultati", "Riepilogo" e "Aggiungi a Manager".
- In "Dimensioni", stimare approssimativamente quanto sono grandi le celle o le particelle di interesse in pixel, poiché verranno inclusi anche il segnale di sfondo o i detriti di celle più piccoli se il parametro Size rimane impostato su 0-Infinity (ad esempio, impostato come 500-Infinity nella Figura 5).
- Una volta impostati tutti i parametri, fare clic su Ok.
NOTA: Appariranno quattro finestre separate, che mostrano (1) i contorni nudi delle celle identificate nell'immagine, (2) le celle contate nell'immagine, (3) i risultati delle misurazioni di ciascuna cella e (4) un riepilogo del numero totale di celle contate e delle loro misurazioni medie.- Se i detriti sono stati contati, le celle sovrapposte sono state contate come una, le singole celle sono state contate come multipli o molte celle sono state omesse dal conteggio, tornare all'immagine originale non modificata, creare un altro duplicato e regolare "Soglia" o altri parametri.
Risultati
Una panoramica del presente protocollo è mostrata nella Figura 1. Le uova incubate sono state aperte e il tuorlo, con l'embrione in superficie, è stato isolato versando delicatamente nel palmo di una mano guantata (Figura 2A,B). Dopo aver eliminato l'albumina (Figura 2C), sono stati applicati telai di carta da filtro sulla membrana del tuorlo che circonda l'embrione per facilitare il taglio e il sollevamento dell'...
Discussione
La tecnica qui descritta fornisce un metodo adattabile per isolare le pieghe neurali dei pulcini e placcarle per creare colture di NCC cranici migratori. Queste colture forniscono condizioni 2D semplificate per una facile analisi della migrazione e della morfologia NCC del pulcino che possono integrare metodi di imaging più tecnicamente impegnativi in ovo 24,25,26. Mentre questo metodo in vitro è relativament...
Divulgazioni
Gli autori non hanno conflitti di interesse.
Riconoscimenti
Ringraziamo Corinne A. Fairchild e Katie L. Vermillion, che hanno partecipato allo sviluppo della nostra versione del protocollo di coltura della piega neurale cranica del pulcino.
Materiali
| Name | Company | Catalog Number | Comments |
| AxioObserver equipped with an LSM710 confocal scan head controlled by ZEN 3.0 SR software | Zeiss | Used alpha Plan-Apochromat 100x/1.46 Oil DIC M27 objective | |
| CaCl2 | Sigma-Aldrich | C3306 | |
| Chamber dishes (glass bottom, single or divided) | MatTek; Cell Vis | P35G-1.5-14-C (MatTek) X000NOJQGX (Cellvis) X000NOK1OJ (Cellvis) | Single chamber 35 mm or 4 chamber 35 mm |
| Cover glass | Carolina Biological Supply Company | 633029, 633031, 633033, 633035, 633037 | circles, 0.13–0.17 mm thickness, available in 12-25 mm diameter |
| DMEM/F12 | ThermoFisher Scientific | 11320033 | Alternative for L15 media |
| Egg incubator | Sportsman | 1502 | |
| FBS | Life Technologies | 10437-028 | |
| Fibronectin | Fisher Scientific | CB-40008A | |
| Filter paper | Whatman | grade 3MM chromatography | |
| Forceps (blunt) | Fisher Scientific; Thomas Scientific | 08-890 (Fisher);1141W97 (Thomas) | |
| Forceps (fine) | Fine Science Tools | 11252-20 | Dumont #5 |
| Image J | https://fiji.sc/ | Free image analysis software | |
| KCl | Sigma-Aldrich | P3911 | |
| KH2PO4 | Sigma-Aldrich | P0662 | |
| L15 media | Invitrogen | 11415064 | |
| L-glutamine | Invitrogen | 25030 | |
| Mounting Media (Vectashield or ProLong Gold) | Vector Laboratories; Thermofisher Scientific | H-1700 (Vectashield); P36930 (ProLong Gold) | |
| Na2HPO4 | Sigma-Aldrich | S9638 | |
| NaCl | Sigma-Aldrich | S9888 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| Penicillin/streptomycin | Life Technologies | 15140-148 | 10,000 Units/mL Penicillin; 10,000 mg/mL Streptomycin |
| Petri Dishes | VWR (or similar) | 60 mm, 100 mm | |
| Phalloidin | Sigma-Aldrich | P1951 | multiple flurophores available |
| Pin holder | Fine Science Tools | 26016-12 | For tungsten needle (alternative for spring scissors) |
| Scissors (dissection) | Fine Science Tools | 14061-10 | |
| Spring Scissors | Fine Science Tools | 15000-08 | 2.5 mm cutting edge (alternative for tungsten needle) |
| Sylgard | Krayden | Sylgard 184 | |
| Syringe Filters | Sigma-Aldrich | SLGVM33RS | Millex-GV Syringe Filter Unit, 0.22 µm, PVDF, 33 mm, gamma sterilized |
| Tissue culture dishes | Sarstedt | 83-3900 | 35 mm culture dishes for bulk neural fold cultures |
| Triton X-100 | Sigma-Aldrich | X100 | |
| Tungsten wire | Variety of sources | 0.01" diameter for tungsten needle (alternative for spring scissors) |
Riferimenti
- Pla, P., Monsoro-Burq, A. H. The neural border: Induction, specification and maturation of the territory that generates neural crest cells. Developmental Biology. 444, 36-46 (2018).
- Tang, W., Bronner, M. E. Neural crest lineage analysis: From past to future trajectory. Development. 147 (20), (2021).
- Piacentino, M. L., Li, Y., Bronner, M. E. Epithelial-to-mesenchymal transition and different migration strategies as viewed from the neural crest. Current Opinion in Cell Biology. 66, 43-50 (2020).
- McLennan, R., et al. Neural crest cells bulldoze through the microenvironment using Aquaporin 1 to stabilize filopodia. Development. 147 (1), 185231 (2020).
- Carmona-Fontaine, C., et al. Complement fragment C3a controls mutual cell attraction during collective cell migration. Developmental Cell. 21 (6), 1026-1037 (2011).
- Giovannone, D., et al. Slits affect the timely migration of neural crest cells via robo receptor. Developmental Dynamics. 241 (8), 1274-1288 (2012).
- Vermillion, K. L., Lidberg, K. A., Gammill, L. S. Cytoplasmic protein methylation is essential for neural crest migration. Journal of Cell Biology. 204 (1), 95-109 (2014).
- Yang, X., Li, J., Zeng, W., Li, C., Mao, B. Elongator Protein 3 (Elp3) stabilizes Snail1 and regulates neural crest migration in Xenopus. Scientific Reports. 6 (1), 1-9 (2016).
- Gonzalez Malagon, S. G., et al. Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nature Communications. 9 (1), 1-15 (2018).
- Bhattacharya, D., Azambuja, A. P., Simoes-Costa, M. Metabolic reprogramming promotes neural crest migration via yap/tead signaling. Developmental Cell. 53 (2), 199-211 (2020).
- Jacques-Fricke, B. T., et al. Profiling NSD3-dependent neural crest gene expression reveals known and novel candidate regulatory factors. Developmental Biology. 475, 118-130 (2021).
- Bronner-Fraser, M., García-Castro, M. Chapter 4 manipulations of neural crest cells or their migratory pathways. Methods in Cell Biology. 87, 75-96 (2008).
- Milet, C., Monsoro-Burq, A. H. Dissection of xenopus laevis neural crest for in vitro explant culture or in vivo transplantation. Journal of Visualized Experiments. (85), e51118 (2014).
- Malagon, S. G. G., et al. Dissection, culture and analysis of primary cranial neural crest cells from mouse for the study of neural crest cell delamination and migration. Journal of Visualized Experiments. (152), e60051 (2019).
- Theveneau, E., Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Developmental Biology. 366 (1), 34-54 (2012).
- Conrad, G. W., Bee, J. A., Roche, S. M., Teillet, M. A. Fabrication of microscalpels by electrolysis of tungsten wire in a meniscus. Journal of Neuroscience Methods. 50 (1), 123-127 (1993).
- Hamburger, V., Hamilton, H. L. A series of normal stages in the development of the chick embryo. Journal of Morphology. 88 (1), 49-92 (1951).
- Gammill, L. S., Jacques-Fricke, B., Roffers-Agarwal, J. Embryological and genetic manipulation of chick development. Methods in Molecular Biology. 1920, 75-97 (2019).
- Vandekerckhove, J., Deboben, A., Nassal, M., Wieland, T. The phalloidin binding site of F-actin. The EMBO Journal. 4 (11), 2815-2818 (1985).
- Bronner-Fraser, M. Analysis of the early stages of trunk neural crest migration in avian embryos using monoclonal antibody HNK-1. Developmental Biology. 115 (1), 44-55 (1986).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Soille, P., Vincent, L. Determining watersheds in digital pictures via flooding simulations. Visual Communications and Image Processing '90: Fifth in a Series. (1360), 240-250 (1990).
- Haupt, A., Minc, N. How cells sense their own shape - mechanisms to probe cell geometry and their implications in cellular organization and function. Journal of Cell Science. 131 (6), (2018).
- Ezin, M., Fraser, S. Chapter 11 time-lapse imaging of the early avian embryo. Methods in Cell Biology. 87, 211-236 (2008).
- Kulesa, P. M., Bailey, C. M., Cooper, C., Fraser, S. E. In ovo live imaging of avian embryos. Cold Spring Harbor Protocols. 5 (6), (2010).
- McKinney, M. C., Kulesa, P. M. Live imaging of the neural crest cell epithelial-to-mesenchymal transition in the chick embryo. Methods in Molecular Biology. 2179, 107-114 (2021).
- Gustafson, C. M., Roffers-Agarwal, J., Gammill, L. S. Chick cranial neural crest cells release extracellular vesicles that are critical for their migration. Journal of Cell Science. , (2022).
- Williams, R., Sauka-Spengler, T. Ex ovo electroporation of early chicken embryos. STAR Protocols. 2 (2), 100424 (2021).
- Moulton, J. D. Using morpholinos to control gene expression. Current Protocols in Nucleic Acid Chemistry. 68 (1), 4-30 (2017).
- Gandhi, S., et al. A single-plasmid approach for genome editing coupled with long-term lineage analysis in chick embryos. Development. 148 (7), (2021).
- Williams, R. M., et al. Reconstruction of the Global Neural Crest Gene Regulatory Network In Vivo. Developmental Cell. 51 (2), 255-267 (2019).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati