
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
セプチンの超微細構造組織、膜再形成、および曲率感度挙動をアッセイするためのボトムアップ インビトロ 法
* これらの著者は同等に貢献しました
要約
セプチンは細胞骨格タンパク質である。それらは脂質膜と相互作用し、ミクロンスケールで膜曲率を感知するだけでなく生成することもできる。このプロトコルでは、膜変形、曲率に敏感なセプチン結合、およびセプチンフィラメントの超構造を分析するためのボトムアップ インビトロ 方法論について説明します。
要約
膜リモデリングは、原形質膜および細胞小器官内で絶えず起こる。環境(イオン条件、タンパク質および脂質組成、膜曲率)と特定の膜再形成プロセスに関連するさまざまなパートナーの役割を完全に解剖するために、in vitro ボトムアップアプローチを実施します。近年、主要な疾患に関連するセプチンタンパク質の役割を明らかにすることに強い関心が寄せられている。セプチンは、原形質膜と相互作用する必須かつ遍在する細胞骨格タンパク質である。それらは、細胞分裂、細胞運動性、神経形態形成、および精子形成、他の機能の中でも特に関与している。したがって、セプチンが膜でどのように相互作用し、組織化してその後膜変形を誘発するか、およびセプチンが特定の膜曲率にどのように敏感であるかを理解することが重要です。本稿は、分子レベルでのセプチンの超構造とミクロンスケールで起こる膜リモデリングとの相互作用を解読することを目的としている。この目的のために、出芽酵母、および哺乳動物セプチン複合体を組換え発現および精製した。次いで、 インビトロ アッセイの組み合わせを用いて、膜におけるセプチンの自己組織化を分析した。支持脂質二重層(SLB)、巨大な単層小胞(GUV)、大きな単層小胞(LUV)、および波状基質を用いて、セプチン自己組織化、膜再形成、および膜曲率の間の相互作用を研究した。
概要
セプチンは、脂質膜と相互作用する細胞骨格フィラメント形成タンパク質である。セプチンは真核生物に遍在しており、多数の細胞機能に不可欠です。それらは、出芽酵母および哺乳動物における細胞分裂の主要な調節因子として同定されている1,2。それらは、膜再形成事象、繊毛形成3、および精子形成4に関与している。哺乳動物細胞内では、セプチンはまた、Rho GTPases(BORG)依存的に結合剤であるアクチンおよび微小管5、6、7と相互作用し得る8。様々な組織(ニューロン9、繊毛3、精子10)において、セプチンは、膜結合成分11に対する拡散障壁の調節因子として同定されている。セプチンはまた、膜ブレビングおよび突出形成12を調節することが示されている。セプチンはマルチタスクタンパク質であり、様々な流行疾患の出現に関与している13。それらの誤った調節は、癌14および神経変性疾患15の出現と関連している。
生物に応じて、いくつかのセプチンサブユニット(Caenorhabditis elegansでは2つ、ヒトでは13個)が集合して複合体を形成し、その組織は組織依存的に変化する16。基本的なセプチンビルディングブロックは、2〜4つのサブユニットを収集し、2つのコピーで存在し、棒状の回文様式で自己組織化する。出芽酵母において、セプチンは八量体17,18である。その場で、セプチンはしばしばマイクロメートル曲率を有する部位に局在する。それらは分裂狭窄部位、繊毛および樹状突起の基部、および精子の環状で見出される19,20。膜では、セプチンの役割は二重であるように思われる:それらは脂質二重層を再形成し、膜の完全性を維持することに関与している21。したがって、膜におけるセプチンフィラメント形成タンパク質および/またはサブユニットの生物物理学的特性を調査することは、それらの役割を理解するために極めて重要である。十分に制御された環境におけるセプチンの特定の特性を解剖するためには、ボトムアップのインビトロアプローチが適切である。これまでのところ、インビトロでのセプチンの生物物理学的特性を記載しているのはごく少数のグループのみである20、22、23。したがって、他の細胞骨格フィラメントと比較して、インビトロでのセプチンの挙動に関する現在の知識は限られたままである。
このプロトコルは、セプチンフィラメントの組織化、膜の再形成、および曲率感度を分析する方法を説明しています19。この目的のために、光学および電子顕微鏡法(蛍光顕微鏡法、クライオ電子顕微鏡[クライオEM]、および走査型電子顕微鏡[SEM])の組み合わせが使用されてきた。マイクロメートルサイズの巨大な単層小胞(GUV)の膜再形成は、蛍光光学顕微鏡を用いて可視化される。脂質小胞に結合したセプチンフィラメントの配置および超構造の解析は、クライオEMを用いて行われる。セピン曲率感度の解析はSEMを用いて行われ、可変曲率の波状基板上に堆積した固体支持脂質二重層に結合したセプチンフィラメントの挙動を研究することにより、正と負の両方の曲率感度の解析が可能になります。以前の解析20,24と比較して、ここでは、セプチンがどのように自己集合し、相乗的に膜を変形し、曲率に敏感であるかを徹底的に分析するために、これらの方法の組み合わせを使用することを提案します。このプロトコールは、膜に対して親和性を示す任意の糸状タンパク質に有用かつ適応可能であると考えられている。
プロトコル
1. 巨大単層小胞(GUV)を用いた膜リフォーミングの決定
注:このセクションでは、GUVは、細胞の文脈でセプチンによって誘発される可能性のある膜変形を模倣するために生成されます。実際、細胞では、セプチンはマイクロメートルの曲率を有する部位で頻繁に見出される。GUVのサイズは数マイクロメートルから数十マイクロメートルで、変形することができます。したがって、マイクロメートルスケールのセプチン誘発変形をアッセイするのに適しています。蛍光脂質、ならびに蛍光標識されたセプチン(緑色蛍光タンパク質[GFP]を使用)は、蛍光顕微鏡 を介して 脂質およびタンパク質の両方の挙動を追跡するために使用される。
- 緩衝液および溶液の調製
- GUV増殖緩衝液(50 mM NaCl、50 mM スクロース、および10 mM トリス[pH = 7.8])および観察用緩衝液(75 mM NaClおよび10 mM Tris [pH = 7.8])を調製する。
- 測定(市販の浸透圧計を使用)し、観察および成長バッファーの浸透圧を調整します(170 mOsmol·L-1、理論的には)それぞれの浸透圧が等しくなるまで少量のNaClを添加することによって。0.2 μmのフィルターを使用してバッファーをろ過します。成長バッファーをアリコートし、さらに使用するために-20°Cで保存します。観察バッファーを4°Cで保存する。
注: 両方のバッファー間の浸透圧の差は 5% を超えてはなりません。 - 観察バッファーに5mg・mL-1 β-カゼイン溶液を調製した。完全な溶解を確実にする(磁気攪拌しながら4°Cで数時間後)。溶液を0.2 μmフィルター、アリコートでろ過し、-20 °Cで保存します。
- クロロホルム中で総脂質濃度3mg・mL-1の脂質混合物を調製した。56.8%卵L-α-ホスファチジルコリン(EggPC)、15%コレステロール、10%1,2-ジオレオイル-sn-グリセロ-3-ホスホエタノールアミン(DOPE)、10%1,2-ジオレオイル-sn-グリセロ-3-ホスホ-L-セリン(DOPS)、8%脳L-α-ホスファチジルイノシトール-4,5-ビスリン酸(PI(4,5)P2)、および0.2%ボディピー-TR-セラミドの組成物(モル%)を使用して、タンパク質-脂質相互作用を増強し、PI(4,5)P2 25の組み込みを支持する。
メモ:ニトリル手袋と安全メガネを使用して、ヒュームフードの下でクロロホルムを取り扱ってください。クロロホルム溶液をガラスシリンジでピペットし、クロロホルムがプラスチックを溶解するため、プラスチックを避けます。使用前後に、クロロホルム5x-10xをピペッティングしてシリンジをすすいでください。交差汚染を防ぐために、特定の蛍光脂質をピペットに別々のシリンジを使用してください。脂質は、テフロン(登録商標)で蓋をした琥珀色のガラスバイアル中で−20°Cでクロロホルム中に保存することができる。バイアルは、脂質酸化を防ぐために、閉じる前にアルゴンで満たされ、パラフィルムで密封されなければなりません。 - 白金線セットアップによるGUVの電化
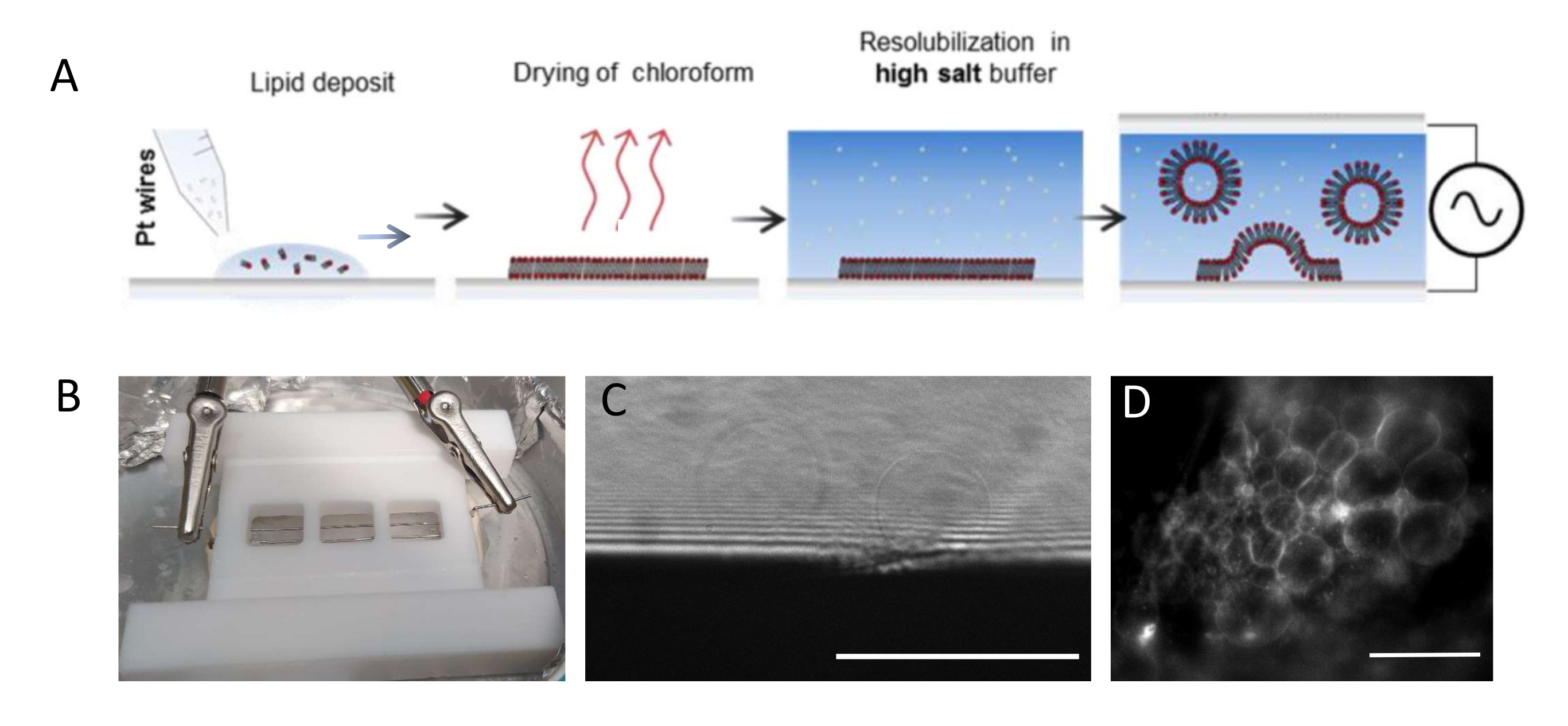
注: 図1 は、実験ステップのスキームとチャンバーの写真を示しています。- チャンバーと白金線を次のように徹底的に清掃し、脂質残渣を除去します。
- ワイヤーとチャンバーをアセトンに突っ込み、10分間超音波処理します。アセトンを使用して紙のティッシュで慎重に拭きます。
- ワイヤーを挿入してチャンバーを組み立て、アセトンに再度突っ込み、10分間超音波処理する。もう一度アセトンで拭き取り、ワイヤーが完全にクリーニングされていることを確認します。チャンバーをエタノールに浸し、10分間超音波処理し、エタノールで拭きます。
- 最後に、チャンバーを脱イオン水に浸し、10分間超音波処理し、窒素または空気の流れで乾燥させる。
注:テフロン(登録商標)チャンバー(図1B)は、社内ワークショップでカスタムメイドされた。これは、ガラスカバースリップを使用して両側に密封することができる3つのコンパートメントを収容する。白金線は、直径1.3mmの穴を通してチャンバに挿入することができます。 - チャンバーを洗浄した後、各白金線上に3mg・mL-1 脂質混合物のコンパートメントあたり3〜4滴(各滴は約0.1μL)を堆積させる。ワイヤーを180°回転させ、各白金ワイヤーの反対側にコンパートメントごとに3〜4個の脂肪滴を堆積させます。滴が互いに接触しないようにします。チャンバー全体あたり約5μLの脂質混合物が必要である。
- 成長チャンバを真空チャンバに30分間置き、クロロホルムの痕跡を除去します。
注:深い真空(0.1 mbar)が最適です。乾燥すると、脂質は酸化に対して脆弱であるため、数分間以上空気中に放置しないでください。 - シリンジを使用して3つのコンパートメントの周囲に沿ってチャンバーの底部(ワイヤに最も近い側)に高真空グリースを堆積させ、グリースに対して清潔な(22 mm x 40 mm)カバースリップを押し付けて、完璧なシーリングを確保します。チャンバーの両端(すなわち、ワイヤの出入り部位)をシーリングペースト(ワックスプレート)を使用してシールする。同様に、チャンバーの反対側に真空グリースを塗布します。
- ピペットを用いて、区画を成長緩衝液(チャンバー当たり〜1mL)で満たした。脂質膜がワイヤーから脱落するのを防ぐために、溶液をあまりにも速くまたは強く攪拌しないでください。チャンバーの上部を 22 mm x 40 mm のカバースリップを使用して、グリースに押し付けて気密にシールします。気泡が形成されないようにするには、ガラスカバースリップを中央から端まで静かに押します。
- チャンバーを4°Cの冷蔵庫に置き、ワイヤを波動関数発生器(500Hzの正弦関数)に接続します。Beberらの研究ですでに提示され最適化されているように、より短い成長期間(すなわち、6時間)では350mV、より長い成長期間(すなわち、12〜16時間)では250mVの実効電圧を設定する。
- カバースリップを取り外し、シーラントとグリースを拭き取り、ワイヤーを取り外します。水とエタノール(≥70%)を交互に使用して、チャンバーを紙ティッシュで洗ってこすります。
注:GUV成長に最適な電圧および時間スケールは、バッファ塩濃度からチャンバ形状(ワイヤ間の距離とチャンバサイズ)まで、多くのパラメータに依存します。再現性を確保するために、実験を繰り返すたびに同じチャンバーを使用してください。ワイヤはチャンバの底部に近接しているため、蛍光顕微鏡を使用して脂質を画像化することができます。各ステップの脂質を画像化して( 図1C、Dを参照)、電気形成プロセスが成功したことを確認します。
- 小胞とのセプチンインキュベーション
- ワイヤーの近くに持ち込まれた事前にカットされたピペットチップ(約1mmの開口部)を使用して、ワイヤーからGUVを収集します。次いで、溶液をワイヤーに沿って全面的にピペット化する。この手順により、GUV を混乱させる可能性のある強いラメラ流の生成が防止されます。このステップの後、ピペットチップを切断する必要はもうありません。実際、ラメラ流は溶液中のGUVを損傷しません。
注:脂質混合物中にPI(4,5)P2 が存在するため、収集されたGUVは実験前に2〜3時間以内に保存する必要があります。実際、PI(4,5)P2 は急速に可溶化され、セプチンは形成後数時間後に膜に結合しなくなります。しかし、セプチンが膜に結合すると、数日間結合したままになります。 - セプチン原液をトリス10mM(pH8)のみで希釈して、成長緩衝液の浸透圧に等しい浸透圧に達する。必要に応じて、セプチン溶液を観察バッファーでさらに希釈する。収集したGUV(200 μLの総容量に対して50-100 μL)の意図した容量を追加します。β-カゼインによる不動態化後に観察チャンバー内で直接インキュベーションを行う(下記参照)。平衡に達するには20〜30分の待ち時間が必要です。
注:セプチン八量体複合体(ヒトまたは出芽酵母)の発現および精製は、他の第17条に広く記載されている。簡単に説明すると、セプチンを大腸菌で発現させ、親和性、サイズ排除、およびイオン交換クロマトグラフィーのステップを使用してラボで精製し、〜1mg·mL-1(3μM)濃度で50mM Tris-HCl(pH8)、300mM KCl、および5mM MgCl2の水溶液中で-80°Cで保存した。セプチン凝集を避けるために、高い塩濃度が使用されます。セプチン複合体は、凝集を誘導し、したがってタンパク質収量を低下させるフィルター遠心分離装置を通して濃縮すべきではない。
- ワイヤーの近くに持ち込まれた事前にカットされたピペットチップ(約1mmの開口部)を使用して、ワイヤーからGUVを収集します。次いで、溶液をワイヤーに沿って全面的にピペット化する。この手順により、GUV を混乱させる可能性のある強いラメラ流の生成が防止されます。このステップの後、ピペットチップを切断する必要はもうありません。実際、ラメラ流は溶液中のGUVを損傷しません。
- 共焦点および/または回転ディスク顕微鏡によるイメージング
- GUVが表面に付着したり爆発したりするのを防ぐために、観察チャンバを5mg・mL-1 βカゼイン溶液で30分間インキュベートして不動態化します。
- βカゼイン溶液を取り出し、ピペットを用いてセプチン-GUV溶液を観察チャンバーに移す(ステップ1.4.2)。GUVの沈殿物をチャンバーの底に10〜15分間入れます。
メモ: GUV の内部と外部バッファーの間にコンポジションの不一致があると、密度と屈折率の両方の不整合が生じます。屈折率の不一致のために、GUVは透過光光学顕微鏡で見ることができます。 - 共焦点顕微鏡を用いて、脂質の蛍光シグナルを可視化し、GUVsの品質および膜ラメラ性状態をチェックする。適切な較正を実施した後にセプチンの蛍光シグナルを記録することによって、GUVに結合したセプチンの密度を評価する25。0.4 μmの空間間隔でZスタック集録を実行して、セプチンと膜の間の相互作用によって誘発される小胞の3D変形を解析および視覚化します。
注:倍率60倍または100倍のオイル浸漬対物レンズを使用しました。ピクセルサイズがそれぞれ250nmおよび110nmの標準的な共焦点または回転ディスク顕微鏡(材料表)を使用した。イメージング条件を特定の機器に適合させる必要があります。特定の抗光漂白剤を溶液に添加しなかった。

図1:GUVの 電鋳 (A) 白金線を用いた電鋳プロセスの模式図。(B)電鋳によるGUVの生成に使用される白金線で組み立てられたテフロン(TEFLON)自家製デバイスの写真。ワイヤの直径は0.5mm、間隔は3mmです。(C)成長過程の間に透過光学顕微鏡によって観察されるGUV(球状物体)。画像の下部にある不透明なゾーンはプラチナ線です。(d)白金線上での成長中に蛍光顕微鏡で観察されるGUV(丸い蛍光物体)。スケールバー = 100 μm。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
2. クライオ電子顕微鏡によるセプチンフィラメントの超微細組織解析
注:小胞は、標準的な電子顕微鏡法によるイメージングには適していません。実際、サンプルは標準的なネガティブ染色法を用いて乾燥される。脱水すると、小胞は非特異的な変形を受ける可能性が高く、しばしば脂質突起を生じる。したがって、クライオ電子顕微鏡は、小胞の特定の変形を観察するためのはるかに優れた戦略です。クライオEMを使用して、サンプルはガラス化氷の薄い(〜100〜200nm)層内に埋め込まれ、サンプルをネイティブ状態に近い状態に保ちます。しかし、GUVは大きすぎる(数十マイクロメートル)ので、薄い氷の中に埋め込むことができず、透過型電子顕微鏡で画像化することができます。したがって、直径が〜50〜500nmの範囲の大きな単層小胞(LUV)が生成され、セプチンが小胞をどのように変形させるか、および小胞上でどのように配置されるかを決定する。
- 大きな単層小胞(LUV)の生成
- 50 μgの脂質ミックスをクロロホルムに可溶化し、57%EggPC、15%コレステロール、10%DOPE、10%DOPS、および8%脳PI(4,5)P2)のモル組成で調製し、ガラスバイアル中の膜とのセプチン相互作用を増強するように最適化した。
- アルゴン流下で溶液を乾燥させ、バイアル中に乾燥脂質膜を生成させた。バイアルを真空下に30分間入れて脂質を完全に乾燥させる。
- 脂質膜を50 μLの水溶液(50 mM Tris-HCl [pH 8]; 50 mM KCl; 2 mM MgCl2)に再可溶化し、終濃度1 mg・mL-1、ボルテックスを10秒間、溶液をチューブに移した。
注:LUVは、セプチンとのインキュベーションのために一度に使用する必要があります。さもなければ、PI(4,5)P2可溶化のためにタンパク質 - 脂質相互作用は弱くなる。この粗再可溶化プロセスは、直径50nm〜500nmの範囲の不均一な小胞集団を生成する。したがって、直径の全範囲、したがって曲率が同時にアッセイされます。
- 高塩緩衝液(50 mM Tris-HCl [pH 8]、300 mM KCl、2 mM MgCl 2)中で、それぞれ最終脂質およびセチン濃度0.1 mg·mL-1 (約300 nM)および20 nMの可溶化脂質と共にセプチンをインキュベートします。サンプルを室温で1時間インキュベートする。
- サンプルをガラス化するためのプランジ凍結
- グロー放電は、プラズマ発生装置を使用してカーボン側のカーボングリッド(300メッシュ)を5mAで30秒間ホーリーし、湿度の高い環境でプランジ凍結機内にグリッドを挿入します。
- 4 μLのサンプルをグリッドのグロー排出カーボン側に吸着します(ステップ2.2)。サンプル吸着の直前に、サンプルを使用してクライオトモグラフィーによって傾斜したシリーズを生成する場合に備えて、5-10 nmの金ビーズを溶液に加えます。
注:金ビーズの密度は、経験的にスクリーニングおよび調整する必要があり、プロバイダーによって異なります。最適化された密度は、視野内で10-15個の金ビーズです。 - 裸側からサンプルをブロットドライし、反対側のサンプルドロップを吸引します。
注:ブロッティング時間は通常4秒で、ろ紙の代替位置をテストおよびスクリーニングして、氷質(厚さ)と材料密度を最適化することにより、ろ紙の位置と吸い取り力が経験的に調整されます。 - グリッドを顕微鏡に移すか、液体窒素容器に保管します。
注:穴の開いたカーボングリッドを使用することは、小胞のサイズの多分散性に対応するために不可欠です。反対側から試料を吸い取ると、グリッド上の生物学的物質の吸着性が高まる。
- クライオ電子顕微鏡イメージング
- グリッドをクライオEM観察用に装備された電子顕微鏡(EM)に挿入します。低倍率(通常は120倍倍)でサンプル全体のマップを生成してグリッド全体をスクリーニングし、より良い氷(すなわち、薄くてガラス化されている)を表示する領域を選択します。
- クライオEM 2Dデータ収集の場合は、1ピクセルあたり約2Åのピクセルサイズで画像を収集し、サンプルの品質を確認します。セプチンフィラメントと脂質二重層の両方が見えることを確認します。
- クライオ電子断層撮影データ収集では、変形した小胞を表示する対象領域を選択します。十分な数の金ビーズ(少なくとも10個)が視野に存在することを確認します。
- 傾斜系列データ収集に使用されるソフトウェアに従って、関心領域から十分に離れるようにフォーカスとトラッキング位置を選択します。傾斜角度を-60°から+60°に変化させ、2°-3°度ごとに画像を収集して、傾斜したシリーズを収集します。
注:総線量は、約100電子/Å2以下でなければなりません。ピクセルサイズは、データ収集に使用される顕微鏡に応じて、1.3 Åから2.1 Åまで変化します。理想的には、データ収集のための角度対称スキームは、0°、-3°、+3°、-6°、+6°、-9°、+9°[...] -60°、+60°として好ましい。しかし、サイドエントリー顕微鏡のゴニオメーターは、これらの対称スキームを達成するのに十分な機械的安定性を提供しません。あるいは、集録を0°~34°で開始し、その後に-2°~-60°の2番目の角度シーケンス、36°~60°の最終シーケンスで終了することもできます。目的は、最も低い角度で最初の画像(放射線損傷が最も低いもの)を収集することです。さらに、小胞および結合したセプチンフィラメントの超微細構造を可視化するために、標準的なクライオEM顕微鏡(200kV、六ホウ化ランタン(LaB6)フィラメント、およびサンプルサイドエントリー)を使用することができる。しかし、さらなる画像処理(例えば、サブトモグラム平均化)を追求する場合は、直下検出器を搭載した前世代のフィールドエミッションガン(FEG)顕微鏡を使用するのが最善です。このプロトコルでは、3D再構成の取得に説明を限定し、サブトモグラム平均化は省略します。
- クライオトモグラフィとセグメンテーションからの3D再構成
- IMODソフトウェアスイート(材料表)を使用して、傾斜したシリーズ画像のアライメントと3D再構成26,27を行います。IMOD内では、基準(金ビーズ)の位置決めに基づいてチルトシリーズアライメントを実行します。さらに、必要に応じて、IMOD26内でコントラスト伝達関数(CTF)決定および補正を行う。最後に、IMODで3D再構成を達成し、各ステップを厳密に実行します。
- IMODソフトウェアスイートの3Dmod27 を使用して脂質二重層とセプチンフィラメントを手動でセグメント化して表示します。
SEMを用いたセプチンの曲率感度の解析
注:セプチンがマイクロメートルの曲率にどのように敏感であるかを理解するために、 in vitro アプローチを使用して、マイクロメートルスケールの波状の波状の起伏パターン上に堆積した固体支持脂質二重層を有するセプチンフィラメント複合体をインキュベートしている。
- 波状ポリジメチルシロキサン(PDMS)パターンからの波状NOA(ノーランド光学接着剤)レプリカの設計
- 250 nmの振幅と2 μmの横方向周期性またはその他の寸法のPDMSうねりパターンを使用して、目的のタンパク質に適した曲率をアッセイします。
注:PDMSの起伏パターンは、Nania et al.28,29に記載されているように設計および生成されます。 - クリーンルーム環境では、直径1cmの円形のガラスカバースリップに液体NOA5μLを堆積させ、PDMSテンプレートを滴の上に置きます。UV光(320nm)で5分間処理し、液体NOAを光重合して薄いポリマーフィルムにします。次に、重合したばかりのNOAとともにPDMSテンプレートをカバースリップから静かに剥がす。
注:NOAは、光学顕微鏡イメージングに適した一般的な光学的に透明な接着剤です。さらに、NOAはSEMイメージングの前に行われる化学固定および染色プロセスに耐性があります。NOA 71 および NOA 81 樹脂はどちらも同様の結果で使用できます。最初のPDMSパターンは、NOAレプリカを生成するために数回使用することができます。得られたNOAレプリカは、室温の箱に数ヶ月間保管することができる。
- 250 nmの振幅と2 μmの横方向周期性またはその他の寸法のPDMSうねりパターンを使用して、目的のタンパク質に適した曲率をアッセイします。
- 支持脂質二重層の生成とタンパク質インキュベーション
- エアプラズマクリーナーでNOAフィルムを5分間処理し、表面を親水性にします。
- 総脂質濃度1mg・mL-1で、57%EggPC、15%コレステロール、10%DOPE、10%DOPS、および8%脳PI(4,5)P2のモル組成を有する小さな単層小胞(SUV)の溶液を調製する。ステップ2.1.3で説明したように、乾燥した脂質膜を観察バッファーに再懸濁することによってSUVを調製する。溶液が透明になるまで、浴用超音波処理機を使用して溶液を5〜10分間穏やかに超音波処理する。
注:SUVの溶液は、-20°Cで数週間凍結保存することができます。 - NOAパターンを支持するカバースリップを細胞培養箱のウェル内に挿入します。1mg・mL-1 SUV溶液100 μLを、新たにグロー排出されたNOAパターン上に堆積させ(ステップ3.2.1)、室温で30分間インキュベートする。このステップは、SUVとNOAパターンの表面との融合を誘導し、支持された脂質二重層を生成する。
- スライドをセプチン緩衝液(50 mM Tris-HCl [pH 8]、50 mM KCl、2 mM MgCl2)で6倍十分にすすぎ、未融着SUVを除去します。各すすぎの後、サンプルを完全に乾燥させないでください。
- セプチン緩衝液(50 mM Tris-HCl[pH 8]、50 mM KCl、2 mM MgCl 2)を用いて、8量体セプチンストック溶液をセプチン高塩緩衝液(50 mM Tris-HCl[pH 8]、300 mM KCl、2 mMMgCl2)で10 nM ~ 100 nMの範囲の最終濃度、および1 mLの容量まで希釈する。スライド上のタンパク質溶液を室温で〜1時間インキュベートする。
メモ: 体積は十分に大きいため、蒸発は問題になりません。
- SEM分析のためのサンプル調製
注:電子顕微鏡では、タンパク質の構造とその組み立てを分析するために、さまざまなプロトコルが開発されています。現在のプロトコルは、Svitkinaらから派生した固定プロトコルを使用して、セプチンフィラメントの組織を保存する。30 実装は簡単です。また、このプロトコルは高解像度SEM観察を最適化します。- 試薬および原液の調製
注:このプロトコルには、事前にまたはインキュベーションの直前に調製できるいくつかの試薬および溶液が必要です。付属の指示に従って、アーティファクトや化学反応性の欠如を避けてください。- カコジル酸ナトリウム0.2 M原液:この倍強度溶液を調製するために、磁気攪拌下で2.14 gのカコジル酸ナトリウム粉末を~40 mLの蒸留水に溶解する。完全に溶解した後、0.1 M HCl(50 mLの溶液の場合は〜1 mL)を穏やかに添加してpHを7.4に調整し、蒸留水で最終容量を構成します。この溶液は、4°Cで24〜48時間保存することができます。
- 0.1 M カコジル酸ナトリウム (固定液) 中の 2% グルタルアルデヒド (GA): 高純度 EM グレード GA を 0.2 M カコジル酸ナトリウム溶液 (上記参照) で希釈して、使用直前にこの溶液を調製します。25%-50%の市販のGAストックソリューションは、4°Cでストレージ容量が最適化されているため、使用してください。 10 mLの固定液溶液を調製するために、0.8 mLの25%市販GA溶液を4.2 mLの蒸留水および5 mLの0.2 Mカコジル酸ナトリウムで希釈する。
- 0.1 M カコジル酸ナトリウム中の 1% 四酸化オスミウム (OsO 4): 市販の4% OsO4 原液を 0.2 M カコジル酸ナトリウム (上記参照) で希釈して、使用直前にこの第 2 固定液を調製します。4 mLのOsO4固定液の場合、1 mLの蒸留水および2 mLの0.2 Mカコジル酸ナトリウムで1 mLの市販の4%OsO4溶液を希釈する。
注:切断可能なガラス球には、その保管および取り扱い特性のために、市販の4%OsO4 ストック溶液を使用してください。OsO4 は反応性が高いことに注意してください。その色がわずかに黄色で、暗くないことを確認してください。 - 水中の1%タンニン酸(TA):使用直前にこの溶液を準備してください。このTA溶液を調製し、室温の蒸留水中で終濃度1%TAを得た。10mgを1mLの蒸留水に溶解し、数分間ボルテックスする。TA溶液は保存できず、使用前に0.2μmフィルターでろ過する必要があります。
- 水中の1%酢酸ウラニル(UA)溶液UA溶液を調製し、蒸留水中の最終濃度1%UAを得た。1mLの蒸留水に10mgを溶解し、室温でボルテックスまたは振とうすることにより、少なくとも30分〜1時間ボルテックスする。この溶液は4°Cで1ヶ月間保存することができますが、容易に沈殿する可能性があるため、使用前に0.2μmフィルターでろ過する必要があります。
- 段階的な一連のエタノール溶液 水中で50%、70%、95%、および100%エタノール溶液を調製する。100%エタノールの新しく開いたボトルから、またはモレキュラーシーブ(公称細孔径= 4Å)で少なくとも24時間脱水された100%エタノールから最終浴を準備し、乾燥およびコーティングを妨げる可能性のあるすべての微量の水をサンプルから除去する。この溶液を振ると、ケイ酸塩粒子の再懸濁を引き起こす可能性があるため、注意してください。
メモ:このプロトコルで使用される化学試薬の取り扱いには注意してください。グルタルアルデヒド、四酸化オスミウム、カコジル酸ナトリウム、酢酸ウラニルはすべて毒性が高く、酢酸ウラニルも放射性です。試薬とその廃棄物のすべての操作は、実験室固有の手順に従って、個々の(手袋、ラボコート、安全メガネ)および集団保護(ヒュームフードおよびプレキシガラスシールド)を使用して行う必要があります。
- サンプル固定
- サンプル(すなわち、融合支持脂質二重層およびインキュベートタンパク質を有するNOAスライド)をPBSで洗浄する。PBSを37°Cで予熱したGA固定液に交換し、15分間反応を進行させた。その後、サンプルを4°Cで保存することができます。
- 固定液を除去し、固定サンプルを0.1 Mカコジル酸ナトリウム(洗浄あたり5分)で3倍、穏やかな振とうで洗浄します。
- 光から最大限の保護を得て、OsO4 固定溶液中でサンプルを10分間インキュベートし、膜状構造の固定を可能にし、サンプルの電気伝導率を高めます。固定液を除去し、サンプルを蒸留水で3倍(洗浄あたり5分)穏やかに振とうしながら洗浄する。
- 洗浄したサンプルを濾過したTA固定液中で最大10分間インキュベートする。TA溶液を取り出し、サンプルを蒸留水で3倍(洗浄あたり5分)穏やかに振とうしながら洗浄する。
- 洗浄したサンプルを、新しくろ過したUA固定液中で10分間インキュベートし、光から最大限の保護を行います。UA溶液を取り出し、サンプルを蒸留水で3倍(洗浄あたり5分)穏やかに振とうしながら洗浄する。
- サンプル脱水と臨界点乾燥
メモ:液体状態から気体状態に変化する際に、不要な表面張力によるサンプルの損傷を避けるために、空気乾燥は許可されていません。EMでは、CO2の超臨界状態に達し、その臨界点(31°C、74バール)をバイパスする物理的方法、または表面張力を低下させた乾燥剤であるヘキサメチルジシラザン(HMDS)を蒸発させる化学的方法の2つの方法が確立されている。CO2およびHMDSは水との混和性が低い。したがって、乾燥プロセス中の後の損傷を避けるために、すべての微量の水を移行溶媒(エタノール)で置換する必要があります。- サンプルを各エタノール溶液中で2〜3分間インキュベートし、50%〜100%(無水)エタノール浴から開始する。
注:浴間のエタノールの蒸発は速いため、空気乾燥を避けるためにサンプルの取り扱いも速くなければなりません。 - スライドガラスをエタノールで予め充填された臨界点乾燥機内に移し、製造元の指示に従ってください。
メモ: このプロトコルでは、自動装置が使用されていましたが、どのシステムでも使用できます。プロトコルは装置ごとに異なる場合がありますが、エタノールを完全に除去し(このプロトコルでは25浴)、異なる溶媒交換(CO2 入口およびエタノール/CO2 出口)および最終減圧(このプロトコルではほぼ1時間)の速度を低下させるように最適化する必要があります。 - 臨界点乾燥が終わったら、取り付けとコーティングまでサンプルを直ちにデシケータに保管します。乾燥したサンプルは吸湿性が高いため、できるだけ早くコーティングしてください(下記参照)。
注:または、HMDSはより安価で高速な方法を提供することができます。HDMSは我々のサンプルでテストされたことがないが、このアプローチは細胞膜の内面におけるタンパク質の観察に良い結果をもたらした31,32。
- サンプルを各エタノール溶液中で2〜3分間インキュベートし、50%〜100%(無水)エタノール浴から開始する。
- サンプルの取り付けとコーティング。
注:生体試料は電気伝導特性が悪いため、SEM観察前に導電性金属膜でコーティングする必要があります。プラズママグネトロンスパッタリングが用いられる。- カバースリップは、将来のSEM観察に使用されるスタブに取り付けます。カーボンディスクと比較して電気伝導性が向上しているため、銀塗料を使用してください。カバースリップの上面に銀色の塗料を加え、スタブとの接続が満足のいくものであることを確認します。塗料の凝集物は避けてください。
メモ:銀色のペイントストリップは、高解像度(すなわち、低い作動距離)で作業するときに、SEMのサンプルと対物レンズとの接触を避けるために薄くなければなりません。 - 溶媒が完全に蒸発するのを待ちます。
メモ: このステップの期間は、銀塗料付着物の量と厚さによって異なる場合があります。このステップは、ベルジャーまたはコーターと一次真空を10〜30分間使用することで短縮できます。 - プラズママグネトロンスパッタリングヘッドと回転惑星ステージを備えた装置を使用し、製造元が提供する標準プロトコルに従ってください。ここでは、装置を2.5 x 10-5 mbarまで真空排気し、高品質のアルゴンで1xをパージし、8.0 x 10-3 mbarに調整しました。
- プレスパッタリング(120mAで60秒)を行い、表面の酸化物層を除去します。次いで、膜厚モニターの助けを借りて、1.5nmのタングステン(90mA、作動距離= 50mm)を堆積させる。
メモ:フィルム蒸発の再現性を確保するために、コーターを完全にクリーニングする必要があります。コーティングは、目標の厚さに達したら停止する必要があります。最終的な膜厚を計算し、後で補正する。約1.5nmのフィルムは、平均して、我々の装置を使用して0.7nmのポスト補正を有する。これらの補正値が目標に近いため、一連のコーティングを実行して評価し、この補正を目標値から減算します。 - サンプルを真空下に保管して、すべてのSEM分析まで、およびすべてのSEM分析を通して周囲空気から保護します。
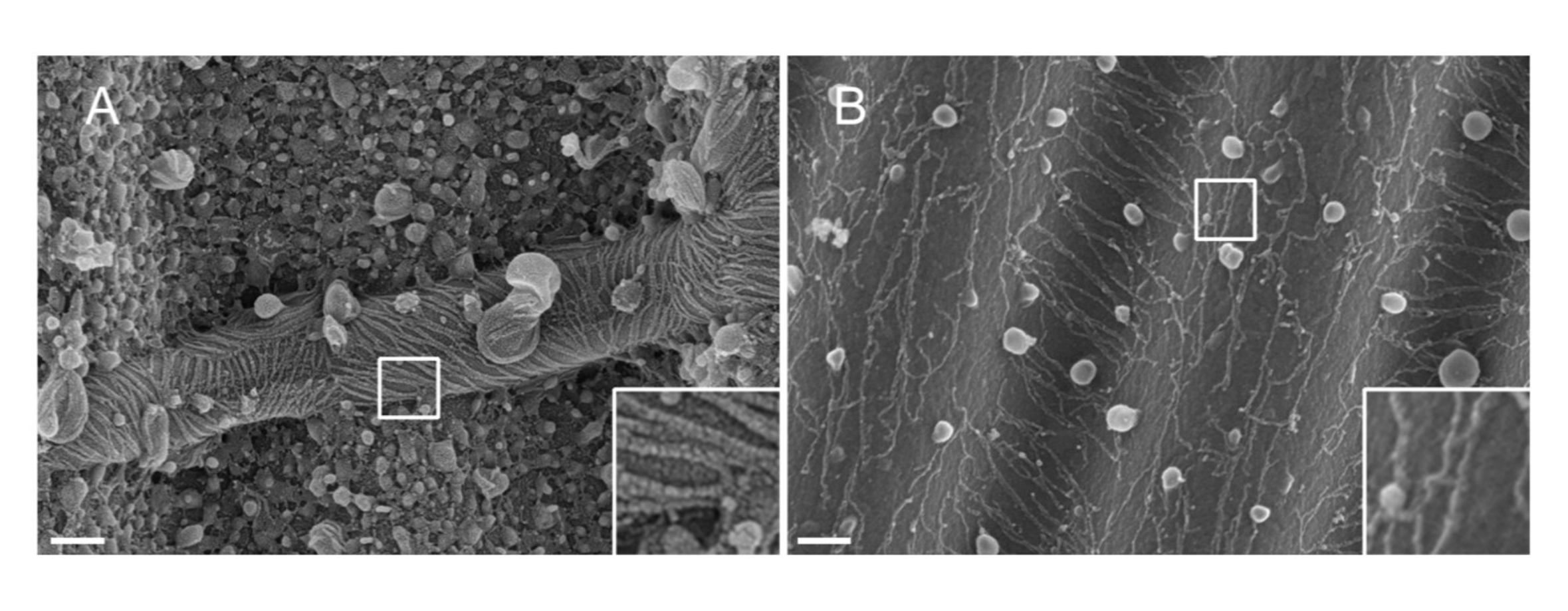
注:スパッタリングに使用される金属の性質。Ptは、一般的な材料であるにもかかわらず、セプチンフィラメント(1.5nm)に適合したPt膜厚を有する低品質のコーティングをもたらす。高解像度では、1.5 nm Pt膜は凝集性に欠けます。Ptクラスターとセプチンフィラメントのサイズはそれによって類似し、フィラメントセグメンテーションプロセス33 の間に誤った解釈につながる(図 2A、差し込み図参照)。タングステンは、より小さな結晶粒径を表示し、高解像度SEMではほとんど見えないため、Ptの優れた代替品です(図 2B、差し込み図を参照)。それにもかかわらず、純粋なタングステンは容易に酸化され、ステップ3.3.4で詳述された手順であればSEM観察中に強い帯電効果アーチファクトにつながる。厳密には守られていません。
- カバースリップは、将来のSEM観察に使用されるスタブに取り付けます。カーボンディスクと比較して電気伝導性が向上しているため、銀塗料を使用してください。カバースリップの上面に銀色の塗料を加え、スタブとの接続が満足のいくものであることを確認します。塗料の凝集物は避けてください。
- 試薬および原液の調製
- 電界放出型SEM(FESEM)顕微鏡を用いて画像を取得します。
注:SEM技術は最近、解像度を向上させるためにアップグレードされており、技術はメーカー(電子光学 、ビーム減速、磁気レンズなど)によって異なります。この分解能の向上(複数のメーカーがアクセス可能)、特に低加速電圧(1kVでナノメートル付近)では、セプチンネットワークに似たナノメートル構造を解くために必要です。- 以下の設定で「レンズ内」検出器で一次二次電子(SE1)を検出し、高解像度イメージングを実現します。
- 加速電圧を3kVに設定します。帯電効果の抑制が必要な場合は、20 μm アパーチャ (ツァイス ジェミニ I カラムの場合、または 34 pA に相当) または 15 μm アパーチャ (ツァイス ジェミニ I カラムの場合、または 18.5 pA に相当) でビーム電流を固定します。
- 観測には21.25 nm/ピクセルから1.224 nm/ピクセルの範囲の解像度を使用し、データ分析には〜5.58 nm/ピクセル(ポラロイド545リファレンスによる倍率20,000倍)の解像度を使用します。
- 作動距離は、高解像度の観察の場合は 1 mm ~ 2 mm、被写界深度を大きくする必要がある場合は約 3 mm に設定します。スキャン速度とライン積分を継続的に調整して、画像あたり約30~45秒の集録時間で一定の信号対雑音比を確保します。

図2:波状PDMSパターンに対するセプチンフィラメントに堆積した材料の影響。 スパッタリングによって被覆されたセプチンフィラメントのSEMは、(A)1.5nmの白金、白金核のクラスター間の凝集性の欠如の典型的な「乾燥ひび割れ土壌」パターン、または(B)滑らかで凝集性層で覆われた1.5nmのタングステンのいずれかを示す。スケール バー = 200 nm。白い四角いボックスは、右下の拡大ビューを表します。球状小球は、セプチンと相互作用する小さな脂質小胞である。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
結果
ギューブの変形
セプチンと共にインキュベートされた後に再形成されたGUVの典型的な共焦点蛍光画像は、セプチンが重合する条件下で、 図3に示されている。裸のGUV(図3A)は完全に球形であった。50nM以上の出芽酵母セプチンフィラメントとのインキュベーションにより、小胞は変形して現れた。100nMの出芽酵母セプチン八量体の濃度?...
ディスカッション
上記のように、脂質二重層内のPI(4,5)P2 の取り込みを増強し、したがってセプチン - 膜相互作用を促進する脂質混合物が使用されている。実際、我々は、出芽酵母セプチンがPI(4,5 )P 2特異的な様式で小胞と相互作用することを他の25で示した。この脂質組成物は、複数の組成物をスクリーニングすることから経験的に調整され、現在、著者らによって広く使用?...
開示事項
著者には利益相反はありません。
謝辞
Patricia Bassereau と Daniel Lévy の有益なアドバイスと議論に感謝します。この作業は、プロジェクト「SEPTIME」、ANR-13-JSV8-0002-01、ANR SEPTIMORF ANR-17-CE13-0014、およびプロジェクト「SEPTSCORT」(ANR-20-CE11-0014-01)に資金を提供するためのANR(Agence Nationale de la Recherche)の支援の恩恵を受けました。B. ショーヴァンは、エコール・ドクターラーレ「ED564: Physique en Ile de France」とFondation pour lea Recherche Médicaleから資金提供を受けています。中澤圭吾はソルボンヌ大学(AAP Emergence)の支援を受けた。G.H. Koenderinkは、Nederlandse Organisatie voor Wetenschappelijk Onderzoek(NWO/OCW)の「BaSyC-Building a Synthetic Cell」を通じて支援を受けた。重力グラント(024.003.019).Labex Cell(n)Scale (ANR-11-LABX0038) と Paris Sciences et Lettres (ANR-10-IDEX-0001-02) に感謝します。我々は、フランス国立研究インフラフランスバイオイメージング(ANR10-INBS-04)のメンバーである細胞組織イメージング(PICT-IBiSA)、Institut Curieに感謝します。
資料
| Name | Company | Catalog Number | Comments |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | 850725 | |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine | Avanti Polar Lipids | 840035 | |
| Bath sonicator | Elma | Elmasonic S10H | |
| Bodipy-TR-Ceramide | invitrogen, Thermo Fischer scientific | 11504726 | |
| Chemicals: NaCl, Tris-HCl, sucrose, KCl, MgCl2, B-casein, chloroform, sodium cacodylate, tannic acid, ethanol | Sigma Aldrich | ||
| Confocal microscope | nikon | spinning disk or confocal | |
| Critical point dryer | Leica microsystems | CPD300 | |
| Deionized water generator | MilliQ | F1CA38083B | MilliQ integral 3 |
| Egg L-α-phosphatidylcholine | Avanti Polar Lipids | 840051 | |
| Field Emission Gun SEM (FESEM) | Carl Zeiss | Gemini SEM500 | |
| Glutaraldehyde 25 %, aqueous solution | Thermo Fischer scientific | 50-262-19 | |
| High vacuum grease, Dow corning | VWR | ||
| IMOD software | https://bio3d.colorado.edu/imod/ | software suite for tilted series image alignment and 3D reconstruction | |
| Lacey Formvar/carbon electron microscopy grids | Eloise | 01883-F | |
| Lipids | Avanti Polar Lipids | ||
| L-α-phosphatidylinositol-4,5-bisphosphate | Avanti Polar Lipids | 840046 | |
| Metal evaporator | Leica microsystems | EM ACE600 | |
| NOA (Norland Optical Adhesives), NOA 71 and NOA 81 | Norland Products | NOA71, NOA81 | |
| Osmium tetraoxyde 4% | delta microscopies | 19170 | |
| Osmometer | Löser | 15 M | |
| Plasma cleaner | Alcatel | pascal 2005 SD | |
| Plasma generator | Electron Microscopy Science | ||
| Plunge freezing equipment | leica microsystems | EMGP | |
| Transmission electron microscope | Thermofischer | Tecnai G2 200 kV, LaB6 | |
| Uranyl acetate | Electron Microscopy Science | 22451 | this product is not available for purchase any longer |
| Wax plates, Vitrex | VWR |
参考文献
- Finger, F. P. Reining in cytokinesis with a septin corral. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 27 (1), 5-8 (2005).
- Barral, Y., Kinoshita, M. Structural insights shed light onto septin assemblies and function. Current Opinion in Cell Biology. 20 (1), 12-18 (2008).
- Hu, Q., et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329 (5990), 436-439 (2010).
- Lin, Y. -. H., Kuo, Y. -. C., Chiang, H. -. S., Kuo, P. -. L. The role of the septin family in spermiogenesis. Spermatogenesis. 1 (4), 298-302 (2011).
- Addi, C., Bai, J., Echard, A. Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Current Opinion in Cell Biology. 50, 27-34 (2018).
- Spiliotis, E. T., Kesisova, I. A. Spatial regulation of microtubule-dependent transport by septin GTPases. Trends in Cell Biology. 31 (12), 979-993 (2021).
- Spiliotis, E. T., Nakos, K. Cellular functions of actin- and microtubule-associated septins. Current Biology: CB. 31 (10), 651-666 (2021).
- Salameh, J., Cantaloube, I., Benoit, B., Poüs, C., Baillet, A. Cdc42 and its BORG2 and BORG3 effectors control the subcellular localization of septins between actin stress fibers and microtubules. Current Biology: CB. 31 (18), 4088-4103 (2021).
- Ewers, H., Tada, T., Petersen, J. D., Racz, B., Sheng, M., Choquet, D. A septin-dependent diffusion barrier at dendritic spine necks. PloS One. 9 (12), 113916 (2014).
- Myles, D. G., Primakoff, P., Koppel, D. E. A localized surface protein of guinea pig sperm exhibits free diffusion in its domain. The Journal of Cell Biology. 98 (5), 1905-1909 (1984).
- Luedeke, C., Frei, S. B., Sbalzarini, I., Schwarz, H., Spang, A., Barral, Y. Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. The Journal of Cell Biology. 169 (6), 897-908 (2005).
- Gilden, J. K., Peck, S., Chen, Y. -. C. M., Krummel, M. F. The septin cytoskeleton facilitates membrane retraction during motility and blebbing. The Journal of Cell Biology. 196 (1), 103-114 (2012).
- Dolat, L., Hu, Q., Spiliotis, E. T. Septin functions in organ system physiology and pathology. Biological Chemistry. 395 (2), 123-141 (2014).
- Angelis, D., Spiliotis, E. T. Septin mutations in human cancers. Frontiers in Cell and Developmental Biology. 4, 122 (2016).
- Takehashi, M., et al. Septin 3 gene polymorphism in Alzheimer's disease. Gene Expression. 11 (5-6), 263-270 (2004).
- Shuman, B., Momany, M. Septins from protists to people. Frontiers in Cell and Developmental Biology. 9, 824850 (2022).
- Bertin, A., et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105 (24), 8274-8279 (2008).
- Iv, F., et al. Insights into animal septins using recombinant human septin octamers with distinct SEPT9 isoforms. Journal of cell science. 134 (15), (2021).
- Beber, A., et al. Membrane reshaping by micrometric curvature sensitive septin filaments. Nature communications. 10 (1), 420 (2019).
- Bridges, A. A., Jentzsch, M. S., Oakes, P. W., Occhipinti, P., Gladfelter, A. S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. The Journal of Cell Biology. 213 (1), 23-32 (2016).
- Patzig, J., et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 5, 17119 (2016).
- Szuba, A., et al. Membrane binding controls ordered self-assembly of animal septins. eLife. 10, 63349 (2021).
- Tanaka-Takiguchi, Y., Kinoshita, M., Takiguchi, K. Septin-mediated uniform bracing of phospholipid membranes. Current Biology: CB. 19 (2), 140-145 (2009).
- Bertin, A., et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of Molecular Biology. 404 (4), 711-731 (2010).
- Beber, A., et al. Septin-based readout of PI(4,5)P2 incorporation into membranes of giant unilamellar vesicles. Cytoskeleton. 76 (4,5), 92-103 (2019).
- Mastronarde, D. N., Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology. 197 (2), 102-113 (2017).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Nania, M., Foglia, F., Matar, O. K., Cabral, J. T. Sub-100 nm wrinkling of polydimethylsiloxane by double frontal oxidation. Nanoscale. 9 (5), 2030-2037 (2017).
- Nania, M., Matar, O. K., Cabral, J. T. Frontal vitrification of PDMS using air plasma and consequences for surface wrinkling. Soft Matter. 11 (15), 3067-3075 (2015).
- Svitkina, T. M., Borisy, G. G. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods in Enzymology. 298, 570-592 (1998).
- Franck, A., et al. Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Molecular Biology of the Cell. 30 (5), 579-590 (2019).
- Elkhatib, N., et al. Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science. 356 (6343), (2017).
- Stokroos, I., Kalicharan, D., Van Der Want, J. J., Jongebloed, W. L. A comparative study of thin coatings of Au/Pd, Pt and Cr produced by magnetron sputtering for FE-SEM. Journal of Microscopy. 189, 79-89 (1998).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved