Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Методы «снизу вверх in vitro » для анализа ультраструктурной организации, изменения формы мембраны и чувствительности к кривизне септинов
* Эти авторы внесли равный вклад
В этой статье
Резюме
Септины являются цитоскелетными белками. Они взаимодействуют с липидными мембранами и могут ощущать, но также генерировать кривизну мембраны в микронном масштабе. В этом протоколе мы описываем методологии снизу вверх in vitro для анализа деформаций мембран, чувствительного к кривизне связывания септина и ультраструктуры септиновой нити.
Аннотация
Ремоделирование мембраны происходит постоянно на плазматической мембране и внутри клеточных органелл. Чтобы полностью проанализировать роль окружающей среды (ионные условия, белковые и липидные композиции, кривизна мембраны) и различных партнеров, связанных со специфическими процессами изменения формы мембраны, мы предпринимаем подходы in vitro снизу вверх. В последние годы наблюдается большой интерес к выявлению роли белков септина, связанных с основными заболеваниями. Септины являются незаменимыми и вездесущими цитоскелетными белками, которые взаимодействуют с плазматической мембраной. Они участвуют в делении клеток, подвижности клеток, нейроморфогенезе и спермиогенезе, среди других функций. Поэтому важно понять, как септины взаимодействуют и организуются на мембранах, чтобы впоследствии индуцировать деформации мембран и как они могут быть чувствительны к определенным кривизнам мембраны. Целью данной статьи является расшифровка взаимодействия между ультраструктурой септинов на молекулярном уровне и ремоделированием мембран, происходящим в микронном масштабе. С этой целью почковые дрожжи и комплексы септина млекопитающих были рекомбинантно экспрессированы и очищены. Комбинация анализов in vitro затем использовалась для анализа самосборки септинов на мембране. Поддерживаемые липидные бислои (SLB), гигантские одноламеллярные везикулы (GUV), большие одноцветные везикулы (LUV) и волнистые субстраты использовались для изучения взаимодействия между самосборкой септина, изменением формы мембраны и кривизной мембраны.
Введение
Септины представляют собой цитоскелетные нитеобразующие белки, которые взаимодействуют с липидными мембранами. Септины повсеместно распространены у эукариот и необходимы для многочисленных клеточных функций. Они были идентифицированы как основные регуляторы деления клеток у почковых дрожжей и млекопитающих 1,2. Они участвуют в событиях изменения формы мембраны, цилиогенезе3 и спермиогенезе4. В клетках млекопитающих септины могут также взаимодействовать с актинами и микротрубочками 5,6,7 в связующем звене Rho GTPases (BORG)-зависимым способом 8. В различных тканях (нейроны9, реснички3, сперматозоиды10) септины были идентифицированы как регуляторы диффузионных барьеров для мембранно-связанных компонентов11. Было также показано, что септины регулируют образование мембран и протрузии12. Септины, являясь многозадачными белками, причастны к возникновению различных распространенных заболеваний13. Их неправильная регуляция связана с возникновением раковых заболеваний14 и нейродегенеративных заболеваний15.
В зависимости от организма несколько субъединиц септина (две у Caenorhabditis elegans до 13 у людей) собираются в комплексы, организация которых изменяется тканезависимым образом16. Основной строительный блок септина собирает от двух до четырех субъединиц, присутствующих в двух копиях и самостоятельно собранных палиндромным способом. В почковых дрожжах септины октамерны17,18. In situ септины часто локализуются на участках с кривизной микрометра; они обнаруживаются в местах сужения деления, у основания ресничек и дендритов, а также в кольце сперматозоидов19,20. На мембране роль септинов, по-видимому, двойственна: они участвуют в изменении формы липидного бислоя и в поддержании целостности мембраны21. Следовательно, исследование биофизических свойств септиновых нитеобразующих белков и/или субъединиц на мембране имеет решающее значение для понимания их роли. Для анализа специфических свойств септинов в хорошо контролируемой среде уместны подходы in vitro снизу вверх. До сих пор только несколько групп описали биофизические свойства септинов in vitro 20,22,23. Следовательно, по сравнению с другими цитоскелетными нитями, текущие знания о поведении септинов in vitro остаются ограниченными.
Этот протокол описывает, как можно проанализировать организацию нитей септина, изменение формы мембраны и чувствительность к кривизне19. С этой целью была использована комбинация методов оптической и электронной микроскопии (флуоресцентная микроскопия, криоэлектронная микроскопия [крио-ЭМ] и сканирующая электронная микроскопия [SEM]). Изменение формы мембраны гигантских одноламельных везикул (ГУВ) размером с микрометр визуализируется с помощью флуоресцентной оптической микроскопии. Анализ расположения и ультраструктуры септиновых нитей, связанных с липидными везикулами, проводится с использованием крио-ЭМ. Анализ чувствительности к кривизне септина проводится с использованием SEM, путем изучения поведения нитей септина, связанных с твердо поддерживаемыми липидными бислоями, нанесенными на волнистые субстраты переменных кривизн, что позволяет анализировать чувствительность кривизны как для положительных, так и для отрицательных кривизн. По сравнению с предыдущим анализом 20,24, здесь мы предлагаем использовать комбинацию методов для тщательного анализа того, как септины могут самособираться, синергетически деформировать мембрану и быть чувствительными к кривизне. Считается, что этот протокол полезен и адаптируется к любому нитевидному белку, который проявляет сродство к мембранам.
протокол
1. Определение изменения формы мембраны с использованием гигантских одноламеллярных везикул (ГУВ)
ПРИМЕЧАНИЕ: В этом разделе ГУВ генерируются для имитации деформаций мембраны, возможно, индуцированных септинами в клеточном контексте. Действительно, в клетках септины часто встречаются на участках с микрометрическими кривизнами. ГУВ имеют размеры от нескольких до десятков микрометров и могут деформироваться. Таким образом, они подходят для анализа любых деформаций, вызванных септином в микрометровом масштабе. Флуоресцентные липиды, а также флуоресцентно меченые септины (с использованием зеленого флуоресцентного белка [GFP]) используются для отслеживания поведения как липидов, так и белков с помощью флуоресцентной микроскопии.
- Приготовление буферов и растворов
- Подготовьте буфер роста ГУВ (50 мМ NaCl, 50 мМ сахарозы и 10 мМ Tris [pH = 7,8]) и буфер наблюдения (75 мМ NaCl и 10 мМ Tris [pH = 7,8]).
- Измерение (с помощью коммерческого осмометра) и регулировка осмолярности буферов наблюдения и роста (170 мОсмоль· L-1, в теории), добавляя небольшие количества NaCl до тех пор, пока их соответствующие осмолярности не будут равны. Фильтруйте буферы с помощью фильтра 0,2 мкм. Aliquot буфер роста и храните его при -20 °C для дальнейшего использования. Храните буфер наблюдения при температуре 4 °C.
ПРИМЕЧАНИЕ: Разница в осмолярности между обоими буферами не должна превышать 5%. - Готовят 5 мг·мл-1 β-казеиновый раствор в буфере наблюдения. Обеспечить полное растворение (через несколько часов при 4 °C при магнитном перемешивании). Отфильтруйте раствор фильтром 0,2 мкм, аликвотой и храните при -20 °C.
- Готовят липидные смеси при общей концентрации липидов 3 мг·мл-1 в хлороформе. Используйте композицию (моль%) из 56,8% Яичного L-α-фосфатидилхолина (EggPC), 15% холестерина, 10% 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин (DOPE), 10% 1,2-диолеоил-sn-glycero-3-фосфо-L-серин (DOPS), 8% L-α-фосфатидилинозитол-4,5-бисфосфата (PI(4,5)P2) и 0,2% Bodipy-TR-Ceramide для усиления белково-липидных взаимодействий и содействия включению PI(4,5)P225.
ПРИМЕЧАНИЕ: Обрабатывайте хлороформ под вытяжным кожухом с помощью нитриловых перчаток и защитных очков. Пипетки хлороформ растворы со стеклянными шприцами и избегайте пластика, так как хлороформ растворяет пластик. До и после использования промыть шприцы пипеткой хлороформ 5х-10х. Используйте отдельные шприцы для пипетки специфических флуоресцентных липидов, чтобы предотвратить перекрестное загрязнение. Липиды могут храниться в хлороформе при -20 °C во флаконе из янтарного стекла, покрытом тефлоном. Флаконы должны быть заполнены аргоном перед закрытием и запечатаны парапленкой, чтобы предотвратить окисление липидов. - Электроформирование ГУВ с помощью установки платиновых проводов
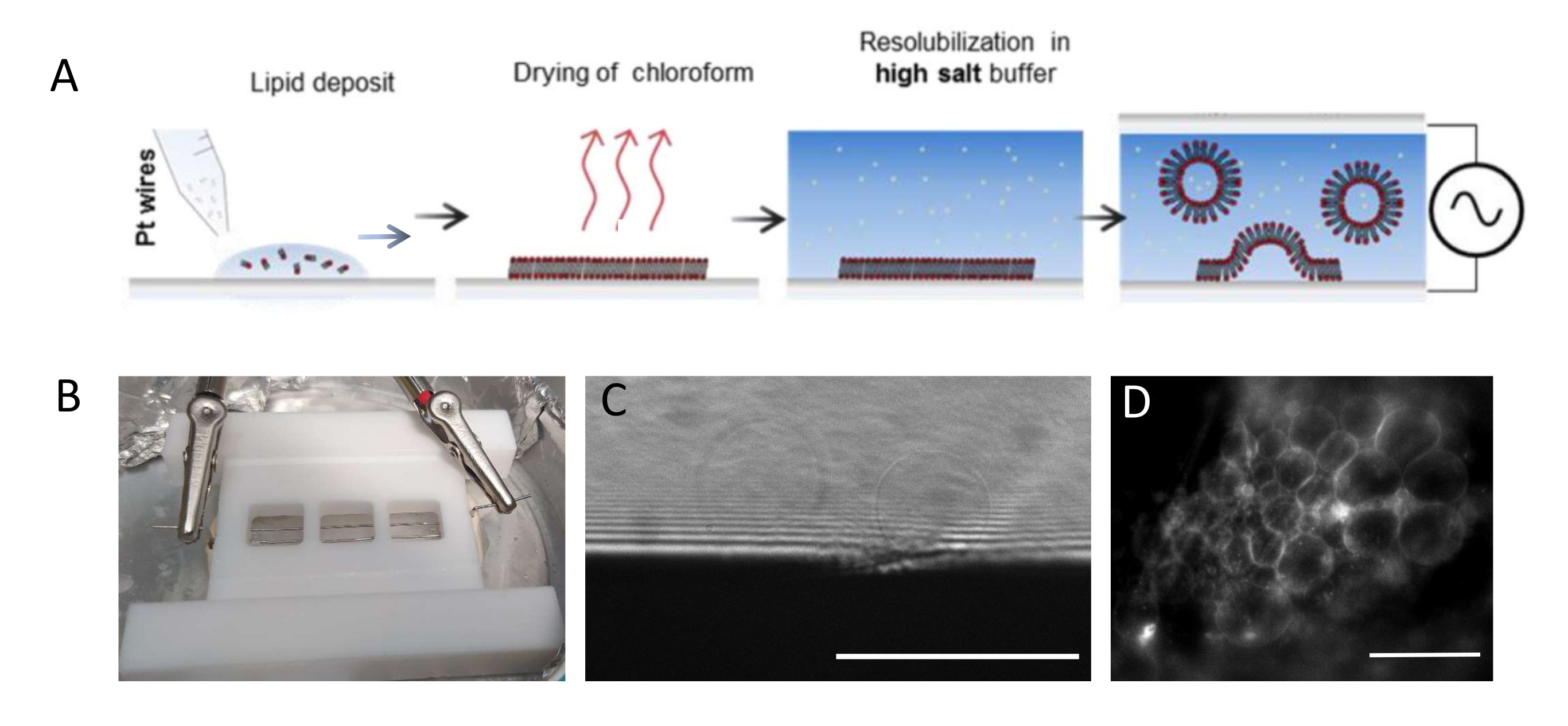
ПРИМЕЧАНИЕ: На рисунке 1 представлена схема экспериментальных этапов и изображение камеры.- Тщательно очистите камеру и платиновые провода следующим образом, чтобы удалить остатки липидов.
- Погрузите провода и камеру в ацетон и обработайте ультразвуком на 10 мин. Тщательно протрите их бумажной салфеткой с использованием ацетона.
- Соберите камеру, вставив провода, снова погрузите в ацетон и храните ультразвуком в течение 10 минут. Протрите еще раз ацетоном, убедившись, что провода полностью очищены. Окуните камеру в этанол, соникуйте на 10 мин и протрите этанолом.
- Наконец, окуните камеру в деионизированную воду, обработайте ультразвуком в течение 10 минут и высушите струей азота или воздуха.
ПРИМЕЧАНИЕ: Тефлоновая камера (рисунок 1B) была изготовлена на заказ в собственной мастерской. Он вмещает три отсека, которые могут быть герметизированы с обеих сторон с помощью стеклянных крышек. Платиновые провода могут быть вставлены в камеру через отверстия диаметром 1,3 мм. - После очистки камеры нанесите 3-4 капли на отсек (каждая капля составляет около 0,1 мкл)) липидной смеси 3 мг·мл-1 на каждую платиновую проволоку. Поверните провода на 180° и положите 3-4 капли липидов на отсек на противоположную сторону каждой платиновой проволоки. Следите за тем, чтобы капли не контактировали друг с другом. На целую камеру требуется около 5 мкл липидной смеси.
- Поместите камеру роста в вакуумную камеру на 30 минут, чтобы удалить следы хлороформа.
ПРИМЕЧАНИЕ: Глубокий вакуум (0,1 мбар) является лучшим. При сушке липиды уязвимы к окислению и, следовательно, не должны оставаться на воздухе более чем на несколько минут. - Нанесите высоковакуумную смазку на дно камеры (сторона, ближайшая к проводам) по периферии трех отсеков с помощью шприца и прижмите чистый (22 мм х 40 мм) закрывающий лист к смазке, чтобы обеспечить идеальное уплотнение. Запечатайте обе оконечности камеры (т.е. в местах входа/выхода проводов) с помощью герметизирующей пасты (восковых пластин). Аналогично нанесите вакуумную смазку на другую сторону камеры.
- Заполните отсеки буфером роста (~1 мл на камеру) с помощью пипетки. Не перемешивайте раствор слишком быстро или сильно, чтобы предотвратить отслоение липидной пленки от проводов. Герметично запечатайте верхнюю часть камеры с помощью крышки размером 22 мм x 40 мм, прижав ее к смазке. Чтобы избежать образования пузырьков воздуха, аккуратно прижмите стеклянную крышку от центра к краям.
- Поместите камеру в холодильник с температурой 4 °C и подключите провода к генератору волновой функции (синусоидальная функция при 500 Гц). Установите эффективное напряжение 350 мВ для более короткого периода роста (т.е. 6 ч) или 250 мВ для более длительного периода роста (т.е. 12-16 ч), как уже представлено и оптимизировано в исследовании Beber et al.25.
- Снимите обшивки, протрите герметик и смазку, а также удалите провода. Мойте и протрите камеру бумажной салфеткой, используя воду и этанол (≥70%) поочередно.
ПРИМЕЧАНИЕ: Оптимальное напряжение и временная шкала для роста ГУВ зависят от многих параметров, от концентрации буферной соли до геометрии камеры (т.е. расстояния между проводами и размера камеры). Используйте одну и ту же камеру каждый раз, когда эксперимент повторяется, чтобы обеспечить воспроизводимость. Провода находятся в непосредственной близости от нижней части камеры, так что липиды могут быть визуализированы с помощью флуоресцентной микроскопии. Изобразите липиды на каждом этапе (см. Рисунок 1C, D), чтобы убедиться, что процесс электрообразования является успешным.
- Инкубация септина в везикулами
- Собирайте ГУВ из проводов, используя предварительно вырезанные наконечники пипеток (отверстие ~ 1 мм), которые приближаются в непосредственной близости от проводов. Затем пипеткой распределите раствор по всей проволоке. Эта процедура предотвращает образование сильных пластинчатых потоков, которые могут нарушить работу ГУВ. После этого шага резка наконечников пипеток больше не требуется. Действительно, пластинчатые потоки не повреждают ГУВ в растворе.
ПРИМЕЧАНИЕ: Из-за наличия PI(4,5)P2 в липидной смеси собранные ГУВ необходимо хранить не более 2-3 ч до начала эксперимента. Действительно, PI(4,5)P2 быстро солюбилизируется, и септины больше не связываются с мембранами через несколько часов после их образования. Однако, как только септины связываются с мембраной, они остаются связанными в течение нескольких дней. - Разбавляют раствор септинов в Tris 10 мМ (рН 8) исключительно для достижения осмолярности, равной осмолярности буфера роста; при необходимости разбавляют раствор септина дополнительно в буфере наблюдения. Добавьте предполагаемый объем собранных ГУВ (50-100 мкл при общем объеме 200 мкл). Проводят инкубацию непосредственно в камере наблюдения после пассивации β-казеином (см. ниже). Для достижения равновесия требуется 20-30 минут времени ожидания.
ПРИМЕЧАНИЕ: Экспрессия и очистка октамерных комплексов септина (человеческих или почковых дрожжей) подробно описаны в других статьях17. Вкратце, септины экспрессировали в кишечной палочке, очищали в лаборатории с использованием стадий аффинности, исключения размера и ионообменной хроматографии и хранили при -80 °C в водном растворе 50 мМ Tris-HCl (рН 8), 300 мМ KCl и 5 мМ MgCl2 при концентрации ~1 мг·мл-1 (3 мкМ). Высокая концентрация соли используется, чтобы избежать агрегации септина. Комплексы септина не следует концентрировать через фильтрующее центрифугирование, которое индуцирует агрегацию и, таким образом, снижает выход белка.
- Собирайте ГУВ из проводов, используя предварительно вырезанные наконечники пипеток (отверстие ~ 1 мм), которые приближаются в непосредственной близости от проводов. Затем пипеткой распределите раствор по всей проволоке. Эта процедура предотвращает образование сильных пластинчатых потоков, которые могут нарушить работу ГУВ. После этого шага резка наконечников пипеток больше не требуется. Действительно, пластинчатые потоки не повреждают ГУВ в растворе.
- Визуализация с помощью конфокального и/или вращающегося дискового микроскопа
- Чтобы предотвратить прилипание ГУВ к поверхности и/или взрыв, пассивируйте камеру наблюдения, инкубируя ее с раствором 5 мг·мл-1 β-казеина в течение 30 мин.
- Удалите раствор β-казеина и перенесите раствор септина-ГУВ (шаг 1.4.2.) в камеру наблюдения с помощью пипетки. Дайте ГУВ отстой на дно камеры в течение 10-15 мин.
ПРИМЕЧАНИЕ: Несоответствие состава между внутренней частью ГУВ и внешним буфером создает несоответствие как плотности, так и показателя преломления. Из-за несоответствия показателя преломления ГУВ видны при проходящей световой оптической микроскопии. - Используя конфокальную микроскопию, визуализируйте флуоресцентный сигнал липидов, чтобы проверить качество ГУВ и состояние пластинчатости мембраны. Оценить плотность септинов, связанных с ГУВ, путем записи флуоресцентного сигнала септинов после выполнения надлежащей калибровки25. Выполняйте сбор Z-стека с пространственным интервалом 0,4 мкм для анализа и визуализации 3D-деформаций везикул, вызванных взаимодействием между септинами и мембраной.
ПРИМЕЧАНИЕ: Использовались масляные погружные объективы с 60- или 100-кратным увеличением. Использовались стандартные конфокальные или вращающиеся дисковые микроскопы (Таблица материалов) с размерами пикселей 250 нм и 110 нм соответственно. Необходимо адаптировать условия визуализации к данной единице оборудования. Никаких специфических антифотоотбеливателей в раствор не добавляли.

Рисунок 1: Электроформирование ГУВ. (А) Схематическое изображение процесса электрообразования с использованием платиновых проводов. (B) Изображение тефлонового самодельного устройства, собранного из платиновых проводов, используемых для генерации ГУВ путем электрообразования. Провода имеют диаметр 0,5 мм и расстояние до 3 мм друг от друга. (C) ГУВ (сферические объекты), наблюдаемые с помощью пропускающей оптической микроскопии в процессе роста. Непрозрачной зоной в нижней части изображения является платиновый провод. (D) ГУВ (круглые флуоресцентные объекты), наблюдаемые с помощью флуоресцентной микроскопии во время роста на платиновой проволоке. Шкала стержней = 100 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
2. Анализ ультраструктурной организации септиновых нитей методом криоэлектронной микроскопии
ПРИМЕЧАНИЕ: Везикулы не подходят для визуализации стандартными методами электронной микроскопии. Действительно, образцы сушат с использованием стандартных методов отрицательного окрашивания. При обезвоживании везикулы, вероятно, подвергаются неспецифическим деформациям, что часто приводит к липидным протрузиям. Таким образом, криоэлектронная микроскопия является гораздо лучшей стратегией для наблюдения за специфическими деформациями везикул. Используя крио-ЭМ, образцы встраиваются в тонкий (~100-200 нм) слой остеклованного льда, который сохраняет образцы, близкие к родному состоянию. ГУВ, однако, слишком велики (несколько десятков микрометров), чтобы их можно было встроить в тонкий лед и, таким образом, визуализировать с помощью просвечивающей электронной микроскопии. Следовательно, большие одноламеллярные везикулы (LUV), диаметры которых варьируются от ~ 50-500 нм, генерируются, чтобы определить, как септины могут деформировать везикулы и как они расположены на везикулах.
- Генерация больших одноламеллярных везикул (LUV)
- Готовят 50 мкг липидной смеси, солюбилизированной в хлороформе с молярным составом 57% EggPC, 15% холестерина, 10% DOPE, 10% DOPS и 8% brain PI(4,5)P2), которая оптимизирована для усиления взаимодействия септина с мембраной в стеклянном флаконе.
- Высушите раствор под потоком аргона, чтобы получить высохшую липидную пленку во флаконе. Поместите флакон под вакуум в течение 30 минут, чтобы полностью высушить липид.
- Повторно растворяют липидную пленку в 50 мкл водного раствора (50 мМ Tris-HCl [рН 8]; 50 мМ KCl; 2 мМ MgCl2) для получения конечной концентрации 1 мг·мл-1, вихрь в течение 10 с и переносят раствор в трубку.
ПРИМЕЧАНИЕ: LUV необходимо использовать сразу для инкубации с септинами. В противном случае белково-липидное взаимодействие будет слабым из-за солюбилизации PI(4,5)P2. Этот грубый процесс ресолюбилизации генерирует гетерогенную популяцию везикул с диаметрами от 50 нм до 500 нм. Следовательно, одновременно анализируется целый ряд диаметров и, следовательно, кривизны.
- Инкубировать септины с солюбилизированными липидами в конечных концентрациях липидов и септинов 0,1 мг·мл-1 (около 300 нМ) и 20 нМ соответственно в буфере с высоким содержанием соли (50 мМ Tris-HCl [рН 8], 300 мМ KCl, 2 мМ MgCl2). Инкубировать образец в течение 1 ч при комнатной температуре.
- Погружная заморозка для остеклования образца
- Тлеющие-разрядные дырявые углеродные решетки (300 меш) на углеродной стороне на 30 с при 5 мА с использованием оборудования плазменного генератора и вставляют сетку внутрь погружной морозильной машины во влажной среде.
- Адсорбировать 4 мкл образца (этап 2.2.) на тлеющей углеродной стороне сетки. Непосредственно перед адсорбцией образца добавьте в раствор 5-10 нм золотые шарики, в случае, если образцы будут использоваться для получения наклонных рядов методом криомографии.
ПРИМЕЧАНИЕ: Плотность золотых бусин должна быть проверена и отрегулирована эмпирически и зависит от поставщика. Оптимизированная плотность составляет 10-15 золотых бусин в поле зрения. - Промокните образцы с голой стороны, чтобы аспирировать каплю образца на противоположной стороне.
ПРИМЕЧАНИЕ: Время промокания обычно составляет 4 с, а положение фильтровальной бумаги и сила промокания регулируются эмпирически путем тестирования и скрининга альтернативных положений фильтровальной бумаги для оптимизации качества (толщины) льда и плотности материала. - Перенесите сетки в микроскоп или храните их в контейнере с жидким азотом.
ПРИМЕЧАНИЕ: Использование дырявых углеродных решеток имеет важное значение для учета полидисперсности в размере пузырьков. Промокание образца с противоположной стороны способствует усиленной адсорбции биологического материала на сетке.
- Криоэлектронная микроскопия
- Вставьте сетки в электронный микроскоп (ЭМ), оборудованный для крио-ЭМ наблюдения. Откройте всю сетку, сгенерировав карту всего образца при низком увеличении (обычно при 120-кратном увеличении), чтобы выбрать области, отображающие лучший лед (т. Е. Тонкий и хорошо остеклованный).
- Для сбора данных cryo-EM 2D соберите изображения размером около 2 Å на пиксель, чтобы проверить качество образца. Убедитесь, что видны как септиновые нити, так и липидный бислой.
- Для сбора данных криоэлектронной томографии выберите интересующие области с деформированными везикулами. Убедитесь, что в поле зрения присутствует достаточное количество золотых бусин (не менее 10).
- В соответствии с программным обеспечением, используемым для сбора данных наклонных рядов, выберите фокусировку и позиции отслеживания, чтобы они находились достаточно далеко от интересующей области. Собирайте наклонные ряды, изменяя угол наклона от -60° до +60°, собирая изображение каждые 2°-3° градусов.
ПРИМЕЧАНИЕ: Общая доза должна быть около или меньше 100 электронов/Å2. Размер пикселя варьируется от 1,3 Å до 2,1 Å, в зависимости от микроскопа, используемого для сбора данных. В идеале предпочтительна угловая симметричная схема сбора данных: 0°, -3°, +3°, -6°, +6°, -9°, +9°[...] -60°, +60°. Однако гониометры боковых микроскопов не обеспечивают достаточной механической стабильности для достижения этих симметричных схем. Альтернативно, захват может быть начат с 0° до 34°, за которым следует вторая угловая последовательность от -2° до -60°, и заканчивается конечной последовательностью от 36° до 60°. Цель состоит в том, чтобы собрать первые изображения (с наименьшими радиационными повреждениями) под наименьшими углами. Кроме того, для визуализации ультраструктуры везикул и связанных нитей септина может быть использован стандартный крио-ЭМ-микроскоп (200 кВ, нить гексаборида лантана (LaB6) и боковой вход образца). Однако, если кто-то стремится к дальнейшей обработке изображений (например, усреднению субтомограмм), лучше всего использовать микроскопы последнего поколения с полевой эмиссией (FEG), оснащенные прямыми детекторами. В этом протоколе мы ограничиваем наше описание получением 3D-реконструкций и исключаем усреднение субтомограмм.
- 3D реконструкция по криомографии и сегментации
- Используйте пакет программного обеспечения IMOD (Таблица материалов) для выравнивания наклонных серий изображений и 3D-реконструкции26,27. В рамках IMOD выполните выравнивание серии наклона на основе позиционирования фидуциалов (золотых бусин). Кроме того, при необходимости проводят определение и коррекцию функции переноса контраста (CTF) в рамках IMOD26. Наконец, достигните 3D-реконструкции с помощью IMOD, строго следуя каждому шагу.
- Вручную сегментируйте липидные бислои и септиновые нити с помощью 3Dmod27 из пакета программного обеспечения IMOD для отображения.
3. Анализ чувствительности кривизны септина с помощью SEM
ПРИМЕЧАНИЕ: Чтобы понять, как септины могут быть чувствительны к кривизне микрометра, был использован подход in vitro для инкубации комплексов нитей септина с твердо поддерживаемыми липидными бислоями, нанесенными на волнистые волнистые волнистые узоры микрометрового масштаба.
- Проектирование волнистой реплики NOA (оптический клей Norland) из волнистых моделей полидиметилсилоксана (PDMS)
- Используйте волнистые паттерны PDMS с амплитудой 250 нм и боковой периодичностью 2 мкм или другими размерами для анализа кривизны, подходящие для интересующего белка.
ПРИМЕЧАНИЕ: Волнообразные паттерны PDMS разрабатываются и генерируются, как описано в Nania et al.28,29. - В помещении для чистых помещений нанесите 5 мкл жидкого NOA на круглый стеклянный покров диаметром 1 см и поместите шаблон PDMS на каплю. Обработать ультрафиолетовым светом (320 нм) в течение 5 мин для фотополимеризации жидкого НОА в тонкую полимерную пленку. Затем аккуратно отклейте шаблон PDMS от крышки со свежеполимеризованного NOA.
ПРИМЕЧАНИЕ: NOA является распространенным оптически прозрачным клеем, подходящим для оптической микроскопии. Кроме того, NOA устойчив к процессам химической фиксации и окрашивания, проводимым до визуализации SEM. Как смолы NOA 71, так и NOA 81 могут использоваться с аналогичными результатами. Исходный шаблон PDMS может быть использован несколько раз для создания реплик NOA. Полученные реплики NOA можно хранить в коробке при комнатной температуре в течение нескольких месяцев.
- Используйте волнистые паттерны PDMS с амплитудой 250 нм и боковой периодичностью 2 мкм или другими размерами для анализа кривизны, подходящие для интересующего белка.
- Генерация поддерживаемого липидного бислоя и белковой инкубации
- Обработайте пленки NOA с помощью воздушного плазмоочистителя в течение 5 минут, чтобы сделать поверхность гидрофильной.
- Готовят раствор небольших одноламеллярных везикул (SUV) с молярным составом 57% EggPC, 15% холестерина, 10% DOPE, 10% DOPS и 8% мозгового PI(4,5)P2 при общей концентрации липидов 1 мг·мл-1. Подготовьте внедорожники путем повторного использования высушенной липидной пленки в буфере наблюдения, как описано на этапе 2.1.3. Аккуратно обработайте раствор ультразвуком с помощью машиниста для ванны в течение 5-10 минут, пока раствор не станет прозрачным.
ПРИМЕЧАНИЕ: Раствор внедорожников можно хранить замороженным в течение нескольких недель при -20 °C. - Вставьте обшивки, поддерживающие узоры NOA, в колодцы ящиков для клеточных культур. Нанесите 100 мкл раствора внедорожника 1 мг·мл-1 на свежее тлеющее разряженное НОА (этап 3.2.1.) и инкубируйте в течение 30 мин при комнатной температуре. Этот шаг индуцирует слияние внедорожников с поверхностью паттерна NOA для создания поддерживаемого липидного бислоя.
- Тщательно промойте слайды 6x с буфером септина (50 мМ Tris-HCl [pH 8], 50 мМ KCl, 2 мМ MgCl2), чтобы удалить несросшийся внедорожник. После каждого смывания никогда не позволяйте образцу полностью высохнуть.
- Разбавляют раствор октамерного септина в высокосолевом буфере септина (50 мМ Tris-HCl [рН 8], 300 мМ KCl, 2 мМ MgCl2) с использованием буфера септина (50 мМ Tris-HCl [pH 8], 50 мМ KCl, 2 мМ MgCl2) до конечных концентраций в диапазоне от 10 нМ до 100 нМ и объемов 1 мл. Инкубировать белковый раствор на слайдах в течение ~1 ч при комнатной температуре.
ПРИМЕЧАНИЕ: Объем достаточно велик, чтобы испарение не было проблемой.
- Пробоподготовка к СЭМ-анализу
ПРИМЕЧАНИЕ: В электронной микроскопии были разработаны различные протоколы для анализа белковых структур и их сборки. Текущий протокол сохраняет организацию септиновых нитей, используя протокол фиксации, полученный от Svitkina et al.30 это легко реализовать. Кроме того, этот протокол оптимизирует наблюдения SEM с высоким разрешением.- Приготовление реагентов и стоковых растворов
ПРИМЕЧАНИЕ: Этот протокол требует нескольких реагентов и растворов, которые могут быть приготовлены заранее или непосредственно перед инкубацией. Следуйте приведенным инструкциям, чтобы избежать артефактов или отсутствия химической реакционной способности.- Раствор какодилата натрия 0,2 М: Для приготовления этого раствора двойной прочности растворите 2,14 г порошка какодилата натрия в ~ 40 мл дистиллированной воды при магнитном перемешивании. После полного растворения отрегулируйте рН до 7,4, осторожно добавив 0,1 М HCl (~1 мл на 50 мл раствора) и восполните конечный объем дистиллированной водой. Этот раствор можно хранить в течение 24-48 ч при 4°C.
- 2% глутаральдегид (GA) в 0,1 М какодилата натрия (фиксирующий раствор): Приготовьте этот раствор непосредственно перед использованием, разбавляя высокочистый ЭМ-класс GA 0,2 М раствором какодилата натрия (см. Выше). Используйте 25%-50% коммерческое решение GA из-за его оптимизированной емкости хранения при 4 °C. Для приготовления 10 мл фиксирующего раствора разводят 0,8 мл 25% раствора коммерческого ГА 4,2 мл дистиллированной воды и 5 мл 0,2 М какодилата натрия.
- 1% тетроксид осмия (OsO4) в 0,1 М какодилата натрия: Приготовьте этот второй фиксирующий раствор непосредственно перед использованием, разбавляя коммерческий 4% раствор OsO4 0,2 М какодилата натрия (см. Выше). Для 4 мл фиксаторного раствора OsO4 разбавляют 1 мл 4% коммерческого раствора OsO4 1 мл дистиллированной воды и 2 мл 0,2 М какодилата натрия.
ПРИМЕЧАНИЕ: Используйте коммерческий 4% раствор OsO4 в расщепляющихся стеклянных колбах из-за их свойств хранения и обработки. Обратите внимание, что OsO4 обладает высокой реакционной способностью. Убедитесь, что его цвет слегка желтый, а не темный. - 1% дубильная кислота (ТА) в воде: Приготовьте этот раствор непосредственно перед использованием. Готовят раствор ТА для получения конечной концентрации 1% ТА в дистиллированной воде комнатной температуры. Растворить 10 мг в 1 мл дистиллированной воды и вихрь в течение нескольких минут. Раствор TA не может быть сохранен и должен быть отфильтрован фильтром 0,2 мкм перед использованием.
- 1% раствор уранилацетата (UA) в воде Готовят раствор UA для получения конечной концентрации 1% UA в дистиллированной воде. Растворить 10 мг в 1 мл дистиллированной воды и вихря в течение не менее 30 мин до 1 ч путем вихря или встряхивания при комнатной температуре. Этот раствор может храниться в течение 1 месяца при 4 °C, но может легко выпадать в осадок и, следовательно, должен быть отфильтрован фильтром 0,2 мкм перед использованием.
- Градуированные серии растворов этанола готовят 50%, 70%, 95% и 100% растворы этанола в воде. Подготовьте окончательную ванну из вновь открытых бутылок со 100% этанолом или из 100% этанола, который был обезвожен в течение не менее 24 ч молекулярными ситами (номинальный диаметр пор = 4 Å), чтобы удалить из образцов все следы воды, которые могут помешать сушке и покрытию. Будьте осторожны, чтобы не встряхнуть этот раствор, так как это может привести к повторному суспендированию силикатных частиц.
ПРИМЕЧАНИЕ: Будьте осторожны при обращении с химическими реагентами, используемыми в этом протоколе. Глутаральдегид, тетроксид осмия, какодил натрия и уранилацетат очень токсичны, а уранилацетат также радиоактивен. Все манипуляции с реагентами и их отходами должны выполняться с использованием индивидуальной (перчатки, лабораторные халаты, защитные очки) и коллективной защиты (вытяжки и щиты из плексигласа), в соответствии с лабораторными процедурами.
- Фиксация образца
- Промывайте образцы (т.е. слайды NOA со слитыми поддерживаемыми липидными бислоями и инкубированным белком) PBS. Замените PBS фиксирующим раствором GA, предварительно расплавленным при 37 °C, и дайте реакции продолжаться в течение 15 мин. Затем образцы могут храниться при температуре 4 °C.
- Удалить фиксирующий раствор и промыть неподвижные образцы 3x 0,1 М какодилата натрия (5 мин на стирку) с легким встряхиванием.
- Инкубируйте образцы в фиксаторном растворе OsO4 в течение 10 мин с максимальной защитой от света, чтобы обеспечить фиксацию мембранных структур и повысить электропроводность образцов. Удалите фиксирующий раствор и промыть образцы 3 раза в дистиллированной воде (5 мин на стирку) с легким встряхиванием.
- Инкубируйте промытые образцы в фильтрованном фиксаторном растворе TA в течение максимум 10 минут. Удалите раствор ТА и промыть образцы 3 раза в дистиллированной воде (5 мин на стирку) с легким встряхиванием.
- Инкубировать промытые образцы в свежефильтрованном фиксаторном растворе UA в течение 10 мин с максимальной защитой от света. Удалить раствор UA и промыть образцы 3x в дистиллированной воде (5 мин на промывку) с легким встряхиванием.
- Обезвоживание проб и сушка в критической точке
ПРИМЕЧАНИЕ: Сушка на воздухе не допускается во избежание повреждения образцов из-за нежелательного поверхностного напряжения при переходе из жидкого в газообразное состояние. В ЭМ установлены два метода: физический метод достижения сверхкритического состоянияСО2 и обхода его критической точки (31 °C, 74 бар) или химический метод испарения гексаметилдисилазана (HMDS), сушильного агента с пониженным поверхностным натяжением. CO2 и HMDS плохо смешиваются с водой. Следовательно, все следы воды должны быть заменены переходным растворителем (этанолом), чтобы избежать каких-либо повреждений в дальнейшем во время процессов сушки.- Инкубируют образцы в течение 2-3 мин в каждом растворе этанола, начиная от 50% до 100% (безводной) этаноловой ванны.
ПРИМЕЧАНИЕ: Поскольку испарение этанола между ваннами происходит быстро, обработка образцов также должна быть быстрой, чтобы избежать высыхания на воздухе. - Переложите стеклянные горки внутрь сушилки критической точки, предварительно заполненной этанолом, и следуйте инструкциям производителя.
ПРИМЕЧАНИЕ: В этом протоколе использовался автоматический аппарат, но можно использовать любую систему. Протоколы могут отличаться для каждого устройства, но должны быть оптимизированы для полного удаления этанола (25 ванн в этом протоколе) и снижения скорости для различных обменов растворителей (входы CO2 и выходы этанола / CO2 ) и окончательной разгерметизации (почти 1 ч в этом протоколе). - В конце критической точки высыхания храните образцы непосредственно в осушителе до монтажа и нанесения покрытия. Поскольку высушенные образцы очень гигроскопичны, покройте их (см. Ниже) как можно скорее.
ПРИМЕЧАНИЕ: С другой стороны, HMDS может предложить более дешевый и быстрый метод. Хотя HDMS никогда не тестировался на наших образцах, этот подход дал хорошие результаты для наблюдения белков на внутренней стороне клеточных мембран31,32.
- Инкубируют образцы в течение 2-3 мин в каждом растворе этанола, начиная от 50% до 100% (безводной) этаноловой ванны.
- Монтаж и покрытие образцов.
ПРИМЕЧАНИЕ: Поскольку биологические образцы обладают плохими свойствами электропроводности, они должны быть покрыты проводящей металлической пленкой перед наблюдением SEM. Таким образом, используется плазменно-магнетронное напыление.- Прикрепите крышку к заглушкам, которые будут использоваться для будущих наблюдений SEM. Используйте серебристую краску из-за ее улучшенной электропроводности, по сравнению с углеродными дисками. Добавьте полоску серебристой краски на верхнюю грань крышки и убедитесь, что соединение с заглушкой удовлетворительное. Избегайте любых агломератов краски.
ПРИМЕЧАНИЕ: Полоса серебристой краски должна быть тонкой, чтобы избежать контакта между образцом и объективом SEM при работе с высоким разрешением (т.е. на низком рабочем расстоянии). - Подождите, пока растворитель полностью испарится.
ПРИМЕЧАНИЕ: Продолжительность этого этапа может варьироваться в зависимости от количества и толщины отложения серебряной краски. Этот этап можно укоротить с помощью колокольчика или коатера и первичного вакуума в течение 10-30 мин. - Используйте аппарат, оснащенный плазменно-магнетронной напыляющей головкой и вращательно-планетарной ступенью, и следуйте стандартным протоколам, предоставленным производителем. Здесь аппарат эвакуировали на 2,5 х 10-5 мбар, продувляли 1х качественным аргоном, а затем доводили до 8,0 х 10-3 мбар.
- Выполните предварительное напыление (120 мА в течение 60 с) для удаления оксидного слоя на поверхности. Затем нанесите 1,5 нм вольфрама (90 мА, рабочее расстояние = 50 мм) с помощью монитора толщины пленки.
ПРИМЕЧАНИЕ: Коатер должен быть идеально очищен для обеспечения воспроизводимости испарения пленки. Покрытие должно быть остановлено после достижения заданной толщины. Окончательная толщина пленки рассчитывается и корректируется после этого. Пленки около 1,5 нм имеют, в среднем, посткоррекцию 0,7 нм с помощью нашего аппарата. Поскольку эти значения коррекции близки к целевому показателю, выполняется серия покрытий для оценки, а затем вычитания этой коррекции из целевого значения. - Храните образцы в вакууме, чтобы защитить их от окружающего воздуха до и во время всех анализов SEM.
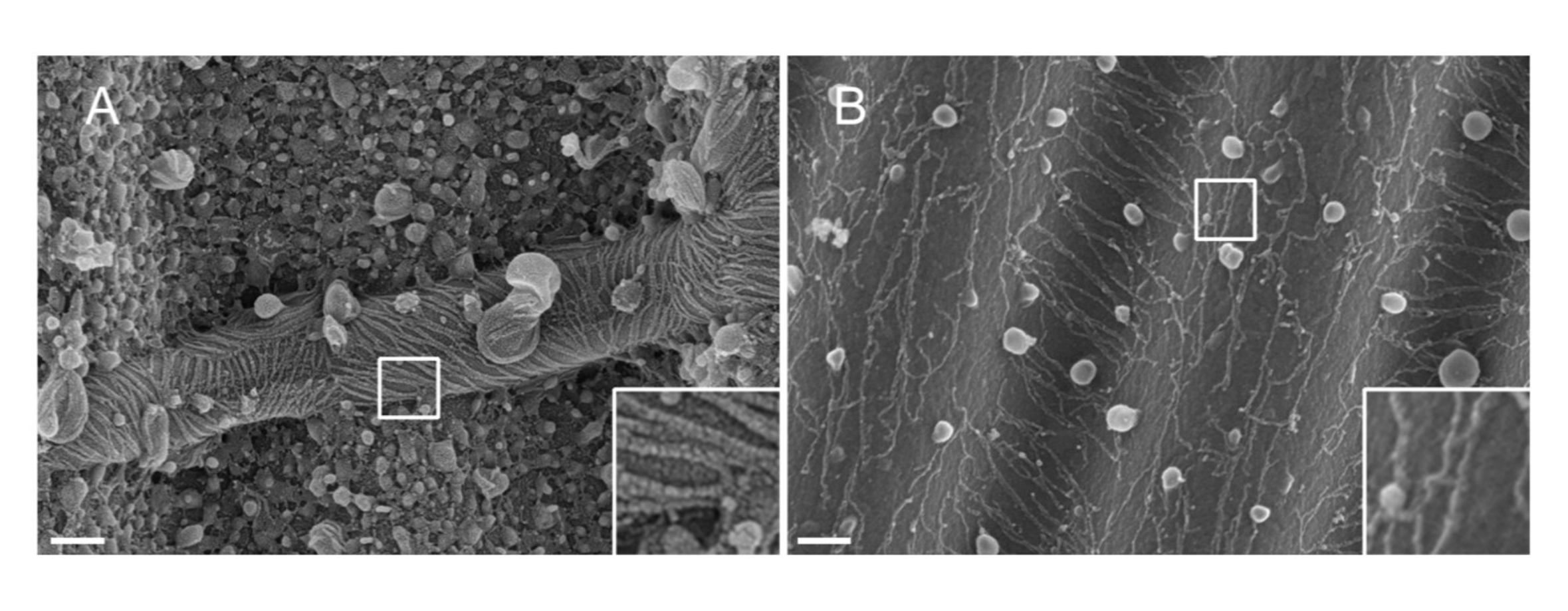
ПРИМЕЧАНИЕ: Природа металла, используемого для распыления, имеет значение. Pt, несмотря на то, что является распространенным материалом, приводит к некачественному покрытию с толщиной пленки Pt, адаптированной к септиновым нитям (1,5 нм). При высоком разрешении пленке Pt 1,5 нм не хватает связности; таким образом, размер кластеров Pt и нитей септина становится одинаковым, что приводит к неправильной интерпретации во время процесса сегментации нитинакаливания 33 (см. Фиг.2A, вставка). Вольфрам является хорошей альтернативой Pt, потому что он отображает меньший размер зерна, едва заметный при высоком разрешении SEM (см. Рисунок 2B, вставка). Тем не менее, чистый вольфрам легко окисляется, что приводит к сильным артефактам зарядного эффекта во время наблюдения SEM, если процедура описана в шаге 3.3.4. не соблюдается строго.
- Прикрепите крышку к заглушкам, которые будут использоваться для будущих наблюдений SEM. Используйте серебристую краску из-за ее улучшенной электропроводности, по сравнению с углеродными дисками. Добавьте полоску серебристой краски на верхнюю грань крышки и убедитесь, что соединение с заглушкой удовлетворительное. Избегайте любых агломератов краски.
- Приготовление реагентов и стоковых растворов
- Получайте изображения с помощью полево-эмиссионного микроскопа SEM (FESEM).
ПРИМЕЧАНИЕ: Технологии SEM недавно были модернизированы для улучшения разрешения, и технологии зависят от производителя (например, электронная оптика, замедление луча, магнитная линза). Это усиление разрешения (доступное у нескольких производителей), особенно при низком ускоряющем напряжении (около нанометра при 1 кВ), необходимо для разрешения нанометрических структур, аналогичных септиновым сетям.- Получение изображений с высоким разрешением путем обнаружения первичных вторичных электронов (SE1) с помощью детектора «в объективе» с использованием следующих настроек.
- Установите ускоряющее напряжение на 3 кВ. Зафиксируйте ток луча с апертурой 20 мкм (для колонн Zeiss Gemini I или эквивалент 34 пА) или с диафрагмой 15 мкм (для колонн Zeiss Gemini I или эквивалент 18,5 пА), если требуется подавление эффектов зарядки.
- Для наблюдений используйте разрешения в диапазоне от 21,25 нм/пиксель до 1,224 нм/пиксель, а для анализа данных используйте разрешение ~5,58 нм/пиксель (увеличение 20 000x в соответствии со ссылкой Polaroid 545).
- Установите рабочее расстояние между 1 мм и 2 мм для наблюдения с высоким разрешением и около 3 мм, если требуется увеличенная глубина резкости. Непрерывная регулировка скорости сканирования и интеграции линий для обеспечения постоянного отношения сигнал/шум со временем захвата около 30-45 с на изображение.

Рисунок 2: Влияние материала, нанесенного на септиновые нити, на волнистые паттерны PDMS. SEM септиновых нитей, покрытых распылением либо (A) 1,5 нм платины, демонстрируя рисунок «засушливой трещины», типичный для отсутствия связности между кластерами ядер платины, либо (B) 1,5 нм вольфрама, покрытого гладким и когезивным слоем. Шкала = 200 нм. Белые квадратные квадраты представляют увеличенные виды в правом нижнем углу. Сферические шарики представляют собой небольшие липидные пузырьки, взаимодействующие с септинами. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Результаты
Деформации ГУВ
Типичные конфокальные флуоресцентные изображения ГУВ, измененные после инкубации с септинами, показаны на рисунке 3 в условиях полимеризации септинов. Голые ГУВ (рисунок 3А) были идеально сферическими. При инкубации с более чем ...
Обсуждение
Как указано выше, была использована липидная смесь, которая усиливает включение PI(4,5)P2 в липидный бислой и, таким образом, облегчает септин-мембранные взаимодействия. Действительно, мы показали в другом месте25 , что почковые дрожжевые септины взаимодействуют с везику?...
Раскрытие информации
У авторов нет конфликта интересов.
Благодарности
Мы благодарим Патрисию Бассеро и Даниэля Леви за их полезные советы и обсуждения. Эта работа была поддержана ANR (Agence Nationale de la Recherche) для финансирования проекта "SEPTIME", ANR-13-JSV8-0002-01, ANR SEPTIMORF ANR-17-CE13-0014 и проекта "SEPTSCORT", ANR-20-CE11-0014-01. B. Chauvin финансируется Ecole Doctorale "ED564: Physique en Ile de France" и Fondation pour lea Recherche Médicale. К. Накадзава был поддержан Университетом Сорбонны (AAP Emergence). Г.Х. Кёндеринк был поддержан Нидерландской организацией вур Ветеншаппелийк Ондерзоек (NWO/OCW) через «BaSyC-Building a Synthetic Cell». Гравитационный грант (024.003.019). Мы благодарим Labex Cell(n)Scale (ANR-11-LABX0038) и Paris Sciences et Lettres (ANR-10-IDEX-0001-02). Мы благодарим Cell and Tissue Imaging (PICT-IBiSA), Институт Кюри, члена Французской национальной исследовательской инфраструктуры France-BioImaging (ANR10-INBS-04).
Материалы
| Name | Company | Catalog Number | Comments |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | Avanti Polar Lipids | 850725 | |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine | Avanti Polar Lipids | 840035 | |
| Bath sonicator | Elma | Elmasonic S10H | |
| Bodipy-TR-Ceramide | invitrogen, Thermo Fischer scientific | 11504726 | |
| Chemicals: NaCl, Tris-HCl, sucrose, KCl, MgCl2, B-casein, chloroform, sodium cacodylate, tannic acid, ethanol | Sigma Aldrich | ||
| Confocal microscope | nikon | spinning disk or confocal | |
| Critical point dryer | Leica microsystems | CPD300 | |
| Deionized water generator | MilliQ | F1CA38083B | MilliQ integral 3 |
| Egg L-α-phosphatidylcholine | Avanti Polar Lipids | 840051 | |
| Field Emission Gun SEM (FESEM) | Carl Zeiss | Gemini SEM500 | |
| Glutaraldehyde 25 %, aqueous solution | Thermo Fischer scientific | 50-262-19 | |
| High vacuum grease, Dow corning | VWR | ||
| IMOD software | https://bio3d.colorado.edu/imod/ | software suite for tilted series image alignment and 3D reconstruction | |
| Lacey Formvar/carbon electron microscopy grids | Eloise | 01883-F | |
| Lipids | Avanti Polar Lipids | ||
| L-α-phosphatidylinositol-4,5-bisphosphate | Avanti Polar Lipids | 840046 | |
| Metal evaporator | Leica microsystems | EM ACE600 | |
| NOA (Norland Optical Adhesives), NOA 71 and NOA 81 | Norland Products | NOA71, NOA81 | |
| Osmium tetraoxyde 4% | delta microscopies | 19170 | |
| Osmometer | Löser | 15 M | |
| Plasma cleaner | Alcatel | pascal 2005 SD | |
| Plasma generator | Electron Microscopy Science | ||
| Plunge freezing equipment | leica microsystems | EMGP | |
| Transmission electron microscope | Thermofischer | Tecnai G2 200 kV, LaB6 | |
| Uranyl acetate | Electron Microscopy Science | 22451 | this product is not available for purchase any longer |
| Wax plates, Vitrex | VWR |
Ссылки
- Finger, F. P. Reining in cytokinesis with a septin corral. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 27 (1), 5-8 (2005).
- Barral, Y., Kinoshita, M. Structural insights shed light onto septin assemblies and function. Current Opinion in Cell Biology. 20 (1), 12-18 (2008).
- Hu, Q., et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329 (5990), 436-439 (2010).
- Lin, Y. -. H., Kuo, Y. -. C., Chiang, H. -. S., Kuo, P. -. L. The role of the septin family in spermiogenesis. Spermatogenesis. 1 (4), 298-302 (2011).
- Addi, C., Bai, J., Echard, A. Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Current Opinion in Cell Biology. 50, 27-34 (2018).
- Spiliotis, E. T., Kesisova, I. A. Spatial regulation of microtubule-dependent transport by septin GTPases. Trends in Cell Biology. 31 (12), 979-993 (2021).
- Spiliotis, E. T., Nakos, K. Cellular functions of actin- and microtubule-associated septins. Current Biology: CB. 31 (10), 651-666 (2021).
- Salameh, J., Cantaloube, I., Benoit, B., Poüs, C., Baillet, A. Cdc42 and its BORG2 and BORG3 effectors control the subcellular localization of septins between actin stress fibers and microtubules. Current Biology: CB. 31 (18), 4088-4103 (2021).
- Ewers, H., Tada, T., Petersen, J. D., Racz, B., Sheng, M., Choquet, D. A septin-dependent diffusion barrier at dendritic spine necks. PloS One. 9 (12), 113916 (2014).
- Myles, D. G., Primakoff, P., Koppel, D. E. A localized surface protein of guinea pig sperm exhibits free diffusion in its domain. The Journal of Cell Biology. 98 (5), 1905-1909 (1984).
- Luedeke, C., Frei, S. B., Sbalzarini, I., Schwarz, H., Spang, A., Barral, Y. Septin-dependent compartmentalization of the endoplasmic reticulum during yeast polarized growth. The Journal of Cell Biology. 169 (6), 897-908 (2005).
- Gilden, J. K., Peck, S., Chen, Y. -. C. M., Krummel, M. F. The septin cytoskeleton facilitates membrane retraction during motility and blebbing. The Journal of Cell Biology. 196 (1), 103-114 (2012).
- Dolat, L., Hu, Q., Spiliotis, E. T. Septin functions in organ system physiology and pathology. Biological Chemistry. 395 (2), 123-141 (2014).
- Angelis, D., Spiliotis, E. T. Septin mutations in human cancers. Frontiers in Cell and Developmental Biology. 4, 122 (2016).
- Takehashi, M., et al. Septin 3 gene polymorphism in Alzheimer's disease. Gene Expression. 11 (5-6), 263-270 (2004).
- Shuman, B., Momany, M. Septins from protists to people. Frontiers in Cell and Developmental Biology. 9, 824850 (2022).
- Bertin, A., et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proceedings of the National Academy of Sciences of the United States of America. 105 (24), 8274-8279 (2008).
- Iv, F., et al. Insights into animal septins using recombinant human septin octamers with distinct SEPT9 isoforms. Journal of cell science. 134 (15), (2021).
- Beber, A., et al. Membrane reshaping by micrometric curvature sensitive septin filaments. Nature communications. 10 (1), 420 (2019).
- Bridges, A. A., Jentzsch, M. S., Oakes, P. W., Occhipinti, P., Gladfelter, A. S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. The Journal of Cell Biology. 213 (1), 23-32 (2016).
- Patzig, J., et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 5, 17119 (2016).
- Szuba, A., et al. Membrane binding controls ordered self-assembly of animal septins. eLife. 10, 63349 (2021).
- Tanaka-Takiguchi, Y., Kinoshita, M., Takiguchi, K. Septin-mediated uniform bracing of phospholipid membranes. Current Biology: CB. 19 (2), 140-145 (2009).
- Bertin, A., et al. Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. Journal of Molecular Biology. 404 (4), 711-731 (2010).
- Beber, A., et al. Septin-based readout of PI(4,5)P2 incorporation into membranes of giant unilamellar vesicles. Cytoskeleton. 76 (4,5), 92-103 (2019).
- Mastronarde, D. N., Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology. 197 (2), 102-113 (2017).
- Kremer, J. R., Mastronarde, D. N., McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 116 (1), 71-76 (1996).
- Nania, M., Foglia, F., Matar, O. K., Cabral, J. T. Sub-100 nm wrinkling of polydimethylsiloxane by double frontal oxidation. Nanoscale. 9 (5), 2030-2037 (2017).
- Nania, M., Matar, O. K., Cabral, J. T. Frontal vitrification of PDMS using air plasma and consequences for surface wrinkling. Soft Matter. 11 (15), 3067-3075 (2015).
- Svitkina, T. M., Borisy, G. G. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods in Enzymology. 298, 570-592 (1998).
- Franck, A., et al. Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Molecular Biology of the Cell. 30 (5), 579-590 (2019).
- Elkhatib, N., et al. Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science. 356 (6343), (2017).
- Stokroos, I., Kalicharan, D., Van Der Want, J. J., Jongebloed, W. L. A comparative study of thin coatings of Au/Pd, Pt and Cr produced by magnetron sputtering for FE-SEM. Journal of Microscopy. 189, 79-89 (1998).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены