
Method Article
Seguindo o destino celular, em
Neste Artigo
Resumo
Este artigo descreve o procedimento para a preparação de uma versão marcada com fluorescência do bacteriófago lambda, infecção de E. coli Bactérias, após o resultado da infecção sob o microscópio, e análise dos resultados de infecção.
Resumo
Compreendendo o sistema bacteriófago (fago) lambda e a bactéria E. coli serviu durante muito tempo como um paradigma para a célula destino, 1,2 determinação. Seguindo a infecção simultânea da célula de um número de fagos, uma de duas vias é escolhido: lítico (virulento) ou lisogênico (latentes) 3,4. Recentemente, desenvolveu um método para etiquetar fluorescentemente fagos individuais, e foram capazes de estudar a decisão de pós-infecção, em tempo real, ao microscópio, a nível de fagos individuais e as células 5. Aqui, descrevemos o procedimento completo para a realização dos experimentos de infecção descritas em nossos trabalhos anteriores 5. Isto inclui a criação de fagos fluorescentes, a infecção das células, sob o microscópio de imagem e análise de dados. O fago fluorescente é um "híbrido", co-expressando as versões de tipo selvagem e YFP-fusão da proteína da cápside gpD. Um ligado fágico bruto é primeiro obtido por indução de um lisogénio de o (gpD-EYFP EnhYellow Fluorescent Protein ANCED fago), portadora de um plasmídeo que expressa tipo selvagem gpD. Uma série de passos de purificação são então realizadas, seguido por DAPI-rotulagem e de imagem sob o microscópio. Isto é feito a fim de verificar a uniformidade, a eficiência de empacotamento do ADN, o sinal de fluorescência e a estabilidade estrutural do stock de fagos. A adsorção inicial de fagos às bactérias é realizada em gelo, seguido por uma incubação breve, a 35 ° C para provocar a injecção do ADN virai 6. O fago / bactérias mistura é, então, transferido para a superfície de uma laje fina agar nutriente, coberta com uma lamela e visualizados sob um microscópio de epifluorescência. O processo de pós-infecção é seguida durante 4 h, a 10 minutos de intervalo. Posições de múltiplos estágios são monitorados tais que ~ 100 infecções celulares podem ser rastreados em um único experimento. Em cada ponto de posição e de tempo, as imagens são adquiridas no contraste de fase e canais de vermelho e verde fluorescente. A imagem de contraste de fase é usado posteriormente para automatizado celreconhecimento l enquanto os canais fluorescentes são usados para caracterizar o resultado de infecção: produção de novos fagos fluorescentes (verde) seguido de lise das células, ou a expressão de factores de lisogenia (vermelho), seguido de crescimento das células é reiniciada e divisão. Os adquiridos lapso de tempo os filmes são processadas utilizando uma combinação de métodos manuais e automatizados. Dados os resultados da análise para a identificação de parâmetros de infecção para cada evento de infecção (por exemplo, número e posições de infectar fagos), bem como resultado da infecção (lise / lisogenia). Os parâmetros adicionais podem ser extraídas se desejado.
Protocolo
1. Criação de um lisado de fago em bruto (fig. 1)
- Num balão de 50 ml, inocular uma colónia fresca de LE392 (λ LZ1) [pPLate * D] (ver Tabela 1 para detalhes) em 6 ml de meio LB suplementado com 10 7 ug / ml e canamicina 100 ng / ml de ampicilina. Crescer durante a noite a 30 ° C com agitação suave (180 rpm).
- Diluiu-se a 1:100 em cultura LBM (LB suplementado com 10 mM de MgSO 4) e crescer a 30 ° C com agitação suave (180 rpm). A fim de optimizar o rendimento de fago, certifique-se de que o volume de cultura não é mais do que um décimo da capacidade de volume do frasco. Nós normalmente preparar dois frascos de capacidade de 2 litros ou 2,5 litros, e adicionar 2,5 ml da cultura durante a noite em 250 ml de meio de LBM em cada frasco.
- Quando a densidade celular atinge OD 600 ≈ 0,6 (~ 2,5-3 hr), induzem a lisogénio movendo a cultura a 42 ° C de um banho de água em agitador durante 18 min com agitação suave (180 rpm) e, em seguida incubate a 37 ° C com agitação suave (180 rpm) até que a lise é visível (cultura torna-se claro, em ~ 60 - 90 min).
- Adicionar clorofórmio a 2% para a cultura, agitar para misturar à mão, e, em seguida, incubadas durante 15 min à temperatura ambiente. Cuidado: Use luvas para lidar com clorofórmio, e evitar respirar ele.
- Transferir a cultura em dois frascos de centrífuga de 250 ml, centrifugar a cultura num rotor Sorvall GSA a 10000 rpm durante 15 min a 4 ° C. Recuperar o sobrenadante que contém as partículas de fago, e descartar o pellet de detritos. Realizar uma segunda centrifugação para se certificar de se livrar dos restos visíveis.
- Usar um protocolo padrão de titulação de fagos 8 para medir a concentração de fagos. O título do fago deve ser ~ 5-10 x 10 9 pfu / ml. Utilizar uma estirpe supF, tais como a estirpe LE392 como indicador, devido à mutação SAM7 no genótipo do fago fluorescente, e usar agar de topo e placas de agar feitas com NZYM rica para se obter placas maiores (Figura 2).
2. Purificação de fagos (Figura 1)
- Verter o lisado para um balão (por exemplo, de 2 litros) grande, adicionar DNase I e RNase (1 ug / ml de cada) com o lisado, a fim de digerir os ácidos nucleicos libertados a partir de bactérias lisadas, e incubar 1 hora à temperatura ambiente.
- Adicionar NaCl 1M ao lisado, transferir o lisado para frascos de centrífuga de 250 ml, e incuba-se 3 horas em gelo. Centrifuga-se o lisado numa Sorvall GSA a 10000 rpm durante 15 min a 4 ° C. Recuperar o sobrenadante. O título do fago deve ser semelhante à do lisado em bruto, que é de aproximadamente 5-10 x 10 9 pfu / ml. A adição de NaCl promove a dissociação das partículas de fago a partir de resíduos bacterianos e é requerido para a precipitação de partículas de fago eficiente por PEG 8.
- Verter o lisado para um balão de grande porte, por exemplo, de 2 litros frasco, adicionar 10% (w / v) de PEG8000 para o lisado, lentamente agitar ou agitar para dissolver PEG8000 à temperatura ambiente. Transferir o lisado em 250 ml centorifuge garrafas e depois incubar durante a noite (~ 16 horas) a 4 ° C. Centrifugar o lisado num rotor Sorvall GSA a 10000 rpm durante 15 min a 4 ° C. Descartar o sobrenadante.
- Embeber o sedimento (partículas de fago precipitado com PEG8000) com fago tampão SM (4 ml de tampão SM por 250 ml de lisado de fagos inicial). Incubar com agitação muito suave ou sem agitação durante 16 horas a 4 ° C.
- Suavemente retire o lisado (tampão SM com as partículas de fago) para um tubo de centrífuga de 50 ml de Eppendorf e, em seguida lavar o sedimento restante com 0,5 - 1 ml de tampão SM.
- Adicionar um volume igual de clorofórmio ao lisado. Misture delicadamente o lisado com clorofórmio até invertendo e para baixo por algumas vezes. Centrifugar a 4000 rpm durante 15 min a 4 ° C num Eppendorf 5804R ou uma centrífuga de bancada similar.
- Repita o passo 2.6 para obter uma melhor lisado. O título do fago deve ser ~ 1-2 x 10 11 pfu / ml.
- Prepare SM / CsCl soluções com três densidades diferentes (ρ) de 1,3 g / ml, 10,5 g / ml e 1,7 g / ml. Medir o índice de refracção (η) para obter uma leitura mais precisa da densidade. A conversão densidade 9 é ρ = 10,8601 η - 13,4974, a 25 ° C. Consulte a Tabela 3 para detalhes.
- Use uma seringa com uma agulha de tempo para carregar a solução para um ultra-claro 14 ml Beckman 40Ti tubo de ultracentrifugação. Para evitar a mistura e formar um gradiente de densidade melhor, underlaying a solução (isto é, soluções de camadas de densidade crescente sob um outro) deve ser utilizado, ou seja, suavemente transferida 2 ml de SM / CsCl soluções na ordem de 1,3 g / ml, 1,5 g / ml e 1,7 g / ml, por inserção da agulha de uma seringa de 3 ml para a parte inferior do tubo.
- Suavemente carregar 8 ml de lisado de fagos através da sobreposição da parte superior do tubo de ultracentrifugação de 14 ml. Preparar um tubo de equilíbrio. Centrifugar numa SW40Ti rotor Beckman a 24.000 rpm durante 4 horas a 4 ° C.
- Suavemente retirar o tubo num quarto escuro e iluminar a partir do topo do tubo contra um fundo negro com aflashlight. A banda do fago deve ser claramente visível no local da interface entre 1,3 g / ml e 1,5 g / ml de SM / CsCl camadas (Figura 3A). Punção através da parede do tubo ligeiramente abaixo da banda com uma agulha de calibre 21,5 com uma seringa de 3 ml. Recolher suavemente ~ 500 uL da suspensão de fagos. O título do fago deve ser ~ 5-10 x 10 11 pfu / ml.
- Colocar a suspensão de fagos a 4 em ultra-claro ultracentrífuga Beckman ml tubo SW60Ti rotor. Encher o tubo com 1,5 g / ml de SM / solução de CsCl. Preparar um tubo de equilíbrio. Centrifugar numa SW60Ti rotor Beckman a 35.000 rpm durante 24 horas a 4 ° C.
- Repetir o mesmo procedimento como no passo 2.11 a recolher o fago a partir da banda visível. A banda deve ser visível como mostrado na Figura 3B.
- Carregue a suspensão de fagos a uma cassete de membrana de diálise (Tabela 2) e dializar três vezes contra um volume 1000 vezes superior de tampão SM a 4 ° C durante períodos de tempo de 3 horas, 3 horas e overnight (~ 16 horas). O objetivo da diálise é livrar-se de presente CsCl na suspensão de fagos. O título de fagos final deve ser ~ 5-10 x 10 11 pfu / ml.
3. Preparar uma placa de gel de agarose (Figura 4)
- Limpar seis lâminas de microscópio (75 x 50 mm, 1 mm de espessura) com etanol a 70%.
- Dispor as lâminas 5 e fixá-lo com fita adesiva, como mostrado na Figura 4.
- Misturar 0,09 g de agarose em 6 ml de meio num copo pequeno coberto com filme plástico (produzindo agarose 1,5%). Aqueça em uma chapa quente até que a solução se torne clara.
- Despeje a solução de agarose para os slides garantidos.
- Coloque o último slide no topo, evitando cuidadosamente as bolhas de ar. Coloque o peso em cima e deixe arrefecer durante ~ 30 min.
- Remover os quatro lâminas do lado, e envolva a laje em conjunto com as lâminas superior e inferior com filme plástico. A laje pode ser armazenado a 4 ° C durante até 3 dias.
4. Testando o stock de fagos purificados
- Prepararuma laje de gel PBS-agarose como descrito acima (secção 3).
- Manchar o fago purificado com DAPI. Misturar 10 ul de fagos (~ 1 x 10 10 pfu / ml) com 10 ul de 10 ug / ml DAPI (DAPI concentração final de 5 ug / ml), incubar durante 30 min a 4 ° C ou 10 minutos a temperatura ambiente.
- Ul lugar 1 da mistura de fagos / DAPI no centro de uma No.1 24 x 50 mm lamela, sobreposição de um pedaço pequeno (~ 10 x 10 mm) da pré-preparados laje PBS-agarose. O pequeno pedaço de laje de agarose é cortado com uma lâmina de barbear após o deslizamento de topo sobre o gel de sanduíche é removido. Imagem da amostra sob o microscópio de epifluorescência, através dos canais de YFP e DAPI. Fagos individuais, deverá ser visível como difracção limitada fluorescentes "spots" em ambos os canais (Figura 5). Use o mesmo microscópio e configurações da câmera como no Passo 6.2.
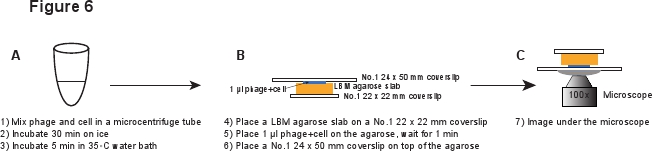
5. Infecção (Figura 6)
- Num tubo Falcon 14 ml, inocular uma colónia fresca de LE392 [pP RE-MCHerry] (ver Tabela 1 para detalhes) em 2 ml de meio LB suplementado com 100 ug / ml de ampicilina, 10 mM de MgSO 4 e 0,2% de maltose. Crescer durante a noite a 37 ° C com agitação moderada (265 rpm).
- Diluiu-se a 1:1000 em cultura LBMM (LB suplementado com 10 mM de MgSO 4 e 0,2% de maltose), ou seja, adicionam-se 5 ul da cultura durante a noite em 5 ml de meio LBMM em um frasco de 50 ml. Crescer a OD 600 ≈ 0,4 a 37 ° C com agitação moderada (265 rpm).
- Use médio LBM para preparar uma laje gel LBM-agarose como descrito na Seção 3 acima.
- Centrifugar 1 ml de células a 2.000 g numa microcentrifuga de bancada durante 2 minutos à temperatura ambiente. Remover o sobrenadante e ressuspender as células suavemente em 20 LBMM gelada ul para chegar a OD 600 & 20.
- Ao manipular o stock de fagos purificados, utilizar uma pipeta de ponta larga ou cortar a ponta da pipeta regular para fazer a ponta de abertura mais amplo, a fim de evitar que as partículas de fagos corte 3. Gentilmente Mix 20 ul de células com 20 ul de fago purificado para atingir uma razão de fagos-a-célula médio na gama de 0,1 - 5. Incubar em gelo durante 30 min para permitir a adsorção do fago e depois incubar em banho de água a 35 ° C durante 5 min para desencadear o fago de DNA de injecção 6.
- Pipetar cima e para baixo algumas vezes para separar qualquer agregados celulares. Novamente utilizar uma ponta de pipeta de largura de corte para evitar os fagos. Dilui-se a mistura em LBMM 01:10, por exemplo, mistura de 5 ul em 45 LBMM ul.
- Coloque um pedaço de LBM-agarose laje (~ 10 x 10 mm) sobre uma lamela de 22 x 22 mm No.1. O pré-preparados laje LBM-agarose deve ser colocada à temperatura ambiente durante pelo menos 1 hora antes de ser utilizado para garantir que a laje de agarose atingir a temperatura ambiente. Ul lugar 1 da mistura de fagos / células na laje de agarose e aguardar 1 minuto para permitir que a mistura de absorver na laje de agarose. Suavemente coloque uma lamela No. 1 24 x 50 mm na parte superior da laje de agarose. Este procedimento destina-se a evitar o corte os fagos a partir da infected célula (Figura 6).
6. Seguindo o destino das células ao microscópio
- Monte cuidadosamente a lamela na fase do microscópio. Para imagiologia, utilize uma objectiva de grande ampliação (por exemplo, 100x) (ver sistema de microscópio em discussão abaixo).
- Adquira uma imagem definida para o período de tempo inicial. Este conjunto de imagem será utilizada para caracterizar os números iniciais e as posições de todos os fagos que infectam. Tomar uma série de 15 imagens em 200 nm eixo Z (vertical) intervalos. Imagem através do canal de YFP. Além disso, o exame de uma imagem em foco único através do contraste de fase e canais mCherry. Optimizar a intensidade da luz e do tempo de exposição para obter um sinal suficientemente minimizando branqueamento e dano celular (ver Aquisição de Imagem em discussão abaixo).
- Adquirir um filme de lapso de tempo do destino da célula pós-infecção. Imagem da amostra em contraste de fase, YFP e mCherrycanais a intervalos de tempo de 10 min a cerca de 4 horas. Durante o filme de lapso de tempo, utilizar uma imagem z posição única por canal por ponto de tempo, a fim de evitar a exposição desnecessária da amostra, o que poderia levar à descoloração e fototoxicidade.
7. A análise de imagem
- Manualmente contar o número de fagos e localização recorde de fago e o comprimento das células no espaço de tempo inicial. Isso pode ser feito usando o software como MetaMorph ou ImageJ. Anote os destinos celulares (lítico, lisogênico ou não infectados), tempo de lise, e qualquer outra informação desejada por reproduzir o filme de lapso de tempo. Para identificar destinos diferentes de células, consulte Time-lapse do filme na seção Resultados Representante abaixo.
- Para além da análise manual acima, mais dados quantitativos (por exemplo, nível de fluorescência ao longo do tempo nas células individuais) pode ser extraído utilizando automatizados célula de reconhecimento e linhagem algoritmos de rastreamento. Nós usamos uma casa construída programa Matlab para tracing a linhagem de célula e os níveis de fluorescência, em conjunto com o código Matlab Schnitzcell para a segmentação das células (escrito por grupo Elowitz Caltech).
8. Os resultados representativos:
Chapeamento Phage:
As placas dos fagos marcadas com fluorescência (em 1,6 passo e Seção 2) são significativamente menores que os do tipo selvagem (Figura 2). Por conseguinte, incubar as placas pelo menos durante 12 horas em 37 ° C para as placas da incubadora a ser visível.
Ultracentrifugação Phage:
Após ultracentrifugação a amostra de fago com o gradiente de CsCl passo (Passo 2.10), duas bandas devem ser visíveis (Figura 3A). A banda superior, na interface entre a suspensão de fagos e SM / CsCl 1,3 camada g / ml, contém fragmentos de células e de fagos cápsides vazias. A banda inferior, na interface entre 1,3 g SM / CsCl / ml e 1,5 g / ml de camadas, é a banda do fago. Thibanda s aparece esverdeado fluorescente para o fago λ LZ2. A banda de tipo selvagem fago λ IG2903 aparece azulada 5. Após a ultracentrifugação de gradiente de equilíbrio de CsCl na Etapa 2.12, uma banda do fago deve ser visível na parte do meio do tubo (Figura 3B). Uma vez que o fago λ LZ2 fluorescente contém uma mistura de gpD-EYFP e cãpsides gpD, a proporção de proteína para o DNA é maior do que o de tipo selvagem. Por conseguinte, a banda do fago λ LZ2 fluorescente é um pouco mais leve (parece estar em uma posição mais elevada dentro do tubo) que o de tipo selvagem λ IG290310.
A coloração DAPI:
A Figura 5 apresenta imagens típicas obtidos após a rotulagem do fago com DAPI (Seção 4). Os sinais YFP e DAPI de um fago com sucesso purificada deve ter cerca de 100% a correspondência. Nós normalmente observam que menos de 1% da mancha YFPs não contêm DAPI (representando cãpsides sem o genoma viral). Menos de 1% dos locais DAPI não contêm YFP (correspondente a não fluorescentes fagos) 5.
Lapso de tempo do filme:
Células líticos são reconhecidos pela acumulação de YFP fluorescência (canal verde) no interior da célula, seguida de lise celular. Células lisogénica são reconhecidos pela acumulação de fluorescência uniforme mCherry (vermelho) no interior da célula e a retoma do crescimento de células normais e de divisão. As células não infectadas (ou células em que a infecção tenha falhado) não apresenta qualquer dos fenótipos acima e vai crescer e dividir-se normalmente. Figura 7 mostra algumas imagens-conjuntos de contraste de fase, YFP e canais mCherry, e as imagens correspondentes sobrepostos destas três canais, a partir de um filme de lapso de tempo típico (Secção 6). Os fagos individuais (pontos verdes) são claramente visíveis no espaço de tempo inicial (Figura 7A). Tipicamente, um númeroOs fagos são vistas de na superfície da célula (presumivelmente infectar essas células) ao passo que outros são os fagos não adsorvido, como mostrado na Figura 7B (painel da esquerda). O resultado da infecção se torna perceptível ao longo do tempo. O ciclo lítico é indicada pela produção intracelular de fagos novos (verde, Figura 7C), seguido de lise celular (células explodidas com fagos libertados verdes, a Figura 7D). Lisogenia é indicada pela produção de mCherry do promotor P RE (vermelho, Figura 7C) e a retoma do crescimento e divisão celular (vermelho, Figura 7D).




Figura 1. gráfico de fluxo que descreve a criação de fagos fluorescentes. Um ligado fágico bruto é primeiro obtido por indução de um lisogénio do fago gpD-EYFP, abrigando um plasmídeo que expressa a proteína do tipo selvagem gpD (painéis AB). O fago é purificado através de uma série de passos (CL painéis).

Figura 2. Placas fágicas. Placas do fago fluorescente (esquerda) são menores do que os do tipo selvagem (direita) após a incubação as placas durante 12 horas a 37 ° C.

Figura 3. Bandas de fagos após ultracentrifugação. D) As duas bandas são visíveis após ultracentrifugação em gradiente de CsCl passo. A de cima corresponde a fragmentos de células e de fagos cápsides vazias, a banda inferior contém o fago desejado. Esquerda: fago fluorescente, a direita:. Selvagem B ) A banda do fago único é visível após ultracentrifugação em gradiente de equilíbrio de CsCl. A banda do fago fluorescente (à esquerda) é esverdeado, em comparação com uma banda azulado para fagos do tipo selvagem (direita).

Figura 4. O procedimento de preparação de placas de gel de agarose.

Figura 5. Imagens fluorescentes de fagos após coloração com DAPI. Fagos individuais são facilmente distinguíveis, e YFP e DAPI sinais co-localizar muito bem.

Figura 6. Descrição esquemática de infecções de fagos e configuração de imagem. Clique aqui para ver uma versão em tamanho dessa imagem.
{kind=link}
gura 7 "src =" files/ftp_upload/3363/3363fig7.jpg / "/>
Figura 7. Imagens típicas de um filme de lapso de tempo de infecção do fago. São mostrados o contraste de fase, YFP e canais mCherry, bem como uma sobreposição dos três canais. (A) YFP canais imagens do prazo inicial. Esquerda, a soma das imagens YFP em diferentes posições z-. As três imagens direita são exemplos de imagens YFP em diferentes posições z-, correspondentes a diferentes áreas da superfície da célula. (B), (C) e (D) imagens sobrepostos (esquerda) da de contraste de fase (meia-esquerda), YFP (meio-direita) e mCherry (direita) canais em diferentes intervalos de tempo. (B) Em t = 0, duas células são vistos, cada infectadas por um fago único (pontos verdes), e uma célula é infectada por três fagos. Também observou que alguns fagos não adsorvido. (C) Em t = 80 min, as duas células infectadas por fagos individuais têm cada um ido para a via lítica, como indicamd pela produção intracelular de fagos novos (verde). A célula infectada por três fagos passou para a via lítica, como indicado pela produção de mCherry do promotor PRE (vermelho). (D) Em t = 2 horas, a via lítica resultou em lise das células (célula explodida), enquanto que a célula lisogénica dividiu §.
§ painéis esquerdo da Figura 7 (C) e (D) são reproduzidos a partir de celular, 141, Lanying Zeng, Samuel O. Skinner, Chenghang Zong, Jean Sippy, Michael Feiss, e Ido Golding, tomada de decisão em um nível subcelular determina o resultado de Infecção Bacteriófago, 682-691, Copyright (2010), com a permissão da Elsevier.
| Coe nome | Genótipo relevante | Fonte / referência |
| Cepas bacterianas | ||
| LE392 | cearF | John Cronan, da Universidade de Illinois |
| Cepas fago | ||
| λ LZ1 | GPD-EYFP, cl857 SAM7 D-eyfp b :: kanR | Zeng et al. 5 |
| λ LZ2 | gpD mosaico genótipo, mesmo que λ LZ1 | Zeng et al. 5 |
| Plasmídeos | ||
| pP RE - mCherry | mCherry sob o controlo de P RE, amp R | Zeng et al. 5 |
| pPLate * D | gpD sob o controlo do promotor tardio λ, amp R | Zeng et al. 5 |
Tabela 1. Estirpes bacterianas,fagos e plasmídeos usados neste trabalho.
| Ρ Densidade (g / ml) | CsCl (g) | SM (ml) | Índice de refração η |
| 1,30 | 39 | 86 | 1,3625 |
| 1,50 | 67 | 82 | 1,3815 |
| 1,70 | 95 | 75 | 1,3990 |
Tabela 3. Soluções de CsCl preparado em tampão SM (100 ml) por passos de gradientes.
Discussão
Estirpes bacterianas, fagos e plasmídeos:
Strain LE392 é supF. Optou-se por suprimir a mutação SAM7 no genoma de fago (ver Tabela 1 para detalhes). Assim, lisogénios induzidas acabará por lise e liberar partículas de fagos, células infectadas como a vontade de que escolheram a via lítica. Células lisogénica são cultivadas a 30 ° C devido à presença do cl 857 sensível à temperatura alelo no genoma do fago. Após a indução de calor, gpD-EYFP e do tipo selvagem gpD são co-expressas a partir do genoma de λ LZ1 eo plasmídeo pPlate * D, respectivamente. Como resultado, a cápside do fago λ recém-criado LZ2 contém uma mistura de gpD-EYFP e proteínas gpd. Este fago mosaico é estruturalmente estável e suficientemente fluorescente para permitir a detecção de fagos individuais 5. pP RE - mCherry é um plasmídeo repórter utilizada para detectar escolha do pathwa lisogénicay. O promotor RE P é ativada pela CII durante o estabelecimento de lisogenia 1,11. pP RE - mCherry 5 foi derivado de pE-gfp 11 substituindo gfp com mCherry 12. Para mais detalhes veja o nosso trabalho anterior 5.
Condição Parâmetros de crescimento:
Durante a indução lisogénio (Seção 1), tremor leve a 180 rpm dá um rendimento bom vírus 13. O uso de glucose no meio de crescimento deve ser evitada, o metabolismo da glicose gera produtos metabólicos acídicos, e as partículas de lambda maduros são instáveis a pH ácido 13. A adição de MgSO 4 destina-se a estabilizar a cápside do fago 3. Para fagos carregando tipo selvagem cI (em vez de cI 857), o lisogénio pode ser induzida usando o C-DNA prejudicial Mitomicina agente 3. No passo 1.3, a incubação a 37 ° C não deve exceder normalmente 90 minutos. É usef ul para verificar a densidade celular por OD 600 a cada 30 min. Para uma boa lisado, OD 600 cai para cerca de 0,2 ou menos, e o restante OD 600 é um resultado de detritos de células. Incubação demasiado longo pode resultar num menor rendimento de fago uma vez que o fago pode começar a recentemente criada para injectar o DNA em fragmentos de células. Para se obter uma banda de fago visível (de pelo menos 1 x 10 11 partículas fágicas) nos passos 2.11 e 2.13, crescem, pelo menos, 500 ml de cultura na etapa 1.2. A adição de maltose a 0,2% para o meio de crescimento nas etapas 5.1 e 5.2 é destinado a induzir a expressão de LamB, o receptor para o fago lambda adsorção 3,14. A diluição de 1000 vezes, em vez de 100 vezes no passo 5.2 é destinado a reduzir o nível de fundo a partir do RE mCherry repórter plasmídeo pP - mCherry. No passo 5.5 para injecção de DNA de fago de disparo, a 35 ° C é escolhido para evitar a indução do alelo sensível à temperatura cI857.
Purificação Phage:
jove_content "> Os passos de purificação de fagos (Passos 2.1 a 2.11) pode ser substituído por outros protocolos de purificação 5, mas a ultracentrifugação de gradiente de equilíbrio final através de CsCl (Passos 2.12 e 2.13), é inevitável. rotores de caçamba móvel são necessários nos passos 2.10 e 2,12 para garantir afiadas bandas fago visíveis. Obtenção de um estoque fago puro pode facilmente levar até uma semana, por isso é necessário verificar o título de fagos ao longo do caminho para garantir que nada dê errado durante as etapas intermediárias.Manuseio Phage:
Durante todos os procedimentos de purificação na Seção 2, é fundamental para lidar com fago lisado com cuidado para evitar corte caudas de fago de cabeças de fagos. Durante a infecção de células na Secção 5 (por exemplo, as etapas de 5,5 através de 5.7), é também crítica para evitar a ruptura das partículas de fago a partir da célula infectada. Note-se que, se o fago é cortado a partir da célula infectada, após injecção de seu DNA, o resultado é uma infecção "escuro", ou seja, a defection resultado será observado na experiência, mas o fago infectando não. Para minimizar esses problemas, usamos uma ponteira larga sempre que manuseia fagos ou a mistura fago / célula.
DAPI Teste:
Manchando o estoque fago com DAPI (Seção 4) é um método rápido e eficiente para controlar a pureza do estoque de fagos. Ele também pode ser usado para testar a possível degradação de um estoque de fago existente ao longo do tempo. Para um estoque puro, a co-localização de sinais YFP e DAPI sob o microscópio de fluorescência deveria ser próximo de 100%. Nós normalmente observam que menos de 1% dos locais YFP não contêm DAPI (representando cãpsides sem o genoma viral), o que indica que estas partículas não êxito empacotar o DNA viral ou já tinham injectado o seu DNA em outro lugar. Menos de 1% dos locais DAPI não contêm YFP (correspondente a fagos não fluorescentes). Se este não for o caso, as etapas de 2,12 a 2,14 necessidade de ser repetido no order para purificar novamente. Com relação aos parâmetros de imagem, a configuração do microscópio no Passo 4.3 não é tão crítica como no capítulo 5, porque não a longo prazo imagem ao vivo de células é necessária aqui. No entanto, mantendo os ajustes de microscopia mesmas como no ponto 5 é útil quando se deseja calibrar a intensidade de fluorescência de uma única partícula fágica. Se a placa de PBS-agarose não é muito limpa, ou muito DAPI corante é usado, alguns pontos DAPI correspondentes ao ADN do fago pode ser rodeada por um "halo". Se for muito pouco corante DAPI é usado, o sinal do canal DAPI pode ser muito fraca.
Sistema de microscópio:
Para a imagem na seção 6, nós usamos um microscópio de epifluorescência comercial invertido (Eclipse TE2000-E, Nikon) com um objetivo 100x (Plano Fluo, abertura numérica 1,40, imersão em óleo) e padrão de conjuntos de filtros (Nikon). A fonte de luz de fluorescência é uma lâmpada de arco com o controlo da intensidade da luz. Os seguintes recursos são controlados por computador: x, y, z posição; campo brilhante e persianas de fluorescência, e opção de filtro de fluorescência. Um recurso de auto-foco é necessária. Caso contrário, o foco pode facilmente afastar-se durante o filme de lapso de tempo (normalmente 4 horas de duração). A capacidade de adquirir várias posições (x, y) em cada ponto de tempo é útil, uma vez que permite acompanhar a eventos de infecções múltiplas em paralelo. Nós normalmente adquirir 8 posições de palco em cada filme, seguindo até 100 ocorrências de infecção. A câmera que usamos é um resfriado 512x512 CCD com 16x16 pixel câmera mM com uma gama dinâmica de 16 bits (Cascade512, Fotometria). Aquisição é realizada usando software MetaMorph (Molecular Devices). O microscópio deve ser colocado numa sala com temperatura controlada, em alternativa, a platina do microscópio deve ser rodeada por uma câmara de temperatura controlada.
Aquisição da Imagem:
Para vivo de células de imagem, que é crítico para evitar a exposição desnecessária da amostra, o que poderia levar à descoloração e phototoxicity. Portanto, o melhor é primeiro caracterizar seu sistema para encontrar uma exposição de luz ideal que permite a detecção de fluorescência, enquanto não levando ao crescimento excessivo de células de branqueamento ou de inibição. Para obter uma boa imagem de fluorescência, jogar com a intensidade de luz emocionante, tempo de exposição e ganho de câmera. Nos Passos 6,2-6,3, o intervalo de quadro 10 min é escolhido com o fim de minimizar a exposição à luz. Em cada quadro, apenas uma imagem em foco único é necessário no contraste de fase (de reconhecimento de célula) e os canais de fluorescência (para determinar o destino da célula). No primeiro momento, no entanto, várias imagens z-posição através do canal de YFP são necessárias para capturar todos os fagos que infectam na superfície da célula. O tempo de exposição YFP no quadro inicial também pode precisar de ser maior do que o utilizado para o filme de lapso de tempo nos períodos de tempo posteriores.
Análise de Imagem:
Muito cuidadosamente contar partículas fágicas em torno da superfície da célula na etapa 7.1. Comomencionado acima, tomamos uma série de z-pilhas através YFP canal no Passo 6.2. No entanto, isso ainda pode deixar algumas partículas de fagos fluorescentes fora de foco, que desafia a contagem. O comprimento das células no espaço de tempo inicial é medida usando o software Metamorph. O comprimento da célula pode também ser medida por ImageJ ou outras ferramentas de software. Além disso, um sistema automatizado de casas construídas programa Matlab pode ser muito útil na obtenção de informações, tais como a mudança de fluorescência ao longo do tempo ao longo de linhagens celulares.
Divulgações
Não há conflitos de interesse declarados.
Agradecimentos
Somos gratos a Michael Feiss e Sippy Jean para a orientação sobre a criação de fagos e purificação. Agradecemos Michael Elowitz para fornecer o software de reconhecimento de célula, Schnitzcell. Trabalho no laboratório de Golding é suportado por subvenções dos Institutos Nacionais de Saúde (R01GM082837), a National Science Foundation (082,265, PFC: Centro de Física de Células Vivas), a Fundação Welch (Grant Q-1759) e Ciências Humanas da Fronteira Programa (RGY 70/2008).
Materiais
| Name | Company | Catalog Number | Comments |
| Nome do reagente | Companhia | Número de catálogo | |
| Clorofórmio | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma | D4527-10KU | |
| RNase | Sigma | R4642-10mg | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| Tampão SM | Teknova | S0249 | |
| NZYM | Teknova | N2062 | |
| CsCl | Sigma | C3011-250G | |
| Seringa | Becton Dickinson | 309585 | |
| Agulha | Becton Dickinson | 305176 | |
| Cassete de diálise | Thermo Scientific | 66333 | |
| Lâmina de microscópio | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti tubo ultra-claro | Beckman Coulter | 344060 | |
| SW60Ti tubo ultra-claro | Beckman Coulter | 344062 | |
| SW40Ti rotor | Beckman Coulter | 331302 | |
| SW60Ti rotor | Beckman Coulter | 335649 | |
| Refratômetro | Fisher Scientific | 13-947 | |
| Microscópio de epifluorescência | Nikon | Eclipse TE2000-E |
Tabela 2. Reagentes e equipamentos.
Referências
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 39, 409-429 (2005).
- Ptashne, M. . A genetic switch : phage lambda revisited. , (2004).
- Hendrix, R. W. . Lambda II. , (1983).
- Hershey, A. D. . The Bacteriophage lambda. , (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. . Current protocols in molecular biology. , (1994).
- Sambrook, J., Russell, D. W. . Molecular cloning : a laboratory manual. , (2001).
- Fasman, G. D. . Practical handbook of biochemistry and molecular biology. , (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Zeng, L. . Personal communication with M. Feiss. , .
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados