
Method Article
Um sangue com base em teste para a detecção de ROS1 e RET transcrições de fusão do ácido ribonucleico, usando a reação em cadeia da polimerase Digital de circulação
Neste Artigo
Resumo
Detecção de ácido ribonucleico (cRNA) do sangue que circula é uma necessidade insatisfeita em diagnósticos clínicos. Aqui descrevemos os métodos que caracterizam cRNA de pacientes de câncer de pulmão não-pequenas células usando a reação em cadeia da polimerase digital sensível e específico. Os testes de requisitos de projeto para detectar variantes de fusão dentro de 72 horas.
Resumo
Desenvolvemos novos métodos para o isolamento e caracterização de derivados de tumor circulante ácido ribonucleico (cRNA) para biópsia líquido à base de sangue. Deteção robusta de cRNA recuperado de sangue constitui uma solução para uma necessidade insatisfeita crítica em diagnósticos clínicos. O teste começa com a coleta de sangue total em tubos de colheita de sangue contendo conservantes que estabilizam cRNA. Sem célula, exosomal e RNA plaquetas associada é isolada do plasma neste sistema de teste. O cRNA é reverso transcrito para DNA complementar (cDNA) e amplificado usando a reação em cadeia da polimerase digital (dPCR). As amostras são avaliadas para o biomarcador de destino, bem como um gene de controle. Validação de teste incluído o limite de detecção, precisão e os estudos de robustez com amostras analíticas. O método desenvolvido como resultado destes estudos reproducibly detectar diversas variantes de fusão para ROS1 (proto-oncogene C-Ros variantes 1; 8) e RET (reorganizados durante proto-oncogene transfeccao; 8 variantes). O fluxo de trabalho do processamento de amostra foi otimizado para que resultados consistentemente podem ser gerados dentro de 72 horas da data de recepção da amostra.
Introdução
Acima de 25% de câncer de pulmão não-pequenas células (NSCLC) pacientes podem não ter tecido suficiente disponível para testes no momento do diagnóstico. Mesmo em casos onde o tecido está disponível, não pode ser de suficiente quantidade ou qualidade para executar recomendado testes moleculares1,2. Em casos onde não há tecido suficiente de uma biópsia para o perfilamento molecular, os pacientes podem ter esperar várias semanas ou mais resultados, ou iniciar o tratamento sem resultados moleculares3,4. No entanto, é essencial que o diagnóstico molecular informativo esteja disponível dado o advento de várias opções de tratamento direcionados para pacientes com NSCLC. Teste de DNA de célula livre circulação (cfDNA) da biópsia líquida é uma solução para os desafios de tecido tradicional teste4,5,6. Opções de testes atuais de mutações acionáveis em NSCLC usando cfDNA e um fluxo de trabalho baseado em dPCR semelhante para geração de resultado rápido, incluem o receptor do fator de crescimento epidérmico (EGFR) sensibilizante mutações ΔE746-A750 e L858R, mutação de resistência do EGFR T790M , Variantes de KRAS Proto-Oncogene (KRAS) e B-Raf Proto-Oncogene (BRAF) variante V600E. Embora não como amplamente adotado pelo setor, circular o tumor derivado de RNA mensageiro (mRNA) isoladas de biópsia líquida também pode fornecer importantes informações clínicas7,8,9. Anteriormente desenvolvemos e informou sobre métodos de detecção multiplexado das Echinoderm Microtubule associado proteína como 4-anaplásico linfoma do Receptor tirosina quinase (EML4-ALK) fusão variantes do plasma de sangue10. Neste estudo, nós estendemos esses métodos para incluir metas de RNA multiplexadas ordem superior para ROS1 e RET, abrangendo oito variantes de fusão dentro de cada ensaio. O objetivo foi desenvolver uma técnica rápida, sensível, específica e reprodutível para a detecção dessas variantes da fusão do plasma de pacientes previamente diagnosticados com NSCLC.
O processo de teste é iniciado em um consultório médico usando o RNA estabilização de tubos de coleta de sangue11. Estes tubos contêm uma célula conservante assim como inibidores de RNase. As amostras são prioridade enviada durante a noite para centralizado faculdade de CAP de patologistas americanos credenciados/clínicos laboratoriais melhoria alterações (CLIA)-certificado de laboratório (laboratório clínico) para processamento por pessoal competente. Uma vez recebida pelo laboratório clínico, cada etapa do processamento é conduzida sob aprovados procedimentos operacionais padrão (SOP). Sangue total é centrifugada para recuperar o plasma, que é usado para isolar o RNA que seja de circulação livre no sangue ou em partes, tais como exosomes e plaquetas7,8,9de encapsulamento. Para isolar o RNA desses compartimentos, nós selecionamos o sistema para recuperação de RNA baseada em comparações de vários métodos de extração. O RNA isolado é concentrada e reverso transcrito de cDNA. Várias enzimas transcriptase reversa e gene-específico primers foram avaliados durante a otimização do método de síntese de cDNA para maximizar ROS1 e RET alvo transcrição conversão10. Isto é crítico para baixa abundância transcrições, como variantes de fusão tumor derivado de circulação. Finalmente, nós aperfeiçoamos dPCR concentrações primer e sonda para permitir detecção multiplexada de RET ou ROS1 variantes de fusão e o gene de controle, glucuronidase-β (GUSB). Então, nós combinamos as melhores condições de cada um dos estudos de otimização em um protocolo final fechado antes de realizar os estudos de validação analítica descritos neste relatório. Este protocolo e estes resultados fornecem a base para um fluxo de trabalho rápido e sensível para a rotina detecção de variantes de fusão rara em circulação.
Protocolo
As instruções dos fabricantes são seguidas para os reagentes listados abaixo, a menos que caso contrário descrito. Os ensaios PCR são comercialmente disponíveis produtos projetados para detectar fusões ROS1 e RET.
1. trabalhar com o RNA em preparação para a transcrição reversa (RT)-dPCR: as melhores práticas de laboratório
- Crie um ambiente livre de RNase ao trabalhar com o RNA.
- Use pulverizadores comercialmente disponíveis projetados para inactivate RNases contaminante.
- Uso certificado livre de RNase reagentes, dicas e tubos. Use dicas de barreira para pipetas para impedir a introdução de RNases ou contaminação de amostras.
- Sempre use um casaco de laboratório para impedir que partículas caindo do vestuário em sua amostra. Designe um laboratório específico de casaco para usar com o processamento do RNA.
- Use luvas para evitar contaminação da amostra de RNases na pele. Luvas mudam com frequência.
Nota: Assumir as superfícies de laboratório estão contaminadas com RNase desde que eles são expostos ao ambiente. As luvas que entre em contato com a pele, cabelo, maçanetas, puxadores de congelador, canetas/marcadores, etc. são consideradas já não RNase-livre. - Descontaminar as pipetas, benchtops do, centrifugadores e outras superfícies de trabalho com um RNase inactivação pulverizador antes da utilização.
- Se possível, manter um conjunto de equipamentos para uso com RNA.
- Minimize a interrupção do fluxo de ar nas áreas de laboratório quando se trabalha com amostras do RNA para impedir que partículas caiam em amostras ou contaminar a área de trabalho.
- Loja purificado RNA no-80 ˚ c.
- Evite congelar-derreta múltiplas de amostras do RNA, pois isso pode causar a degradação.
2. geração de Material de RNA analítica para o controlo positivo
- O projeto DNA sintético usando sequências de mRNA publicado por variantes de fusão de interesse10.
- Para uma variante de fusão determinado, selecione uma sequência de fusão de mRNA que inclui a fusão local mais comprimento suficiente de acompanhamento de cada lado para cobrir o PCR amplicons.
- Selecione sequências nucleotídicas entre 50-250 nt para imitar o tamanho do RNA capturado usando plasma enriquecido em plaquetas de circulação.
- Adicionar uma sequência de promotor T7 (5'-CAGAGATGCATAATACGACTCACTATAGGGAGA-3') à extremidade 5' da sequência alvo.
- Ordenar sequências sintéticas como fragmentos de ácido desoxirribonucleico de encalhado dobro (DNA).
- Reconstitua o DNA sintético em tampão Tris-EDTA (TE) a uma concentração final de 10 ng / µ l.
- Converta 60 ng DNA sintético para RNA usando a transcrição em vitro .
- Purifica as transcrições de RNA usando o reagente fenol/guanidina-com base em12.
- Incluem a DNase eu, RNase-livre para remover o modelo residual DNA.
- Medir a concentração de purificada em vitro RNA usando um dados comercialmente disponíveis com padrões e corantes de RNA específicas. Certifique-se do que RNA está dentro do intervalo aceitável para os padrões escolhidos. Diluição pode ser necessária.
- Confirme a transcrição bem sucedida pela electroforese do gel usando um gel de agarose 2% misturado com RNA gel mancha e uma gama alta escada de RNA, incluindo a faixa de tamanho de nt 50-250.
- Carga 500 ng de cada em vitro RNA em um gel.
- Funcione o gel a 5 V/cm.
- Visualizar bandas única usando iluminação e documentar os resultados.
- Confirme tamanho transcrição esperados para cada uma das variantes fusão (baseados no projeto na etapa 2.1.2).
- Confirmar a deteção de cada em vitro RNA por RT-dPCR usando o ensaio PCR variante específica correspondente (veja as etapas 5 a 8 do presente protocolo).
- Opcional: Prepare uma mistura equimolar em vitro RNA que contém cada uma das variantes a fusão e o gene de controle GUSB.
- Se passo 2.9 é executada: confirmar a deteção de cada uma das variantes fusão incluídos na mistura de controle por dPCR utilizando ensaios PCR de variante específica (consulte as etapas 5 a 8 do presente protocolo).
- Determine a concentração desejada de entrada para controles analíticos positivos através do teste de concentrações que variam de 0,25 a 2,5 fg10. Escolha a concentração, com base na saída número de cópia desejada.
- Após confirmação, prepare alíquotas de uso único 10 µ l de RNA analítica para uso no controle positivo (etapa 4.4) e loja-80 ˚ c.
3. doador espécimes
- Colete as amostras de sangue total humano 10 mL em tubos de colheita de sangue de 10 mL (BCT) que contém um conservante de RNA celular livre.
Nota: Todos os doadores humanos devem consentir a utilização de pesquisa e nenhuma informação de identificação do doador específicos será coletada ou usada durante o teste. - Processar amostras de sangue total, dentro do prazo especificado pelo fabricante do BCT.
- Pool de plasma humano normal pode ser comprado de uma fonte comercial para uso dentro do controle positivo analítico. Prepare-se single-use, alíquotas de 1 mL do pool de plasma humano normal e loja-80 ˚ c para uso com o controle positivo (etapa 4.4).
4. recuperação de RNA do Plasma que circula
Nota: É importante trabalhar rapidamente durante este procedimento.
- Centrifugar tubos de sangue total a 200 x g por 20 min.
- Colete de 4 mL de plasma de tubo de coleta de sangue centrifugado com uma pipeta sorológica. Tenha cuidado para não perturbar ou aspirar a camada de revestimento buffy.
- Isole a circulação do RNA usando um kit comercialmente disponível que pode capturar exosomes, RNA sem célula de plasma e plaquetas. Isole o RNA da amostra de controlo positivo ao lado de cada lote.
-
Prepare o controlo positivo para cada lote de amostras clínicas, como segue:
- Descongele 1 mL pool de plasma humano normal alíquota (passo 3.3).
- Descongele a 10 µ l analítica RNA alíquota (passo 2.12).
- Prepare o controle positivo, adicionando 10 µ l RNA analítica para a amostra de plasma humano normal uma vez que o etanol foi adicionado ao plasma lisado.
- Eluir amostras com água de nuclease livre 100 µ l. Proceder imediatamente com RNA limpar e concentração.
- Amostras podem ser armazenadas no gelo molhado e cobertas, por até uma hora.
- Concentre-se RNA usando o método de coluna com base e eluir em 9 µ l de água livre de RNase.
- Proceder de imediato à etapa 5, ou manter as amostras no gelo molhado por até uma hora.
5. inverter a transcrição do RNA de cDNA
- Converta concentrado circulante amostra de RNA em cDNA usando um kit de reação de transcrição reversa comercialmente disponíveis, incluindo primers aleatórios (ver tabela 1 para componentes).
Nota: Gene primers específicos são opcionais e podem ser projetados para variantes do teste. As primeiras demão são projetadas com base no destino de sequência de RNA. Use sequências de variante de fusão de passo 2.1.- Incluir nenhuma amostra de controle de transcriptase reversa e nenhuma amostra de controle de RNA (ver tabela 1).
- Isole o cDNA da reação de transcrição reversa usando uma coluna de rotação de concentrador comercialmente disponível do DNA.
Nota: Este passo facilita a remoção de enzimas, as primeiras demão e livre deoxynucleotide trifosfatos (dNTPs). - Use o cDNA imediatamente em reações de PCR ou armazenar no-80 ˚ c.
6. digital PCR
Nota: Este PCR é específico para gotícula PCR digital (ver Tabela de materiais).
-
Precauções de mistura PCR.
- Uso uma descartável bata e nitrilo luvas.
- Uso do PCR misturar os reagentes em uma área de preparação de reagente dedicado. Não manuseie o cDNA na área de preparação de reagente-somente.
- Cobrir sondas enquanto trabalhava para protegê-los da luz. Luz em excesso pode foto descorar o corante fluorescente ligado à sonda.
- Transporte de misturas, cobertas e protegido da luz, em uma área de Pre-amplificação separada antes do cDNA é a ser adicionado.
- Adicionar o cDNA a ser testado para mistura PCR em uma capa PCR limpa localizado em área de Pre-amplificação.
- Prepare a mistura PCR para um volume final de reação de 20 µ l de acordo com a tabela 2.
- Distribua a mistura PCR + cDNA para placas PCR.
Nota: Utilização de um layout de placa como um guia é recomendado. - Cubra o prato com um aferidor de placa removível.
- Centrifugar as placas brevemente para coletar as amostras no fundo dos poços.
- Mix agitador de placa em um baixo ajuste para 10 s.
- Centrifugar as placas brevemente para coletar a amostra no fundo do poço.
- Remova o aferidor de placa. Execute a geração de gotículas para PCR-cDNA mistura com qualquer um sistema de geração de gotículas manual ou automatizada.
- Para a geração de gotículas manual, transferi mistura PCR 20 µ l para poços de amostra no cartucho de geração de gotículas. Adicione 70 µ l de óleo de geração da gota. Cobrir com cartucho de borracha gaxeta e transferência para gerador manual da gota para iniciar a geração de gotículas. Após a geração de gotículas, transferi as gotas para um prato fresco do PCR usando dicas recomendadas pelo fabricante. Aspirar e dispensar as gotas lentamente, ao longo de 5 a 6 s cada um, sem tocar a abertura da ponta para o cartucho de gotículas ou placa.
- Para a geração automatizada da gota, sele a placa com um selo de alumínio e a transferência para o gerador da gota. Certifique-se de todas as dicas, cartuchos, e as placas estão no lugar antes de iniciar a geração de gotículas.
- Seguinte geração de gotículas e transferência de gotas para uma placa PCR fresca, selar com um aferidor de chapa de alumínio e placas de ciclo térmico usando as configurações na tabela 3.
- Após o termociclador run é completa, leia a placa usando um leitor de gotículas. Criar um layout de placa para software de leitor que identifica a localização dos controles, amostras, etc.e carga em software para começar a leitura.
7. dados análise e revisão e geração de resultados
- Analise a placa lê os resultados usando o software disponível comercialmente.
- Navegue ao menu Analyze para ver terrenos bidimensional (2D) de amplitude.
- Avalie a qualidade geral dos dados, examinando os dados da gota.
- Avalie dados para números de evento aceito total usando o menu de eventos. Se houver menos de 10.000 eventos por bem, avalie cuidadosamente os dados para problemas adicionais.
- Verificar dados para amplitudes de fluorescência aberrante. Diferenças de amplitude significativa e concentração entre amostras indicam má manipulação ou mistura de amostras.
- Fazer anotações de clusters da gota com testes padrões de pulverizador em um eixo de 45 graus, que é indicativo de gotículas de má qualidade ou amostras problemáticas.
- Examine os dados de controle positivo, No reverso do Transcriptase (RT n) e controle de RNA n (NRC) primeiro. Selecione todas as amostras de controle e examinar a qualidade aglomerado por parcela 2D. Para adequada limiarização, uma clara separação entre clusters de gota deve ser aparente.
- Para cada variante de ensaio, defina o limiar baseado em controle de wells.
- Definidos limiares em parcelas 2D usando a ferramenta de mira para separar a população de gota dupla negativa a população de gene de controle (rotulado com sonda phosphoramidite 5'-hexacloro-fluoresceína-CE), eixo y e população de gene variante, se apresentam ( marcado com fluoresceína amidite sonda de 6-carboxyfluorescein OR), eixo x.
- Soma de cópias de cada poço replicar para uma única amostra.
- Expresse os resultados de teste como o número de cópias variantes detectada.
Nota: Para determinar o valor de corte analítico para chamar uma amostra positiva ou negativa, executar um normal doador saudável amostra definida (pelo menos 10 amostras individuais) com o processo finalizado e estabelecer o corte acima de qualquer sinal detectável de plano de fundo para a mutação de interesse. Além disso, estabelece o número de cópias de gene de controle necessárias para chamar um resultado positivo ou negativo. Este corte de gene de controle funciona como um controle interno de qualidade (QC) para avaliar a quantidade e a qualidade de cada amostra de RNA que é processada.
8. verificação das condições de reação de RT-dPCR usando a linha de celular (opcional)
- Para verificar se a deteção das variantes de fusão, usar comercialmente disponíveis-linhas de células expressando a fusão ROS1 ou RET mRNA do interesse. Proceda do seguinte modo:
- Homogeneizar as células congelado em uma solução baseada em guanidínio lise diretamente do estado congelado. Até breve descongelar antes da homogeneização pode causar a perda e degradação de RNA.
- Isole o RNA usando colunas de rotação de sílica-membrana projetadas para o RNA.
- Medir a concentração de amostras do RNA usando um dados com padrões e reagentes de RNA específicas.
- Dilua o RNA isolado em um fundo do selvagem-tipo RNA do plasma ou outra fonte comercial.
- Realizar as etapas de transcrição reversa do RNA do cDNA, PCR Digital e análise de dados e revisão, e geração de resultados listados neste protocolo para confirmar a deteção da variante desejada.
Resultados
Este protocolo descreve um sistema de teste desenvolvido para a detecção de variantes de fusão de RNA para uso na medição de mutações de motorista dentro do plasma de pacientes NSCLC (Figura 1A). Fusão mRNA produtos da expressão das rearranjos RET e ROS1 mais comuns na população de NSCLC foram identificados13,14,15,16,17. Ensaios PCR multiplexados então foram projetados para detectar as oito variantes mais comuns de transcrição para cada destino em NSCLC dentro de uma única reação. As translocações mais comuns para o locus ROS1 geram associações com as porções 5' do CD74, SDC4, SLC34A2, EZR ou genes TPM3 (Figura 1B). As translocações mais comuns para o locus RET levam a justaposição com KIF5B, para os quais o ensaio abrange seis junções exon. Parceiros RET adicionais que são cobertos incluem aqueles com CCDC6 e TRIM33 (Figura 1C). No total, os ensaios cobrem cerca de 88% da ROS1 e 99% das alterações de RET conhecidas para ocorrer na população de pacientes de NSCLC17.
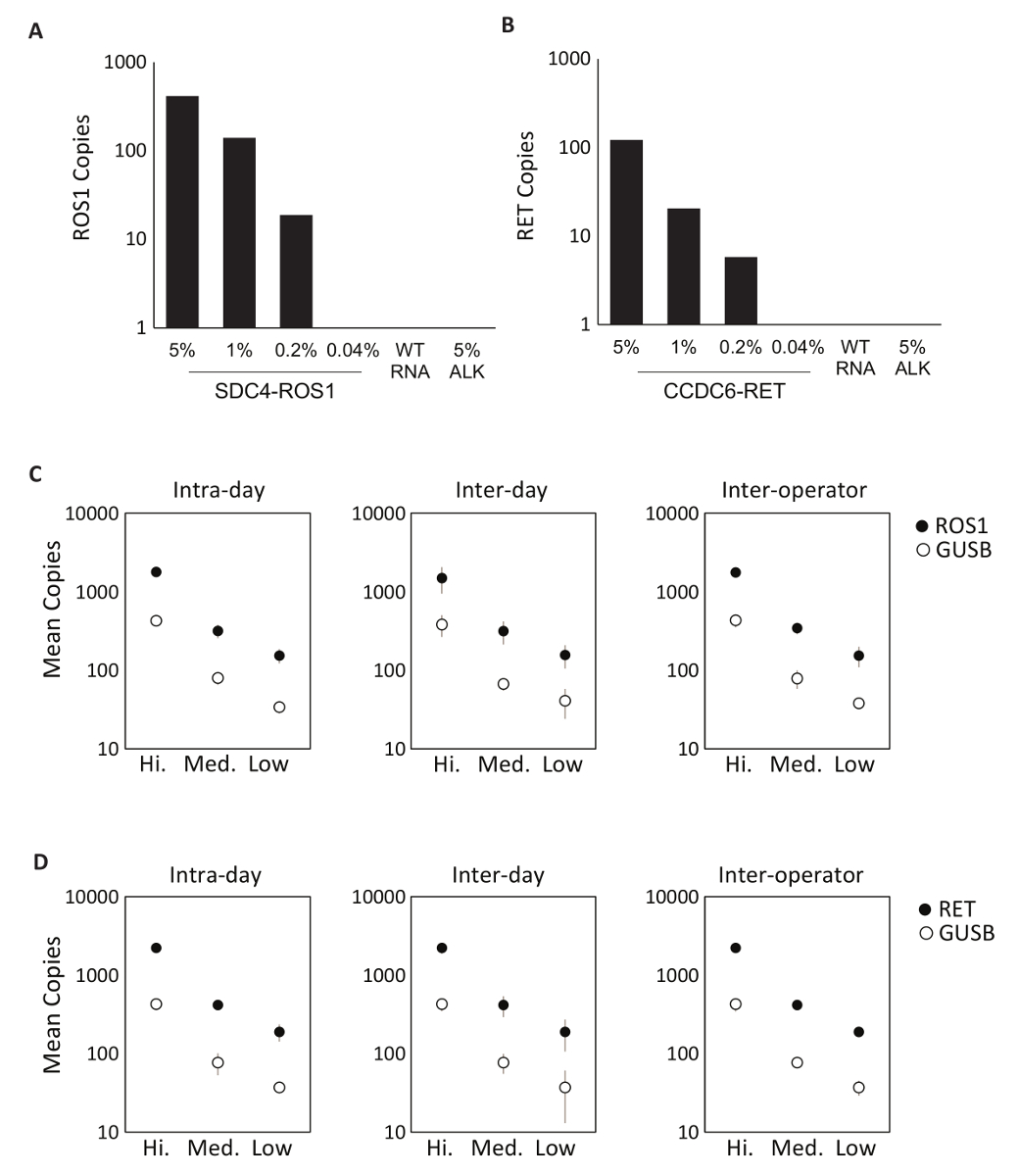
A especificidade dos componentes do ensaio primeiro foi avaliada usando oito individuais em vitro RNAs que contêm a sequência do mRNA para as transcrições de fusão abrangidas pelo ROS1 ou RET multiplexagem ensaios. Cada espécie de RNA foi testado contra cada variante do ensaio individual que compreende a versão multiplexada. Não havia nenhuma reatividade cruzada destes ensaios, demonstrando assim 100% especificidade analítica dentro do projetado multiplexado ensaios (dados não mostrados). Para determinar o limite inferior de detecção do protocolo teste, RNA total derivado de linhagens celulares expressando uma variante de fusão incluída no ensaio foram misturadas em um fundo de RNA normal em concentrações de 5%, 1%, 0,2% e 0,04%. Os multiplexado RET ROS1 variante PCR ensaios e detectados tão pouco como variante de fusão de 0,2% (Figura 2A-B). Além disso, uma preparação de 5% fora do alvo linhagem celular derivada RNA (expressando uma transcrição de fusão EML4-ALK) não foi detectado com os ensaios ROS1 e RET multiplexados, demonstrando ainda mais a especificidade (Figura 2A-B).
Foram realizados testes de precisão do processo RT-dPCR para ambos ROS1 e RET Analytic controle material composto equimolar em vitro RNAs foi processado em três concentrações (baixa, média e alta) através da transcrição reversa e dPCR em três ocasiões diferentes dentro do mesmo dia (intradia), em três dias consecutivos (inter dia) e com dois operadores (inter operador). Resultados dos testes de precisão demonstraram a detecção precisa da transcrição de fusão de interesse, bem como um gene de controle, GUSB, que é incluído como uma métrica QC interna (Figura 2-D).
Além do controle interno GUSB, cada lote de amostras clínicas foi executado com um conjunto de controles de lote. Um controle positivo foi desenvolvido de uma mistura de analítica em vitro RNA que cada uma das variantes fusão testados em RT-dPCR representado, bem como analítica em vitro RNA para GUSB. Este RNA foi cravado em plasma humano normal lisado durante a extração do RNA e foi processada juntamente com as amostras clínicas em todo o protocolo. O não controle da transcriptase reversa (RT n) era um controlo negativo para confirmar a ausência de material contaminante no fluxo de trabalho de extração do RNA e demonstrar a especificidade dos primers de RNA. O controle de RT não foi gerado usando o mesmo material como o controle positivo, mas não inclui enzima dentro da reação de síntese do cDNA. A sem controle de RNA (NRC) é um controle negativo para confirmar a ausência de contaminantes transcrições nos componentes de reação de transcrição reversa. Esse controle foi introduzido o fluxo de trabalho na etapa de síntese do cDNA, e água foi adicionada na reação ao invés de um modelo de RNA. Os controles não RT e NRC devem ser negativos em ambos os canais, se resultados precisos devem ser entregues. A tabela 1 lista os componentes de reação de transcrição reversa para cada controle. Exemplos de 2D parcelas para cada um desses controles são mostrados para o ROS1 (Figura 3 A-C) e ensaios de multiplex RET (Figura 3 E-G). Variantes de fusão foram detectadas usando uma sonda de amidite (FAM) de fluoresceína e são representadas ao longo do eixo y, enquanto o gene de controle, GUSB, foi detectado usando uma sonda de 5'-hexacloro-fluoresceína-CE phosphoramidite (HEX) e no eixo x. Esses controles de lotes foram avaliados ao longo de 21 dias para determinar a robustez do ensaio. Gotículas de fusão positivas e gotículas de gene GUSB controle foram observadas para ROS1 e RET em todas as execuções de 21, executadas ao longo do estudo (Figura 3D, H). Todos os controles negativos (n RT e NRC) produziram resultados negativos entre os dias 21 (dados não mostrados).
A capacidade de solucionar problemas é um componente crítico de qualquer protocolo de teste a ser executado na configuração de laboratório clínico. Aqui, nós fornecemos exemplos do mundo real dos resultados abaixo do ideal, usando o protocolo de RT-dPCR. O primeiro é um plano 2D exemplo demonstra a importância de nenhum controle de transcriptase reversa(Figura 4). Neste exemplo, gotículas positivas mutantes estiveram presentes, mesmo que não havia nenhuma conversão de cDNA devido à falta da enzima. Este resultado foi provavelmente devido dPCR primers amplificar DNA genômico fora do alvo. Neste caso, projeto de um ensaio que mede intrão impedirá a amplificação do DNA genômico. Alternativamente, uma enzima DNase RNase-livre pode ser usada para eliminar o DNA contaminante, mas isso não é recomendado para a deteção de alvos raros, como alguma degradação do RNA pode ocorrer durante a incubação com a enzima. A próxima trama 2D exemplo era um NRC com gotículas positivas em ambos os canais (Figura 4B). Isto indica contaminação em algum momento da configuração do RT-dPCR. Neste caso, a recomendação é descartar qualquer potencialmente contaminados os reagentes utilizados nos testes, completamente descontaminar todo o equipamento e re-teste com componentes de reação de fresco. O terceiro lote 2D exemplo apresentado como um spray de gotas ao longo de uma linha de 45° (Figura 4C). Isto é frequentemente causado por cisalhamento e coalescência das gotículas. Gotícula cuidadosa manipulação antes da ciclagem térmica é essencial, como as gotas são propensas a danos. Recomendamos o uso da geração automatizada da gota, quando disponível. Se transferir manualmente gerado gotículas, é certo que escolher as dicas de largo diâmetro recomendadas e empregam a técnica de pipetagem cuidadosa. Transferência da gota requer lenta aspiração e dispensação, com cada ocorrendo em 5-6 segundos, e é essencial que a abertura de ponta de pipeta não toque o cartucho da gota ou bem. Quando dispensar, manter a ponta da pipeta no nível do líquido e levantá-lo lentamente, como as gotas são dispensada (exibição do vídeo de demonstração). O exemplo de plotagem 2D final demonstra uma falta de separação entre as populações de gotículas positivos e negativos (Figura 4). Isso pode ter várias causas. Inibidores da PCR fortes, como detergentes usados em buffers de Lise e excesso de DNA altamente degradada, podem causar a perda da separação. Neste caso, considere a adição de uma etapa de limpeza entre a síntese do cDNA e dPCR (como descrito no passo 5 deste protocolo). Finalmente, falta de separação também pode ser devido a condições de amplificação sub-ótimo e otimização da etapa PCR também deve ser considerada.
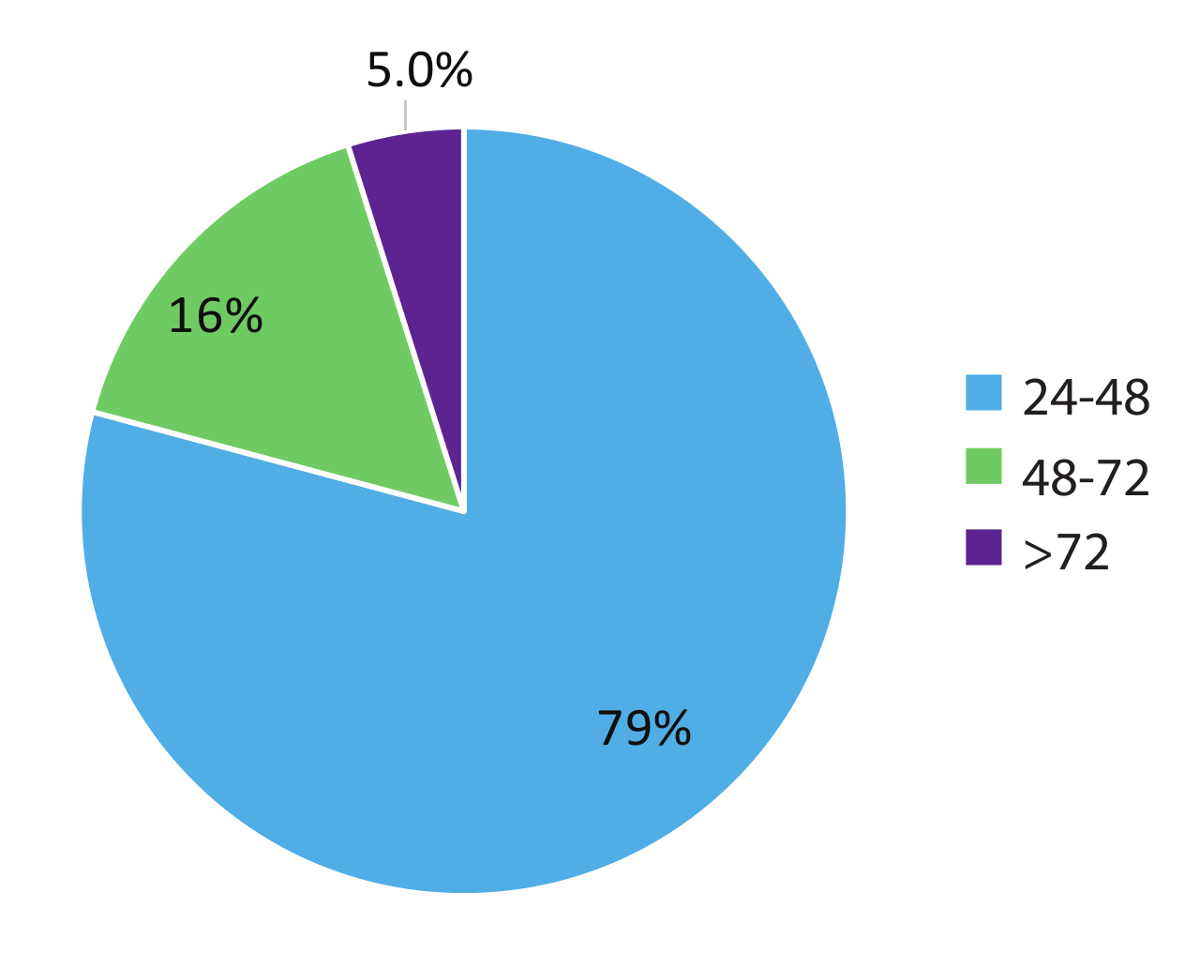
Dados no paciente 984 do mundo real de Figura 5 representam vezes reviravolta da amostra e demonstra a natureza rápida do fluxo de trabalho teste. Os resultados foram relatados para o médico de tratamento tão cedo como dentro de 48 horas (79% dos casos) do recebimento da amostra e em 95% dos casos, no prazo de 72 horas. Em conclusão, a utilização de estabilizado circulando RNA tubos de colheita de sangue, procedimentos de extração de RNA otimizados de sangue e RT-dPCR executar de acordo com um protocolo otimizado com o apropriado interno e controles do lote, pode fornecer um sistema de teste rápido para a detecção precisa de variantes de RNA de fusão relevantes em NSCLC.

Figura 1 : Visão geral das etapas processamento de amostra de sangue para detecção de variantes de fusão usando ensaios específicos para os mais prevalentes RET e ROS1 variantes em NSCLC. (A) amostra de teste é iniciado quando o sangue total é desenhado e uma BCT é enviado dentro do kit de coleção de amostra para o laboratório clínico. RNA de circulação é recuperado de múltiplas fontes dentro do plasma enriquecido em plaquetas, reverso transcrito com escorva específica do gene e purificada para uso em dPCR. As amostras são processadas usando um sistema comercialmente disponível que consiste de geração de gotas (emulsão), amplificação e contando com gota. Dados são analisados utilizando o software disponível comercialmente. Os resultados do teste são então documentados e reportados volta ao médico, solicitando-teste. O processo foi concebido para funcionar dentro de um prazo de 72 horas da data de recepção da amostra para liberação do resultado. Oito variantes para ROS1 (B) e (C) RET são abrangidas os ensaios multiplexados. Adaptado do site Biodesix com permissão. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2 : Validação analítica. Linhagens celulares expressando (A) SDC4-ROS1 fusão e fusão (B) CCDC6-RET foram diluídas em um fundo do RNA do selvagem-tipo humano total (WT RNA). Com cada variante de fusão, o limite de detecção foi estabelecido em 0,2% frequência variante usando critérios pré-definidos para cada variante ensaio. Todas as amostras acima desse limite também continham pelo menos 21 cópias do gene de controle. Padrão de EML4-ALK (ALK) 5% em um fundo do selvagem-tipo RNA foi testado para demonstrar a especificidade do ensaio, que foi confirmada por um resultado negativo. Normas analíticas de RNA multiplexadas foram medidas em alta, média e baixas concentrações ROS1 (C) e (D) RET. precisão foi avaliada sobre três corridas no mesmo dia (Intradia), três corridas em três dias consecutivos (inter dia) e com dois operadores independentes (inter operador). Os meios de cópia número e desvios-padrão são mostrados. Adaptado do site Biodesix com permissão. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3 : Lote processamento exemplos de controle e robustez dados. Terreno 2D do ROS1 multiplexado ensaio dPCR não resultados para controle positivo (A), (B) sem controle de transcriptase reversa e (C) nenhum controle do modelo de RNA. (D) controles foram executados em 21 dias consecutivos (excluindo sábados, domingos e feriados). Quer dizer cópia número + /-desvios-padrão para ROS1 positivo controle eram 439 + /-141. Não transcriptase reversa e sem controles de modelo também foram executados em cada dia, e estes eram todos negativos (dados não mostrados). Terreno 2D do RET multiplexado ensaio dPCR não resultados para controle positivo (E), (F) sem controle de transcriptase reversa e (G) nenhum controle do modelo de RNA. (H) controles foram executados em 21 dias consecutivos (excluindo sábados, domingos e feriados). Quer dizer cópias + /-desvios-padrão para RET positivo de controle foram 586 + /-182. Não mostrados são não transcriptase reversa e sem controles de modelo também foram executados em cada dia e todos negativos. Adaptado do site Biodesix com permissão. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4 : Solucionando problemas de RT-dPCR. 2D parcelas representando sub-ótimo dPCR os resultados obtidos quando há contaminação (por) dentro do controle não transcriptase reversa, contaminação (B) dentro do controle sem RNA, (C) cisalhamento e coalescência das gotículas e (D ) mal otimizado condições do PCR ou inibição de PCR. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5 : Tempo de retorno (TAT). TAT (em horas) foi compilado para testes, solicitando uma variante de RNA (n = 984). Dados exclui fins de semana, feriados e amostras realizadas por > 24 h devido à incompleta informação clínica sobre o formulários de solicitação de teste de laboratório. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Tabela 1: Preparação de reagentes de transcrição reversa para controles de processo.
| Componente de | Volume de |
| 2 x dPCR supermix para sondas (nenhum desoxiuridina 2'-5'-trifosfato) | 10 Μ l |
| conjunto de 20 x variante alvo cartilhas/sondas (450 nmol/L primeiras demão, sonda de FAM 250 nmol/L) | 1 Μ l |
| 20 x controle conjunto de primers/sonda de destino (450 nmol/L primeiras demão, sonda HEX 250 nmol/L) | 1 Μ l |
| água livre de nuclease | 1 Μ l |
| cDNA | 7 Μ l |
Tabela 2: Preparação da mistura mestre dPCR.
| Etapa de ciclismo | Temperatura | Tempo | # Ciclos | Taxa de rampa |
| Ativação da enzima | 95 ° C | 10 min | 1 | ~ 2 oC/s |
| Desnaturação | 94 ° C | 30 s | 40 | |
| Recozimento/extensão | 55 ° C | 1 min | ||
| Desativação da enzima | 98 ° C | 10 min | 1 | |
| Segure (opcional) | 4 ° C | infinita | 1 | ~ 1 óC/s |
Tabela 3: Condições de ciclagem térmicas.
Discussão
Rearranjos RET e ROS1 juntos compõem ~ 3% das mutações motorista dentro da população de NSCLC18. Apesar de rara, a detecção dessas alterações genéticas é vital. Pacientes NSCLC com essas alterações podem beneficiar de terapêutica alvo que inibir a atividade da quinase aberrante que resulta do onco-proteína13. Algumas dessas terapias já são aprovados pela FDA para uso em ROS1 NSCLC positiva, enquanto outros têm demonstrados para ser eficaz contra RET em ensaios clínicos,19.
Tecnologia digital de PCR fornece a sensibilidade que é ideal para aplicações de biópsia líquido20. Houve significativa adoção dessa tecnologia para uso com circulação sem célula de DNA para a medição de mutações tumor em pacientes com NSCLC4,6,21,22,23 . Além de cfDNA, desenvolvemos um protocolo projetado para medição robusta das variantes fusão mais prevalentes em pacientes com NSCLC circulem tumor RNA (Figura 1A)10.
Nosso protocolo estabelecido permite limites analíticos de detecção até 0,2% (Figura 2). Enquanto o RT-dPCR é excepcionalmente específico e sensível, os ensaios limitam-se ao painel de variantes conhecidas de fusão que são escolhidos e multiplexados para detecção no ensaio de PCR. Assim, fusões a serem incluídos em ensaios multiplexados devem ser cuidadosamente selecionados para garantir uma cobertura adequada no seio da população de pacientes com NSCLC. Nós projetamos com êxito ensaios para RET e ROS1 que simultaneamente detectar oito resultante de variantes de fusão de rearranjos dos loci RET ou ROS1 e cobrem 99% e 88% da população RET e ROS1 positiva, respectivamente (Figura 1-B-C )17.
O fluxo de trabalho do teste final conforme descrito neste estudo inclui controles de lote para garantir a consistência dos resultados. Esses controles incluem um padrão positivo de analítico, bem como dois controles negativos, que juntos garantem que não há contaminação ou inibição de PCR ocorrem dentro do lote (Figura 3). Para garantir a robustez do ensaio, um estudo foi realizado utilizando os controles de lote durante um período de 21 dias (Figura 3D, H). Estes dados demonstram a consistência do processo de RNA, conforme estabelecido neste protocolo.
Boas práticas laboratoriais e adequada manipulação de RNA são componentes-chave de garantir resultados robustos e precisos. Espaço de laboratório e equipamentos dedicados para usar com RNA, limpeza do equipamento após cada utilização, utilizando consumíveis e reagentes RNase-livre e aplicar um spray de inactivação de RNase para o espaço de trabalho, todos ajudam a reduzir a contaminação RNases. Manipulação consciente de amostras do RNA por técnicos, incluindo uma bata de laboratório dedicado, frequentes mudanças de luva, trabalhando rapidamente através do procedimento de extração de RNA, e manter as amostras no gelo são de extrema importância para preservar a integridade da amostra. Uma vez que o RNA tem sido reverso transcrito de cDNA, a amostra é em um formulário mais estável que é menos propenso a degradação. Além de práticas que oferecem suporte a integridade do RNA, amostras e componentes do PCR devem ser mantidas em áreas separadas para evitar a contaminação cruzada que pode levar a resultados falso-positivos. O estoque do PCR reagentes e preparação de misturas de mestre de PCR devem ser mantidos separados de modelos PCR e grande cuidado de segregar o modelo amplificado (post-PCR) de todos os materiais pre-amplificados incluindo reagentes, RNA e amostras de cDNA. Finalmente, geração adequada e manipulação de misturas emulsionadas de PCR antes da amplificação é central para a manutenção da integridade da gota e condições dPCR ideal. Precauções como estes são fundamentais durante a execução do presente protocolo para obter resultados precisos e consistentes. Todos os dados devem ser examinados por pessoal treinado antes do lançamento dos resultados para ter certeza que foram cumpridas todas as métricas QC. No caso de resultados de qualidade inferior (Figura 4), o lote deve ser revisado por técnicos e o diretor do laboratório e pode exigir re-processamento.
Resultados de RT-dPCR podem ser produzidos tão cedo quanto 24 horas a partir do recebimento da amostra e 95% dos resultados da amostra dentro de ensaio utilizado neste estudo (n = 984) foram reportados ao médico a encomenda em menos de 72 horas desde o momento da recepção (Figura 5). Este tempo de rotação fornece aos médicos muito necessária informação molecular em um período de tempo que permite a iniciação da terapia adequada. Estes resultados estão normalmente disponíveis mais cedo do que aqueles obtidos usando uma biópsia de tecido convencional. Biomarcadores adicionais para NSCLC e outros tipos de câncer poderiam ser desenvolvidos utilizando abordagens semelhantes baseado em RNA circulantes e se beneficiaria com os mesmos tempo-para-resultados rápidos. Por exemplo, medição da transcrição mRNA ligante de morte programada 1 (PD-L1) usando RT-dPCR poderia informar os médicos sobre as opções de imunoterapia. Há também um interesse crescente no utilitário de biópsia líquida e dPCR no monitoramento de eficácia terapêutica. Indicações anteriores do ressurgimento do tumor usando genômica de testes para variantes específicas poderiam permitir que médicos ajustar esquemas de tratamento antes que os pacientes são sintomáticos pelo padrão de medidas de cuidados como imagem24. Protocolos como o relatado neste estudo são ideais para monitoramento devido à sua não-invasividade, sensibilidade, tempo de rotação rápida e custo-eficácia. O ensaio descrito aqui fornece resultados dentro de 72 horas da data de recepção de amostra, com taxas de detecção de falso-positivo mínimo, que facilita as decisões de tratamento rápido e contorna algumas limitações que experimentou com testes baseados em tecido4.
Nosso protocolo e dados demonstram um sistema robusto de teste para a identificação de variantes de RNA baixa abundância, bem como o potencial de mutação à base de sangue, teste na prática clínica. Para aqueles pacientes que não possuem um driver acionável mutação identificada por biópsia líquida alvo rápida se aproxima como este, a adição de mais extensa do genoma e proteoma teste do sangue e do tecido pode fornecer informação clínica ainda mais ampla para apoiar o planejamento do tratamento.
Divulgações
H.M, L.J., K.A. e Gap são empregados da e mantenha ações em H.M Biodesix, Inc., L.J. e Gap são co-inventores em um pedido de patente apresentado por Biodesix, cobrindo um sistema de teste de diagnóstico para a detecção de variantes genéticas no não-pequenas células de circulação câncer de pulmão.
Agradecimentos
Agradecemos a nossos colaboradores, Stephen Jones, Nia Charrington, Dr. Dianna Maar e Dr. Samantha Cooper do centro de biologia de Digital (Bio-Rad Inc. CA) para seu projeto de ensaio suportam; Nezar Rghei e Dr. Moemen Abdalla (Norgen biotecnologia, Canadá) para conselhos crítico ao otimizar o protocolo de extração de RNA; e Shannon Campbell, Scott Thurston, Jeff Fensterer, Shannon Martello e Joellyn Enos para obter assistência com teste requisitos e acompanhamento comercial.
Materiais
| Name | Company | Catalog Number | Comments |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1604071 |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1809353 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1604071 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1606077 |
| Phosphate Buffered Saline 1X, Sterile | Amresco | K812-500mL | 1446C189 |
| Phosphate Buffered Saline 1X, Sterile – 500 mL | Invitrogen | 10010023 | 1916C092 |
| RNase Zap (Life Tech) (250 mL) | Ambion | AM9780 | 353952 |
| Beta-Mercaptoethanol (BME) (250 mL) | CalbioChem | 6050 | W105B |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56054611 |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56238638 |
| Isopropyl Alcohol | VWR | 0918-4L | 2116C416 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 403648 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 288461 |
| DNase I | Thermo | K0441 | 371299 |
| QIAzol Lysis Reagent | Qiagen | 79306 | 54809699 |
| 20x TE buffer pH 8.0 | Alfa Aesar | J62388 | R13C548 |
| UltraPure Agarose | Invitrogen | 16500-100 | 552730 |
| 10x TBE buffer | Invitrogen | AM9863 | 353065 |
| Cell-Free RNA BCT | Streck | 218976 | 60110327 |
| Cell-Free RNA BCT | Streck | 218976 | 61900327 |
| Cell-Free RNA BCT | Streck | 218976 | 61480327 |
| Cell-Free RNA BCT | Streck | 218976 | 62320327 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 585849 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 588308 |
| Lysis Buffer | Norgen | 21205 | A5F61E |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 585848 |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 588309 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC186976 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188077 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188413 |
| Collection Tubes 500 pack | Zymo | C1001-500 | N/A |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 391657 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 392504 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 448001 |
| SuperScript IV Reverse Transcriptase | Life Technologies | 18090200 | 451702 |
| Qubit HS RNA Assay Kit (500) | Life Technologies | Q32854 | 1745264 |
| Qubit assay tubes (500) | Life Technologies | Q32856 | 13416Q311 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64031651 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64063941 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065740 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065741 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64079083 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64025320 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64052358 |
| gBlock KIF5B-RET K15:R12 | IDT | 151004172 | 4-Oct-16 |
| gBlock KIF5B-RET K16:R12 | IDT | 151004173 | 4-Oct-16 |
| gBlock KIF5B-RET K22:R12 | IDT | 151004174 | 4-Oct-16 |
| gBlock KIF5B-RET K23:R12 | IDT | 151004175 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R11 | IDT | 151004176 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R8 | IDT | 151004177 | 4-Oct-16 |
| gBlock CCDC6-RET C1:R12 | IDT | 151004178 | 4-Oct-16 |
| gBlock TRIM33-RET T14:R12 | IDT | 151004179 | 4-Oct-16 |
| RET exon 8 RT Gene Specific Primer | IDT | 150554385 | 28-Sep-16 |
| 5’-CTCCACTCACACCTG-3’ | IDT | 150554385 | 28-Sep-16 |
| RET exon 11 RT Gene Specific Primer | IDT | 150554384 | 28-Sep-16 |
| 5’-GCAAACTTGTGGTAGCAG-3’ | IDT | 150554384 | 28-Sep-16 |
| RET exon 12 RT Gene Specific Primer | IDT | 150554383 | 28-Sep-16 |
| 5’-CTGCCTTTCAGATGGAAG-3’ | IDT | 150554383 | 28-Sep-16 |
| gBlock CD74-ROS1 C6:R34 | IDT | 152324366 | 15-Nov-16 |
| gBlock CD74-ROS1 C6:R32 | IDT | 152324367 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R32 | IDT | 152324368 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R34 | IDT | 152324369 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R32 | IDT | 152324370 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R34 | IDT | 152324371 | 15-Nov-16 |
| gBlock EZR-ROS1 E10:R34 | IDT | 152324372 | 15-Nov-16 |
| gBlock TPM3-ROS1 T8:R35 | IDT | 152324373 | 15-Nov-16 |
| ROS1 exon 34 RT Gene Specific Primer | IDT | 152704983 | 21-Nov-16 |
| 5’-CCTTCCTTGGCACTTT-3’ | IDT | 152704983 | 21-Nov-16 |
| ROS1 exon 35 RT Gene Specific Primer | IDT | 152704985 | 21-Nov-16 |
| 5’-CTCTTGGGTTGGAAGAGTATG-3’ | IDT | 152704985 | 21-Nov-16 |
| ALK Gene Specific Primer | IDT | 140035422 | 26-Aug-16 |
| 5’-CAGTAGTTGGGGTTGTAGTCG-3’ | IDT | 140035422 | 26-Aug-16 |
| EML4-ALK Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| SLC34A2-ROS1 Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| CCDC6-RET Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| Human Brain Total RNA | Ambion | AM7962 | 1703548 |
| PrimePCR ddPCR Expert Design Assay: K15:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K16:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K22:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K23:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R11 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R8 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C1:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: T14:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: E10:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: T8:R35 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| (version 2) | Bio-Rad | 12003909 | 213939881 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 13-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 20170112v3.2 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851151 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 207383915 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 195995635 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851152 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 213949301 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 20160914 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 211383227 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 1065C220 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052953 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052358 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 1065C320 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64052952 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64064127 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000065883 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084276 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000079928 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084395 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084634 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20160627 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161107 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161206 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161216 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20170125 |
| Pipet Tips for Automated Droplet Generator | Bio-Rad | 1864120 | PR125340 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206894 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206893 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 1409850 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 100402 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 145851 |
| Microseal 'B' seals | Bio-Rad | MSB1001 | BR00428490 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64039089 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049253 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049255 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64081870 |
| DNA Lo Bind Tube 0.5 mL | Eppendorf | 22431005 | E1629620 |
| DNA Lo Bind Tube 1.5 mL | Eppendorf | 22431021 | F16698K |
| DNA Lo Bind Tube 2 mL | Eppendorf | 22431048 | E160610I |
| 50 mL Conicals, Polypropylene (25) | Thermo | 339652 | G5ZF5W8118 |
| TempAssure PCR 8-Strips, Optical Caps, Natural, polypropylene (120) | USA Scientific | 1402-4700 | 16202 |
| For Rainin LTS Pipettors 0.5-20 µL tips | Pipette.com | LF-20 | 40155-642C4-642C |
| For Rainin LTS Pipettors 5-200 µL tips | Pipette.com | LF-250 | 40154-642C4-642B |
| Tips LTS 200 ul Filter 960/10 RT-L200F (10 boxes) | Rainin | 17002927 | 1635 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F1175551-1108 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F118054L-1720 |
| Pipet Tips, 100 ul TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1180-1740 | 0014961Q-2501 |
| Pipet Tips, 200 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1180-8710 | E116684P-1540 |
| Pipet Tips, 1000 ul XL TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1182-1730 | F118815P |
| 5 mL Standard Racked Gilson-fit Reference Tips | Scientific Specialties | 4411-00 | 14312 |
| Combitips advanced, 0.1 mL Biopur | Eppendorf | 003 008 9618 | F165414H |
| Combitips advanced, 0.2 mL Biopur | Eppendorf | 0030 089.626 | F166689J |
| Combitips advanced, 5 mL Biopur | Eppendorf | 0030.089 669 | F166054J |
| Combitips advanced, 50 mL Biopur | Eppendorf | 003.008.9693 | F166055I |
| Reagent Reservoir | VWR | 89094-680 | 141500 |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165029I |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165028G |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Green | Eppendorf | 951020346 | F166183K |
| Equipment Type | Equipment ID | ||
| Analytical Balance | EQP0125 | ||
| Cryogenic Freezer 1, -80oC | EQP0095 | ||
| Refrigerator 6.1 cu ft GP06W1AREF | EQP0139 | ||
| -20oC Freezer | EQP0140 | ||
| Beckman Coulter Microfuge 22R | EQP0025 | ||
| Beckman Coulter Microfuge 22R | EQP0124 | ||
| Thermo Scientific Hereaus Megafuge 8 | EQP0104 | ||
| Mini Centrifuge | EQP0131 | ||
| Mini Centrifuge | EQP0136 | ||
| Mini Centrifuge | EQP0134 | ||
| Mini Centrifuge | EQP0235 | ||
| Mini Centrifuge | EQP0216 | ||
| Thermo Scientific HeraTherm Incubator | EQP0105 | ||
| Pipette 0.1 - 2.5 μL | EQP0182 | ||
| Pipette 0.1 - 2.5 μL | EQP0072 | ||
| Pipette 0.1 - 2.5 μL | EQP0070 | ||
| Pipette 0.5-10 μL | EQP0218 | ||
| Pipette 0.5-10 μL | EQP0075 | ||
| Pipette 0.5-10 μL | EQP0169 | ||
| Pipette 0.5-10 μL | EQP0074 | ||
| Pipette 0.5-10 μL | EQP0147 | ||
| Pipette 2 - 20 μL | EQP0128 | ||
| Pipette 2 - 20 μL | EQP0160 | ||
| Pipette 2 - 20 μL | EQP0018 | ||
| Pipette 2 - 20 μL | EQP0146 | ||
| Pipette 10 - 100 μL | EQP0079 | ||
| Pipette 10 - 100 μL | EQP0181 | ||
| Pipette 10 - 100 μL | EQP0085 | ||
| Pipette 10 - 100 μL | EQP0077 | ||
| Pipette 20 - 200 μL | EQP0088 | ||
| Pipette 20 - 200 μL | EQP0087 | ||
| Pipette 20 - 200 μL | EQP0231 | ||
| Pipette 100 - 1000 μL | EQP0050 | ||
| Pipette 100 - 1000 μL | EQP0158 | ||
| Pipette 100 - 1000 μL | EQP0217 | ||
| Pipette 100 - 1000 μL | EQP0082 | ||
| Pipette 100 - 1000 μL | EQP0183 | ||
| Pipette 100 - 1000 μL | EQP0083 | ||
| Pipette 5 mL | EQP0153 | ||
| Timer | S/N 140623950 | ||
| Hamilton SafeAire VAV Fume Hood | EQP0206 | ||
| Biosafety Cabinet | EQP0205 | ||
| Biosafety Cabinet | EQP0204 | ||
| Qubit 3.0 | EQP0102 | ||
| Benchmark Digital Heat Block | EQP0108 | ||
| Benchmark Digital Heat Block | EQP0231 | ||
| Polaroid Z2300 Instant Print Digital Gel Camera with WiFi and 16GB SDHC memory card | EQP0111 | ||
| Electrophoresis Power Unit | EQP0113 | ||
| Electrophoresis Small Gel Box | EQP0116 | ||
| Maestro Transilluminator | EQP0118 | ||
| Microwave | EQP0215 | ||
| Multichannel 8-well Pipette 2 - 20 μL | EQP0207 | ||
| Multichannel 8-well Pipette 10 - 100 μL | EQP0090 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0094 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0161 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0162 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0163 | ||
| Vortex Genie 2 | EQP0052 | ||
| Vortex Genie 2 | EQP0007 | ||
| Vortex Genie 2 | EQP0132 | ||
| Vortex Genie 2 | EQP0137 | ||
| Vortex Genie 2 | EQP0135 | ||
| Air Clean PCR Workstation | EQP0203 | ||
| Air Clean PCR Workstation | EQP0096 | ||
| Air Clean PCR Workstation | EQP0148 | ||
| Air Clean PCR Workstation | EQP0097 | ||
| QX200 Droplet Generator | EQP0202 | ||
| QX200 Droplet Generator | EQP0121 | ||
| Automated Droplet Generator | EQP0179 | ||
| PX1 PCR Plate Sealer | EQP0123 | ||
| PX1 PCR Plate Sealer | EQP0186 | ||
| C1000 Touch Cycler w/96W FS RM | EQP0120 | ||
| S1000 Cycler w/96W FS RM | EQP0174 | ||
| S1000 Cycler w/96W FS RM | EQP0173 | ||
| T100 Thermal Cycler | EQP0180 | ||
| T100 Thermal Cycler | EQP0175 | ||
| QX200 Droplet Reader | EQP0194 | ||
| QX200 Droplet Reader | EQP0122 |
Referências
- Ignatiadis, M., Lee, M., Jeffrey, S. S. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin Cancer Res. 21 (21), 4786-4800 (2015).
- Alix-Panabieres, C., Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. , (2016).
- Paxton, A. Is Molecular AP testing in sync with guidelines. CAP Today. , (2014).
- Sacher, A. G., et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. , (2016).
- Sozzi, G., et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 21 (21), 3902-3908 (2003).
- Oxnard, G. R., et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 20 (6), 1698-1705 (2014).
- Best, M. G., et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 28 (5), 666-676 (2015).
- Rodriguez, M., et al. Different exosome cargo from plasma/bronchoalveolar lavage in non-small-cell lung cancer. Genes Chromosomes Cancer. 53 (9), 713-724 (2014).
- Kalluri, R. The biology and function of exosomes in cancer. J Clin Invest. 126 (4), 1208-1215 (2016).
- Mellert, H., et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non-Small Cell Lung Carcinoma. J Mol Diagn. 19 (3), 404-416 (2017).
- Qin, J., Williams, T. L., Fernando, M. R. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res Notes. 6, 380 (2013).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 15 (3), 532-537 (1993).
- Kohno, T., et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 4 (2), 156-164 (2015).
- Takeuchi, K., et al. ROS1 and ALK fusions in lung cancer. Nat Med. 18 (3), 378-381 (2012).
- Rimkunas, V. M., et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 18 (16), 4449-4457 (2012).
- Tsuta, K., et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 110 (6), 1571-1578 (2014).
- Forbes, S. A., et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (1), 777-783 (2017).
- Salgia, R. Diagnostic challenges in non-small-cell lung cancer: an integrated medicine approach. Future Oncol. 11 (3), 489-500 (2015).
- Cagle, P. T., Raparia, K., Portier, B. P. Emerging Biomarkers in Personalized Therapy of Lung Cancer. Adv Exp Med Biol. 890, 25-36 (2016).
- Vogelstein, B., Kinzler, K. W. Digital PCR. Proc Natl Acad Sci U S A. 96 (16), 9236-9241 (1999).
- Oxnard, G. R., et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 34 (28), 3375-3382 (2016).
- Reckamp, K. L., et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 11 (10), 1690-1700 (2016).
- Yanagita, M., et al. A prospective evaluation of circulating tumor cells and cell-free DNA in EGFR mutant non-small cell lung cancer patients treated with erlotinib on a phase II trial. Clin Cancer Res. , (2016).
- Abbosh, C., et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 545 (7655), 446-451 (2017).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques. 15 (3), 532-534 (1993).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados