
Method Article
Combinando a análise de DNA em uma extração do Virion bruto com a análise do RNA das folhas infectadas para descobrir novos vírus de genomas
Neste Artigo
Resumo
Aqui nós apresentamos uma nova abordagem para identificar a planta vírus com genoma de DNA dobro-costa. Utilizamos métodos padrão para extrair o DNA e RNA de folhas contaminadas e realizar o sequenciamento de próxima geração. Ferramentas de Bioinformatic montam sequências em contigs, identificam contigs representando genomas do vírus e atribuir genomas de grupos taxonômicos.
Resumo
Esta abordagem de metagenome é usada para identificar a planta vírus com genoma de DNA circular e seus transcritos. Muitas vezes planta vírus de DNA que ocorrem em baixa concentração de acolhimento ou não poderem ser inoculadas mecanicamente para outro host são difíceis de propagar para alcançar uma maior concentração de material infeccioso. Folhas infectadas são terreno em um buffer suave com pH ideal e a composição iônica, recomendado para a maioria dos retrovírus de pará bacilliform de purificação. A ureia é usada para quebrar os corpos de inclusão que armadilha virions e para dissolver os componentes celulares. Centrifugação diferencial fornece uma separação de virions de contaminantes de planta. Tratamento de proteinase K, em seguida, remove os capsids. Em seguida, o DNA viral é concentrado e usado para sequenciamento de próxima geração (NGS). Os dados NGS são usados para montar contigs que são submetidos à NCBI-BLASTn para identificar um subconjunto de sequências de vírus no dataset gerado. Em um pipeline paralelo, RNA é isolado de folhas contaminadas, usando um método de extração de RNA baseado na coluna padrão. Depleção de ribossoma é realizada para enriquecer para um subconjunto de transcrições de mRNA e vírus. Montadas sequências derivadas de RNA (RNA-seq) de sequenciamento foram submetidas à NCBI-BLASTn para identificar um subconjunto de sequências de vírus neste dataset. Em nosso estudo, identificamos dois genomas relacionados badnavirus completos em dois conjuntos de dados. Este método é preferível à outra abordagem comum que extrai a população agregada de pequenas sequências de ARN para reconstituir a planta vírus sequências genomic. Este último metagenomic gasoduto recupera vírus relacionados com sequências que são elementos retrô-transcrevendo inseridos no genoma da planta. Este é acoplado para ensaios bioquímicos ou moleculares mais discernir os agentes infecciosos ativamente. A abordagem documentada neste estudo, recupera sequências representativas da replicação de vírus que provavelmente indicam infecção pelo vírus ativo.
Introdução
Doenças emergentes de planta conduzir os investigadores a desenvolver novas ferramentas para identificar o agente causal correta (s). Os relatórios iniciais de viroses novo ou recorrente baseiam-se geralmente ocorrem sintomas como mosaico e malformações da folha, veia de compensação, nanismo, murcha, lesões, necrose, ou outros sintomas. O padrão para um novo vírus de relatórios como o agente causal de uma doença é para separá-lo de outros patógenos contaminantes, propagá-la em um hospedeiro adequado e reproduzir a doença por inoculação em plantas saudáveis da espécie hospedeiro original. A limitação desta abordagem é que muitos géneros de vírus de planta dependem de um inseto ou outros vetores para a transmissão para um hospedeiro adequado ou voltar para as original espécies de hospedeiros. Neste caso, a busca para o vetor apropriado pode ser prolongada, pode haver dificuldades para estabelecer colônias de laboratório do vetor e mais esforços são necessários para elaborar um protocolo para transmissão experimental. Se as condições para estudos de transmissão bem sucedida de laboratório não podem ser alcançadas, então o trabalho fica aquém do padrão para relatar uma nova doença de vírus. Para vírus que ocorrem em seus hospedeiros naturais em títulos muito baixos, pesquisadores devem identificar hospedeiros alternativos de propagação para manter reservas suficientes de infecciosas para realização de pesquisas. Para as espécies de vírus que infectam apenas algumas plantas isso também pode ser um obstáculo para o cultivo de culturas-mãe1.
Nos últimos anos, os cientistas estão mais frequentemente empregando NGS elevado-throughput e abordagens metagenomic para descobrir sequências de vírus que estão presentes no ambiente, que pode existir relacionados com uma doença conhecida, mas pode ser atribuído a gêneros e espécies taxonômicas 2 , 3 , 4. tais abordagens para a descoberta e a categorização de materiais genéticos em um ambiente distinto fornecem uma maneira para descrever a diversidade de vírus na natureza ou sua presença em um determinado ecossistema, mas não necessariamente confirma um enquadramento para a definição agentes causais para uma doença aparente.
O gênero Badnavirus pertence à família Caulimoviridae de pararetroviruses. Estes vírus são bacilliform em forma com genoma de DNA circular de fita dupla de aproximadamente 7 a 9 kb. Todos os pararetroviruses replicar através de um intermediário de RNA. Pararetroviruses existe como episomes e replicar independentes da planta cromossômico DNA5,6. Estudos de populações do vírus de campo indicam que estas populações de vírus são geneticamente complexas. Além disso, as informações obtidas através de uma gama de genomas de plantas por sequenciamento de alto rendimento tem descoberto inúmeros exemplos de fragmentos de genoma badnavirus inseridos por eventos de integração ilegítimo em genomas de planta. Essas sequências de badnavirus endógena não estão necessariamente associadas com infecção7,8,9,10,11. Posteriormente, o uso de NGS para identificar novos badnaviruses como o agente causal da doença é complicado pela diversidade de genomas epissomal subpopulação, bem como a ocorrência de sequências endógenas12,13.
Enquanto há não um pipeline ideal para a descoberta de genomas de romance pararetrovirus, existem duas abordagens comuns para identificar estes vírus como agentes causais de doença. Um método é enriquecer para pequenas sequências de RNA das folhas infectadas e em seguida montar essas sequências para reconstituir o vírus genome(s)14,15,16,17. Outra abordagem é a amplificação de círculo rolante (RCA) para amplificar a vírus de DNA circular genomas18. O sucesso da RCA depende da idade da folha e do título do vírus no tecido selecionado. Os produtos RCA são submetidos a digestão da limitação e clonados em plasmídeos para direct sequenciamento19,20,21.
Vírus mottle amarelo de canna (CaYMV) é um badnavirus e é descrito como a causa etiológica da doença mottle amarelo em canna, embora tenha sido apenas um fragmento de bp 565 do genoma anteriormente isoladas de cannas infectados22. Um estudo contemporâneo identificado CaYMV em Alpinia purpurata (gengibre de floração; CaYMV-Ap)23. O objetivo deste estudo foi recuperar sequências do genoma completo badnavirus de lírios de canna infectados. Descrevemos um protocolo para a purificação de vírus de contaminantes de planta e depois isolar ADN viral, a partir desta preparação e preparar uma biblioteca de DNA para uso em NGS. Essa abordagem elimina a necessidade de etapas de amplificação molecular intermediário. Nós também isolar mRNA de plantas infectadas para RNA-Seq. NGS, que inclui RNA-seq foi realizada usando cada preparação de ácido nucleico. Contigs montados foram encontrados para relacionar o táxon Badnavirus em ambos os conjuntos de dados usando o centro nacional de biotecnologia e Information (NCBI) ferramenta de busca de alinhamento local básico de ácidos nucleicos (dos). Identificamos os genomas de duas badnavirus espécies24.
Protocolo
1. general vírus purificação por centrifugação diferencial usando o método padrão por Covey et al 25
- Primeiro, corte de 80-100 g de folhas de plantas doentes e moer no liquidificador waring a 4 ° C, usando 200 mL de tampão de moagem (0,5 M NaH2PO4, 0,5 M Na2HPO4 (pH 7,2). e 0,5% (p/v) Na2SO3). Use um casaco de laboratório e luvas para todas as etapas deste procedimento.
- Em seguida, transferi o homogeneizado (300 mL) para um béquer de 1,0 L. Adicione 18 g de ureia e 25 mL de 10% de detergente não iônico (t-Oct-C6H4-(OCH2CH2)9OH) para o homogenate dentro de uma capa de química.
Nota: Para esta etapa é melhor usar óculos de segurança e uma máscara de respiração simples para proteção pessoal. - Agitar com um agitador magnético brevemente no capô e cobrir o béquer com a folha. Em seguida, transferir o copo de alumínio coberto para um quarto frio e mexa com um agitador magnético durante a noite a 4 ° C.
- Transferir o homogeneizado para centrifugar garrafas de rotor (recipientes de 250 mL) e centrifugar em um rotor de ângulo fixo a 4.000 x g durante 10 minutos a 4 ° C. Em uma coifa química, recupere o sobrenadante e filtrar através de 4 camadas de gaze.
- Dividir o homogenate entre tubos de centrífuga de polipropileno 38,5 mL e centrifugar para 2,5 h a 40.000 x g a 4 ° C. Normalmente, verifique a presença de uma bolinha verde na parte inferior do tubo e uma bolinha branca ao longo do comprimento do tubo. Decantar o sobrenadante e manter ambas as pelotas; coloca as amostras no gelo.
Nota: A pelota verde contém cloroplastos, amido e outros organelos. - Trabalhando em uma capa de química, use um policial de borracha para separar as bolinhas. Ressuspender o precipitado branco em cada garrafa de rotor em 1 mL de DDQ2O ao longo de 1-2h, mantendo que as suspensões durante a noite a 4 ° C, para permitir que os materiais de dissolver totalmente a solução. Centrifugar a suspensão em 6.000 x g e a 4 ° C por 10 min remover os detritos remanescentes.
- Centrifugar a suspensão concentrada de 136.000 x g, durante 2 h a 4 ° C para virions de Pelotas. Resuspenda as pelotas em 1 mL de tampão (50 mM Tris-HCl, pH 7,5, 5 mM MgCl2).
Nota: Um passo opcional é tratar virions com DNAse I (10 µ g/mL) por 10 min a 37 ° C para remover o DNA não-encapsidated, ou seja, contaminando o cloroplasto e DNA mitocondrial. Em seguida, desativar o DNAse eu adicionando EDTA 1 mM. - Perturbar os virions com 40 µ l de 2 µ g / µ l proteinase K a 37 ° C por 15 min.
- Trabalhar dentro de uma capa de química para recuperar o DNA do virion por extração orgânica. Use um protetor facial, luvas e um casaco de laboratório durante a extração para proteção contra potenciais efeitos graves para a saúde. Adicionar 1 volume de álcool isoamílico-fenol-clorofórmio (49:50:1) para a amostra e agitar manualmente durante 20 s. centrífuga à temperatura ambiente por 5 min à 16.000 g. x retire a fase aquosa superior e transferência para um novo tubo. Repita esta extração de duas ou mais vezes. Descarte a fase orgânica, colocando-o em uma garrafa de vidro de resíduos para eliminação de químicos institucional adequada26.
- Concentre-se o DNA utilizando a precipitação do álcool etílico. Use a concentração final de 0,3 M de acetato de sódio (pH 5.2) e 2,5 volumes de etanol a 95%. Colocar as amostras a-20 º C durante 30-60 min e centrifugar a 13.000 x g por 10-20 min para o ADN26de Pelotas.
- Trabalhando em uma bancada de laboratório, resuspenda o pellet de DNA em 1 mL de tampão de TE de 0,1 mM (pH 8.0). Limador a suspensão através de uma coluna de filtração de gel de comercial (normalmente usada para reação em cadeia da polimerase (PCR) limpar) para eliminar sais e material de baixo peso molecular que pode impedir a NGS.
- Analise as amostras pela electroforese do gel de agarose a 1% usando brometo de etídio coloração para ver a qualidade das preparações. Avalie a qualidade do DNA usando um espectrofotômetro nanodrop.
Nota: Uma relação de absorvância da amostra em λ 260 e 280 λ entre 1,85 e 2.0 normalmente indica que a preparação é "limpo" de impurezas e com a qualidade desejada. - Analisar a qualidade do DNA (use pg 5-10 ng) usar um chip com base em instrumento de eletroforese capilar.
Nota: A saída de qualidade mostra picos limpos, que representam fragmentos de DNA distribuídos pelo tamanho ao longo de um eixo x. Altura do pico indica abundância do fragmento. Picos irregulares indicam fragmentos parcialmente degradados ou contaminantes químicos. Rodada curvas representam um esfregaço de ADN indicando má qualidade
2. Biblioteca preparação usando DNA e baseada em emulsão Amplification Clonal (emPCR amplificação)
Nota: A biblioteca é tipicamente preparada por uma instalação NGS que realiza trabalho orientado ao cliente.
- Uma solução de DNA (> 200 ng) usando um nebulizador que converte o DNA em fragmentos de cisalhamento. Ligar os adaptadores comerciais de acordo com instruções27 do manual.
- Realize emPCR amplificação da amostra de DNA de acordo com instruções28,29,30 as indicações do fabricante. Repita a etapa de lavagem três vezes e depois de cada lavagem, granular os grânulos em um minicentrifuge por 10 s. descartar o sobrenadante após cada lavagem.
Nota: O procedimento começa com a preparação dos grânulos captura por lavagem no tampão de lavagem comercial fornecido com o kit. emPCR é comumente usado para a amplificação de modelo para NGS. - Calor desnaturar o DNA ou RNA a 95 ° C por 2 min e, em seguida, a 4 ° C até que esteja pronto para usar. Uso 200 milhões de moléculas de DNA/RNA para captura de 5 milhões de contas em um volume final de 30 µ l. preparem uma amostra de simulação juntamente com a amostra de DNA/RNA e realizar as etapas a seguir com a amostra de ácido nucleico, bem como a amostra simulada.
- Executar a emulsificação pela utilização do Vortex o tubo do óleo de emulsão para 10 s velocidade máxima, em seguida despeje todo o conteúdo (4 mL) em um plástico agitando o tubo que é compatível com um homogeneizador de plataforma. Coloque o tubo de agitação na plataforma para misturar a emulsão a 2.000 rpm durante 5 min.
- Distribuir 100 alíquotas µ l de emulsão para tubos de 8-faixa de tampa ou em uma placa de 96 poços. Os tubos do tampão ou selar a placa e realizar emPCR usando programa recomendado28 do fabricante.
Nota: Após a PCR é completa, verifique os poços para ver se a emulsão está intacta e prossiga. Descarte o poço inteiro se a emulsão está quebrada. - Usar um jaleco e um capuz químico para coletar os grânulos de DNA amplificados (ADB). Vácuo aspirar a emulsão dos poços e recolher as gotas em um tubo de 50 mL. Lavar os poços duas vezes com 100 µ l de isopropanol e aspire o enxágue para o mesmo tubo de 50 mL.
- Vórtice o coletados emulsões e ressuspender o ADB com isopropanol até um volume final de 35 mL. De Pelotas a ADB no 930 x g durante 5 min. Retire o sobrenadante e adicionar 10 mL de reforçar a reserva. Vórtice do ADB e lavar em seguida adicionando isopropanol para volume final de 40 mL centrífuga e descartar o sobrenadante após cada lavagem e repita a etapa de lavagem, duas vezes.
- Realize uma lavagem final usando etanol no lugar de isopropanol. Adicionar aumentando o buffer para o volume final de 35 mL, vórtice e pelota os grânulos em 930 x g, durante 5 min. Retire o sobrenadante mas deixar 2ml de reforçar a reserva.
- A suspensão de transferência para um tubo de microcentrifugadora e brevemente centrifugar para pelotas o ADB. Após descartar o sobrenadante, enxágue a pelota ADB duas vezes com 1 mL de tampão de reforço. Centrifugar e desprezar o sobrenadante após cada lavagem.
- Para se preparar para o enriquecimento de grânulo de biblioteca de DNA, adicione 1 mL de NaOH de N 1 aos talões. Vórtice do ADB e então incubar por 2 min à temperatura ambiente. Centrifugar e desprezar o sobrenadante. Repita essa etapa de lavagem uma vez.
- Adicionar 1 mL de tampão de recozimento, então vórtice do ADB e incubar por 2 min à temperatura ambiente. Brevemente, centrifugar e desprezar o sobrenadante. Repita este passo novamente usando 100 µ l de tampão de recozimento.
- Para recozer primeiras demão de sequenciamento do DNA, adicione 15 µ l do Seq Primer A e 15 µ l de Seq Primer B fornecido no kit. Brevemente, misture vortexing e coloque o tubo de microcentrifugadora em um bloco de calor a 65 ° C por 5 min. transferência de gelo por 2 min.
- Lavar três vezes com 1,0 mL de tampão de recozimento. Vórtice para 5 s e descartar o sobrenadante de cada vez.
- Antes de sequenciamento, medir o número de grânulos usando um contador comercial do grânulo. Deve haver pelo menos 500.000 grânulos enriquecidos.
Nota: O contador de grânulo é um dispositivo especial que mede os grânulos em um tubo de microcentrifuga fornecido.
3. general mRNA isolamento e dsDNA síntese começando com infectados Canna deixa o teste por RT-PCR para CaYMV usando relatou diagnóstico Primers
- Use um casaco de laboratório e luvas de látex para proteção pessoal em todas as etapas subsequentes. Trabalhando em uma bancada de laboratório, coletar 12 amostras das folhas e mergulhe as amostras em nitrogênio líquido. Use um moinho de esfera para homogeneização. Use um kit comercial que fornece um método padrão baseados em colunas para planta total isolamento de RNA. Adicionar a lise de isotiocianato de guanidina, fornecido pelo kit para a amostra e agitar por 20 s.
- Adicionar etanol e misture bem, de acordo com as instruções do kit. Adicione cada homogeneizado para uma coluna de rotação que vincula o RNA para a membrana. Lavar três vezes e eluir o RNA em um tubo de recuperação24.
- Quantificar o RNA usando um espectrofotômetro para medir a proporção de absorvância a λ 260 e 280 λ. Verifique se o RNA integridade usando a eletroforese em gel de agarose 1% corado com brometo de etídio.
Nota: Uma relação de absorbância entre 1,85 e 2.0 indica que a preparação é com a qualidade desejada. Tratar de RNA com DNase I (10 µ g/mL) por 10 min a 37 ° C. Use uma coluna de rotação comercial para concentrar o RNA em de água livre de RNase31. Amostras de RNA de piscina antes de prosseguir. - Use um kit de remoção de rRNA para remover a planta do RNA ribossomal. Alíquotas grânulos magnéticos para o tubo de microcentrifugadora e lavar duas vezes com RNase-livre da água. Vórtice do tubo alíquota para Ressuspender, coloque o tubo num suporte magnético e esperar o líquido limpar. Desprezar o sobrenadante e substituir com a solução de ressuspensão magnética do grânulo. Vórtice Resuspenda e adicionar 1 µ l de RNase inibidor.
Nota: Esses kits de usam oligo-dT acoplado a grânulos magnéticos que hibridizam para mRNA. O método usa tecnologia de separação de microesferas de ímã padrão para recuperar as transcrições24. - Combine 500 ng para 1,25 µ g de RNA, água livre de RNase e buffers de reação fornecidos pelo kit. Coloque a mistura por 10 min a 50 ° C. Retire do fogo e adicione grânulos magnéticos lavados em água livre de RNAse. Vórtice brevemente e conjunto em temperatura ambiente por 5 min.
- Coloque em um suporte magnético e aguarde até que o liquido limpar. Transferi o sobrenadante para um tubo de microcentrifugadora fresco. Definido no gelo.
- Usar um método de captura baseados em solução para enriquecimento de exosomes e 200 ng do RNA para preparar a biblioteca de cDNA.
Nota: A biblioteca de cDNA de fita dupla é tipicamente preparada por uma instalação NGS que realiza trabalho orientado ao cliente. - Fragmento do RNA usando uma solução comercial para a fragmentação do RNA (0,136 g ZnCl2 e 100 mM Tris-HCl pH 7.0). Adicionar 2 µ l da solução de 18 µ l de RNA (total de 200 ng). Tubos de centrifugação brevemente na microcentrifuga, coloque as amostras, a 70 ° C por 30 s e a transferência para o gelo. Pare a reação usando 2 µ l de 0,5 M EDTA pH 8.0 e 28 µ l de pH 10 mM Tris-HCl 7.5.
- RNA de vincular a grânulos magnéticos pela mistura à temperatura ambiente durante 10 min. Use um concentrador magnético para coletar os grânulos e descartar o sobrenadante. Lave os grânulos três vezes com 200 µ l de etanol 70%. Descarte cada um lavar e depois secar os grânulos peletizados em temperatura ambiente por 3 min. Resuspenda em 19 µ l de pH 10 mM Tris-HCl 7.5.
- Recoze primers aleatórios para RNA fragmentado por aquecimento a 70 ° C durante 10 minutos e em seguida, coloque o tubo no gelo por 2 min. Prepare a primeira vertente e cDNA da costa segunda, usando um kit de síntese do cDNA comercial padrão.
- Purifica o cDNA da dobro-Costa usando um concentrador magnético do grânulo. Lave com 800 µ l de etanol a 70% três vezes. Descarte cada um lavar e secar as bolinhas em temperatura ambiente por 3 min. Resuspenda em 16 µ l de pH 10 mM Tris-HCl 7.5. Use o concentrador magnético do grânulo para separar os grânulos do cDNA double-stranded, que agora está em solução. Remova o cDNA pipetando para um novo tubo de PCR de 200 µ l.
- Efectue a reparação de fim do fragmento usando Taq polimerase e uma mistura de deoxyribonucleotides fornecido por um kit de preparação comercial de biblioteca. O kit comercial fornece adaptadores pre-diluídos a cada participante do cDNA dobro-Costa usando comercial ligase a 25 ° C por 10 min.
4. NGS de DNA Biblioteca preparados a partir de preparação de vírus bruto e dsDNA biblioteca preparados a partir de mRNA
- Usar um instrumento de pyrosequencing padrão alto throughput e seguir protocolos dos fabricantes todos recomendados para gerar leituras diretas de sequências de ADN. Use os reagentes comerciais sequenciamento, incluindo nucleotídeos fluorescente etiquetados.
Nota: Para detalhes consulte as instruções do fabricante, fornecidas com o instrumento. - Realizar análise pós-sequenciamento utilizando software de montagem do genoma que monta automaticamente lê para produzir o primeiro conjunto de contigs com um comprimento médio de < 700 BP usar o software de FastQC no site iPlant/CyVerse que executa controle de qualidade controlos de dados da sequência primas32. Selecione sequências com PQV escores ≥ 30 continuar a reconstruir mais sequências de menor sequência lê24 usando mapeamento e amplicon software.
Nota: Para obter detalhes, consulte instruções do fabricante. - Submeter estes contigs montados para análise de NCBI-BLASTn usando o módulo padrão de MEGARAJADA, bem como Viridplantae (CNPJ: 33090) e vírus (CNPJ: 10239) como a limitação dos nomes de33. Juntem-se a subpopulação de contigs que mostram alta similaridade de genomas de Badnavirus relatadas em um relatório.
- Verifique se que os andaimes associados que representam um ou mais genomas completos vírus candidato, corretamente produzir sequências em-frame que têm a mesma organização do genoma de badnavirus padrão. Para fazer isso, entrada da genoma do vírus de comprimento total de candidato em um plasmídeo, software de desenho. Em seguida, confirme os primeiros 15 nucleotídeos consiste de um tRNAconheceu (TGGTATCAGAGCGAG) que é altamente conservado entre badnaviruses. Localize o sinal de poliadenilação potencial no final 3' do genoma. Anote o genoma completo para identificar a presença de duas ORFs pequenos e um grande ORF codificação uma poliproteína. Em seguida, use o portal ExPASy traduzir ferramenta para identificar o badnavirus ORF1, ORF2 e ORF3 de produtos de tradução34.
Nota: Este software científico é gratuito e irá gerar DNA circular, identificar leitura tudo aberto frames e fornece uma saída imediata para verificar se a sequência representa o genoma de DNA circular de comprimento total. - Use código aberto várias ferramentas de comparação de sequência, músculo e CLUSTALW, para comparar os genomas de vírus obtidos de DNA e RNA análises35,36.
- Pesquise o banco de dados do NCBI nucleotídeo para obter as sequências do genoma completo de 30 badnavirus espécies e exportá-los como um documento no formato de .fasta. Upload de sequências para um software que realiza a análise genética evolutiva das sequências juntamente com as sequências do genoma vírus obtidas por NGS. Gere vários alinhamentos de sequência e árvores de probabilidade máxima usando músculo37.
5. qualidade avaliação de sequenciamento De Novo por amplificação por PCR do vírus genomas de plantas infectadas
- Entrada as sequências de genoma de badnavirus completos recentemente identificado (formato .fasta) na ferramenta online gratuita de Primer3 para derivar PCR primers38. Identifica os conjuntos de cartilha que produzirão produtos escalonados de 1.000-1.500 bp ao longo de todo o comprimento do genome(s) vírus. Envie as sequências para uma instalação de serviço que irá sintetizar e entregar iniciadores de PCR.
Nota: A saída identifica pares aceitável da primeira demão com temperaturas de derretimento comuns e aceitáveis e locais precisos primer junto as sequências introduzidas. - Trabalhando em uma bancada de laboratório e vestindo um jaleco e luvas, isole a 5 µ g de DNA das folhas infectadas por vírus e saudável controle usando um método automatizado que envolve as partículas de celulose paramagnético padrão para isolar o DNA da planta material39 . Congelar o material da folha (20-40 mg) em nitrogênio líquido em um tubo de microcentrifugadora e moer usando um moinho do grânulo. Combine a amostra com tampão de Lise em um tubo de microcentrifugadora e adicionar RNase A para cada amostra. Vórtice da amostra para 10-20 s e brevemente girar a amostra para remover partículas sólidas.
Nota: As partículas de celulose paramagnético têm alta capacidade de ligação a DNA e isolar os rendimentos elevados do DNA puro. Os métodos de coluna de sílica comerciais padrão para isolamento de DNA não eficientemente extrair o DNA de uma grande variedade de espécies de plantas. Consequentemente, existem dezenas de métodos são modificações desses procedimentos para melhorar a eficiência para espécies de plantas individuais. O método de partícula de celulose paramagnético automatizado foi escolhido porque rende mais e maior qualidade DNA de mais de 25 espécies de angiospérmicas herbáceas40. - Use cartuchos de reagente comercial para isolamento de DNA automatizado paramagnético. Adicione 300 µ l de água livre de nuclease para cada reagente comercial cartucho e transferência planta lisada para o cartucho mesmo. Coloque o cartucho no rack cartucho, coloque um êmbolo no poço mais próximo ao tubo de eluição e colocar o tampão de eluição para o tubo de eluição. Carregar cartuchos em máquina automatizada de ácidos nucleicos isolamento e executar a planta DNA isolamento protocolo41,42.
- Realize o PCR para derivar um conjunto de produtos PCR sobrepostos. Use 5 µ m de cada primer frente e verso com 35 ciclos de amplificação por PCR. Use as seguintes condições de ciclismo: desnaturação a 95 ° C por 60 s, recozendo a 50 ° C, durante 45 segundos e extensão a 72 ° C, durante 1-2 min com uma extensão final a 72 ° C por 7-10 min. Use uma coluna de filtração de gel de pré-embalados para eliminar sais e material de baixo peso molecular como na etapa 1.231.
- Calcular uma proporção de 3:1 molar do produto PCR para vector para determinar a quantidade de produto PCR para ligate para 50 ng do pGEM linear do plasmídeo43. Uso um controle inserir DNA para determinar se a ligadura trabalha eficientemente. Realizar a ligadura durante a noite usando T4 DNA ligase (3 U / µ l) a 4 ° C. Então transformar comercialmente preparado JM109 competente Escherichia coli células. Uso controlar 100 pg sem cortes do ADN do plasmídeo como um controle positivo para transformação eficiente. 100 µ l de células transformadas em placas LB-ágar da placa com antibiótica e azul/branco seleção recuperar plasmídeos ligados26. Incubar as placas por 16-24 h a 37 ° C.
Nota: O vetor pGEM tem um lacZ gene que codifica a β-galactosidase. Transformado as bactérias cultivadas em uma placa contendo 100 ampicilina µ g/mL, 0,5 mM IPTG, 80 µ g/mL 5-bromo-4-chloro-3-indoyl-β-D-galactopyranosidase (X-gal) ficará azul devido à atividade de β-galactosidase. O plasmídeo pGEM é linearizado de uma forma que interrompe o gene lacZ . Colônias que contêm as inserções de produto PCR perturbam o gene lacZ e não metabolizam X-galão. Estas colônias são brancas. Assim, colônias com uma inserção podem ser diferenciadas daqueles sem uma inserção pela cor da colônia (branco e azul)26. - Isole DNA de três colônias usando um padrão baseado em coluna plasmídeo isolamento kit39. Sequência de três plasmídeos de produto de transformação. Compare cada sequência de ADN com os genomas de vírus de novo reunidos produzidos por NGS. Use o CLUSTALW para alinhar as sequências e certifique-se de que elas são ordenadas adequadamente.
Resultados
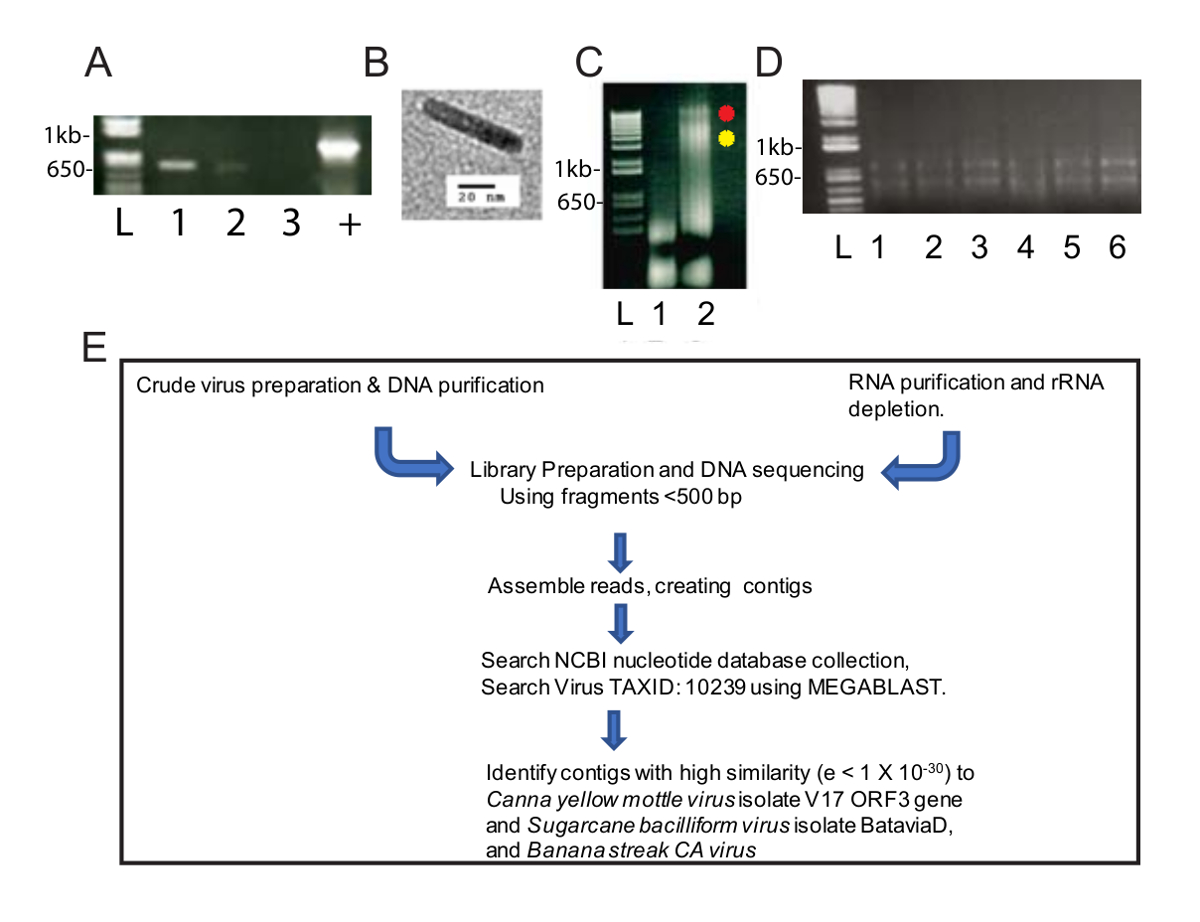
Este método de purificação de vírus modificados desde um enriquecimento do vírus DNAs útil para identificar duas espécies de vírus por NGS e bioinformática. Depois o homogeneizado foi centrifugado a 40.000 x g durante 2,5 h, havia uma bolinha verde na parte inferior do tubo e uma bolinha branca ao longo do comprimento. A pelota verde foi resuspended em um tubo de microcentrifugadora e o sedimento branco foi resuspended em dois tubos de microcentrifuga. PCR foi realizado utilizando primers de diagnóstico padrão CaYMV PCR, e produtos foram detectados na pelota solubilized branca e não a pelota verde (Figura 1A). Uma amostra da preparação bruta foi examinada por microscopia eletrônica de transmissão e observamos as partículas bacilliform medindo 124-133 nm de comprimento (Figura 1B). Isto está dentro do previsto comprimento modal da maioria dos badnaviruses. DNA foi extraído as bolinhas brancas e verdes e resuspended separadamente. Na Figura 1C, nós carregamos a 5 µ l de DNA extraído de verde e amostra de sedimento branco (1,6 µ g de DNA para a fração verde) e 3,1 µ g de DNA para a fração branca de agarose 0,8% electroforese do gel e analisou o DNA seguindo o brometo de etídio a coloração. A fração verde contido baixo peso molecular, DNA, Considerando que a fração branca produzido duas bandas de maior peso molecular DNA, bem como o baixo peso molecular de DNA (Figura 1C). O gel apresentado na Figura 1C foi executado por 40 min a 100 V e o esfregaço na raia 3 sugere que o gel de tensão deve ser diminuída para produzir bandas mais claras. Estes dados sugerem que o sedimento branco foi enriquecido por virions. A concentração de (0,6 µ g/mL) do DNA extraída da amostra branca foi baixa, mas suficiente para NGS, que exige um mínimo de 10 ng de DNA para prosseguir. DNAs fragmentados foram usados para preparar uma biblioteca para NGS.
Em paralelo, RNA foi extraído das plantas de canna infectada (Figura 1D) para alta produtividade RNA-Seq Realizou-se um fluxo de trabalho padrão para a preparação de biblioteca, NGS, criando contigs e identificando sequências do genoma viral (Figura 1E). Foram comparados os resultados da saída do uso de DNA e RNA como matérias-primas.
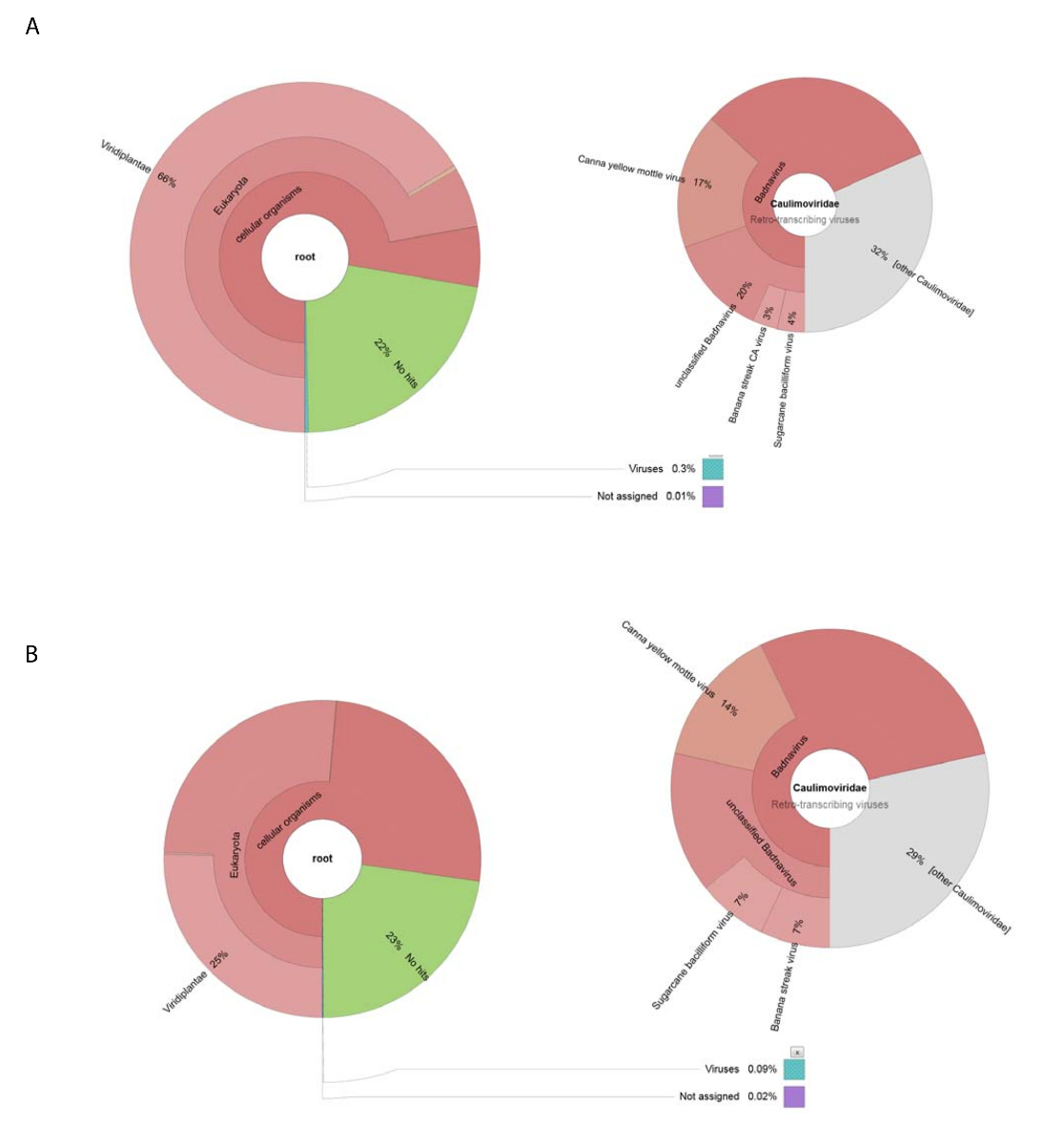
Obtivemos o 188.626 crus leituras de DNA por NGS usando DNA isolado de preparação vírus bruto. Leituras foram montadas em 13.269 contigs e BLASTn foi usado para pesquisar o NCBI dataset de sequências nucleotídicas (usando o Viridplantae TaxID: 33090 e vírus CNPJ: 10239 como os organismos limitantes) (Figura 1E). Os resultados do NCBI-BLASTn revelaram que 93% dos contigs de novo reunidos eram sequências celulares, 22% eram desconhecidos e 0,3% eram vírus contigs(Figura 2). A maioria dos contigs categorizados como sequências de celulares foram identificadas como mitocondrial ou do cloroplasto DNA. Dentro do dataset de vírus contigs, 32% de contigs o vírus estavam relacionados com membros do Caulimoviridae (que não eram Badnavirus sequências) e 58% destes estavam relacionados com Badnavirus. Do vírus contigs, 29% eram altamente semelhantes (e < 1 x 10-30) para CaYMV isolar V17 ORF3 gene (EF189148.1), vírus de Sugarcane bacilliform isolar D Batavia, genoma completo (FJ439817.1), e Banana raia CA virus completa do genoma ( KJ013511). Dentro desta população, havia contigs longo que se assemelhasse a dois genomas completos.
Alto rendimento produzido RNA-seq 153.488 sequência individual limpo leituras com uma média de ler comprimento de < 500 BP Contig montagem esta reduziram a 8.243 contigs. Estes foram submetidos à NCBI-BLASTn (usando o Viridplantae TaxID: 33090 e vírus CNPJ: 10239 como os organismos limitantes) e as saídas colocadas 76% do contigs em uma categoria de celulares sequências de planta, 23% eram desconhecidas, e 0,1% foram categorizados como vírus contigs ( Figura 2 B). uma análise mais aprofundada da população da população de 0,1% de vírus contigs determinado que 68% destes foram atribuídos a Caulimoviridae (Figura 2B). Três grandes contigs dentro desta população foram identificados com alta similaridade (e < 1 X 10-30) para CaYMV isolado gene V17 ORF3 (EF189148.1), vírus de Sugarcane bacilliform isolar D Batavia, genoma completo (FJ439817.1) e Banana streak Vírus de CA completa do genoma (KJ013511). Examinando as três contigs, aderimos manualmente dois para produzir um genoma vírus completos.
Comparamos o contigs de comprimento do genoma vírus produzido por sequenciamento de DNA e RNA como um andaime mútuo para confirmar a presença de dois genomas completos de vírus. Um genoma vírus completos de 6.966 bp foi tentativamente denominado vírus mottle amarelo associado de Canna 1 (CaYMAV-1)(Figura 3). O segundo genoma foi 7.385 bp e uma variante do CaYMV infectando Alpinia purpurata (CaYMV-Ap01)(Figura 3).
Finalmente, iniciadores de PCR que foram projetados para fragmento de bp clone ~ 1.000 de cada vírus, foram usados para detectar diferencialmente os dois genomas em uma população de 227 plantas de canna, representando nove variedades comerciais. Em muitos casos, plantas individuais foram infectadas com ambos os vírus. Nós fornecemos um exemplo de detecção de RT-PCR do CaYMAV-1 e CaYMV-Ap01 em 12 plantas. Três destes foram positivos apenas para CaYMV-Ap01 e nove eram positivos para ambos os vírus (Figura 3B).

Figura 1 : Preparações de ácido nucleico do vírus e fluxo de trabalho NGS. Eletroforese de fragmentos PCR bp 565 de genomas de CaYMV do gel de Agarose (A) (1,0%). Dois produtos PCR foram detectados em amostras preparadas a partir do sedimento branco (faixas 1, 2), mas não na amostra da pelota verde (faixa 3). Controle positivo (+) representa um produto PCR amplificado da planta infectada DNA que foi isolada usando um método automatizado que envolvem as partículas de celulose paramagnético padrão. Lane L contém a escada de DNA usada como um padrão para medir o tamanho das bandas de DNA lineares em pistas de amostra. (B) exemplo de partícula do vírus visualizado por microscopia eletrônica de transmissão no sedimento branco recuperado pelo fracionamento bruto das folhas de canna infectados. (C) Agarose (0,8%) gel de electroforese de DNA recuperado desde o verde (pista 1) e branco (pista 2) pelotas que testado positivo por PCR em painel A. Os pontos vermelhos e amarelos ao lado da pista 2 identificam duas bandas de DNA de alto peso molecular que ocorrem na fração de branco. Electroforese do RNA total recuperado pela purificação de RNA baseado na coluna de gel de Agarose (D) (1%). Lane L contém a escada de DNA usada como um padrão para medir o tamanho das bandas lineares em pistas de amostra. Pista 1-6 contém RNA isolado de folhas de canna infectados que foram agrupadas para uma única amostra para ribo-esgotamento e RNA-Seq. (E) esquemático pipeline de isolamentos de ácido nucleico, preparação de biblioteca, sequenciamento, contig assembly e genoma do vírus descoberta. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2 : Gráficos de krona visualizando as categorias taxonômicas de contigs. (A) o gráfico na esquerda mostra a abundância e distribuição taxonômica de contigs montados desde a preparação do vírus bruto. O mesmo gráfico retrata as proporções de contigs vírus associado com a família Caulimoviridae , gênero Badnavirus e três espécies estreitamente relacionadas. (B) o painel da esquerda mostra a abundância de contigs derivado de RNA-seq baseia sua distribuição taxonômica. À direita está o gráfico representando a abundância de contigs dentro da população de contigs vírus associado com a família Caulimoviridae , gênero Badnavirus e três espécies estreitamente relacionadas. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3 . Caracterização dos genomas CaYMAV-1 e CaYMV-Ap01. (A) representação diagramática de mottle amarelo Canna associar vírus 1 (CaYMAV) e o vírus mottle amarelo de Canna semelhante ao genoma isolado de Alpinia purpurata (CaYMV-Ap01). Posições de nucleotídeo 1-10 é identificado como o início do genoma e contém um tRNAconheceu anticodão site típico da maioria dos genomas de badnavirus. As posições de paragem e arranque para tradução do quadro de leitura aberta (ORF) 1 e 2 são adjacentes. Estas proteínas têm desconhecido funções. ORF3 é uma poliproteína contendo dedo de zinco (ZnF), protease (Pro), transcriptase reversa (RT) e domínios de RNAse H. Uma sequência de sinal 3' poli é conservada para ambos os genomas do vírus. (B) análise de RT-PCR foi realizada utilizando RNA isolado de primers que detectam CaYMAV e CaYMV-Ap01 e folhas infectada pelo vírus. Na mesma população de 12 plantas, três foram infectados com CaYMV-Ap01 apenas, enquanto os restantes foram infectados com CaYMAV e CaYMV-Ap01. (+) indica o controle positivo e (-) indica o controle negativo. Esta figura é reproduzida/modificado de Wijayasekara et al . 24 com permissão. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Nos últimos anos uma variedade de métodos têm sido empregadas para o estudo da biodiversidade de vírus de planta em ambientes naturais, que incluem enriquecedor para partículas vírus-like (VLP) ou vírus específico do RNA ou DNA2,3,44, 45,46 . Esses métodos são seguidos por NGS e bioinformatic análise. O objetivo deste estudo foi encontrar o agente causal de uma doença comum em uma planta cultivada. A doença foi relatada para ser o resultado de um vírus desconhecido, que tem partículas não-envelopado bacilliform, e para que apenas um fragmento de bp 565 tem sido clonado47. Esta informação foi suficiente para os investigadores prévios, hipoteticamente, atribuir o vírus para o gênero Badnavirus dentro da família Caulimoviridae. Enquanto relatórios anteriores a hipótese de que a doença de mottle canna em lírios de canna foi o resultado de um único badnavirus, usando a abordagem de metagenômica descrita neste estudo, determinamos que a doença era causada por badnavirus provisório duas espécies24. Assim, a força do uso de uma abordagem metagenome para descobrir o agente causal de uma doença é que agora podemos identificar situações onde pode haver mais de uma causa.
Nossa abordagem combina dados de sequenciamento de DNA e RNA é completa e também demonstra que os resultados usando duas abordagens produziram resultados consistentes e confirmaram a presença de dois vírus relacionados. Nós empregado um procedimento modificado para isolamento de caulimoviruses e produzido uma amostra que foi enriquecido por ácidos nucleicos de vírus associado e que estavam protegidos dentro do capsídeo do vírus. Um laboratório de serviço foi contratado para realizar o sequenciamento de DNA. O conceito essencial para sequenciamento de novo é que a DNA polimerase incorpora o fluorescente etiquetado nucleotides em uma cadeia de DNA modelo durante ciclos sequenciais de síntese de DNA. O contigs montado seguido por NGS foram apresentados em um fluxo de trabalho de bioinformatic produzindo contigs alguns que foram identificadas como contigs de vírus. Mais uma confirmação do vírus dois genomas10,24,,48,49,50 foi obtida através da análise de bioinformatic da RNA-seq dados obtidos de preparações de RNA ribo-esgotada. Um resultado interessante era aprender que as populações de sequências recuperaram por sequenciamento de DNA e RNA fornecidos distribuições similares dos ácidos nucleicos virais e não virais. Para o sequenciamento de DNA e RNA, < 0.5% das sequências foram de origem do vírus. No seio da população de vírus sequências 78-82% pertencia à família Caulimoviridae. Comparando o vírus montado contigs de sequenciamento de DNA e RNA, confirmamos que os dois genomas montados ocorreram em ambos os conjuntos de dados.
Uma preocupação de usar apenas sequenciamento de DNA para identificar os genomas de vírus novo é que o genoma de badnavirus é um DNA circular aberto. Supõe-se que sequências sobrepondo descontinuidades no genoma podem apresentar obstáculos para a montagem do genoma de contigs. Exame inicial dos resultados de sequenciamento de DNA revelou dois genomas similares do vírus. Formulamos a hipótese que estes genomas também representavam a diversidade genética de uma espécie que não tem sido estudada, ou representados duas espécies co infectando a mesma planta24. Portanto, a análise de bioinformatic coletiva de conjuntos de dados obtidos pelo DNA NGS e sequenciação do ARN, habilitado a confirmação da presença de dois genomas completos.
Há um outro relatório que desenvolveu um método alternativo para extrair VIP e ácidos nucleicos de planta homogenates para estudos de metagenomic, com base em procedimentos para recuperar o DNA do vírus do mosaico da couve-flor (CaMV; um caulimovirus)3. Esta abordagem identificada romance RNA e sequências de DNA vírus em plantas não cultivadas. As etapas derivadas do procedimento de isolamento caulimovirus usado neste estudo para descobrir o agente causal de uma doença de plantas cultivadas são ao contrário as etapas derivadas para extrair VIP de plantas infectadas naturalmente24. O sucesso de ambos os métodos modificados sugere que o procedimento de quadro para caulimovirus isolamento pode ser um valioso ponto de partida para estudos de metagenomic de vírus de plantas em geral.
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Pesquisa foi financiada pelo centro de Oklahoma para o avanço da ciência e tecnologia aplicada pesquisa programa fase II AR 132-053-2; e pelo departamento de agricultura especialidade culturas de Oklahoma programa de bolsas de investigação. Agradecemos o Dr. HongJin Hwang e a instalação de núcleo de Bioinformática OSU que foi apoiada por concessões do NSF (EOS-0132534) e NIH (2P20RR016478-04, 1P20RR16478-02 e 5P20RR15564-03).
Materiais
| Name | Company | Catalog Number | Comments |
| NaH2PO4 | Sigma-Aldrich St. Louis MO | S5976 | Grinding buffer for virus purification |
| Na2HPO4 | Sigma-Aldrich | S0751 | Grinding buffer for virus purification |
| Na2SO3 | Thermo-Fisher Waltham, MA | 28790 | Grinding buffer for virus purification |
| urea | Thermo-Fisher | PB169-212 | Homogenate extraction |
| Triton X-100 | Sigma-Aldrich | X-100 | Homogenate extraction |
| Cheesecloth | VWR Radnor, PA | 21910-107 | Filter homogenate |
| Tris | Thermo-Fisher | BP152-5 | Pellet resuspension& DNA resuspension buffers |
| MgCl2 | Spectrum, Gardena, CA | M1035 | Pellet resuspension buffer |
| EDTA | Spectrum | E1045 | Stops enzyme reactions |
| Proteinase K | Thermo-Fisher | 25530 | DNA resuspension buffer |
| phenol:chloroform:isoamylalcohol | Sigma-Aldrich | P2069 | Dissolve virion proteins |
| DNAse I | Promega | M6101 | Degrade cellular DNA from extracts |
| 95% ethanol | Sigma-Aldrich | 6B-100 | Virus DNA precipitation |
| Laboratory blender | VWR | 58984-030 | Grind leaf samples |
| Floor model ultracentrifuge &Ti70 rotor | Beckman Coulter, Irving TX | A94471 | Separation of cellular extracts |
| Floor model centrifuge and JA-14 rotor | Beckman Coulter | 369001 | Separation of cellular extracts |
| Magnetic stir plate | VWR | 75876-022 | Mixing urea into samples overnight |
| Rubber policeman | VWR | 470104-462 | Dissolve virus pellet |
| 2100 bioanalyzer Instrument | Agilent Genomics, Santa Clare, CA | G2939BA | Sensitive detection of DNA and RNA quality and quantity |
| 2100 Bioanalyzer RNA-Picochip | 5067-1513 | Microfluidics chip used to move, stain and measure RNA quality in a 2100 Bioanalyzer | |
| 2100 Bioanalyzer DNA-High Sensitive chip | 5067-4626 | Microfluidics chip used to move, stain and measure DNA quality in a 2100 Bioanalyzer | |
| Nanodrop spectrophotometer | Thermo-Fisher | ND-2000 | Analysis of DNA/RNA quality at intermediate steps of procedures |
| Plant total RNA isolation kit | Sigma-Aldrich | STRN50-1KT | Isolate RNA for RNA-seq |
| RNase-free water | VWR | 10128-514 | Resuspension of DNA and RNA for NGS |
| RNA concentrator spin column | Zymo Research, Irvine, CA | R1013 | Prepare RNA for RNA-seq |
| rRNA removal kit | Illumina, San Diego, CA | MRZPL116 | Prepare RNA for RNA-seq |
| DynaMag-2 Magnet | ThermoFisher | 12321D | Prepare RNA for RNA-seq |
| RNA enrichment system | Roche | 7277300001 | Prepare RNA for RNA-seq |
| Agarose | Thermo-Fisher | 16500100 | Gel analysis of DNA/RNA quality at intermediate steps of procedures |
| Ethidium bromide | Thermo-Fisher | 15585011 | Agarose gel staining |
| pGEM-T +JM109 competent cells | Promega, Madison, WI | A3610 | Clone genome fragments |
| pFU Taq polymerase | Promega | M7741 | PCR amplify virus genome |
| dNTPs | Promega | U1511 | PCR amplify virus genome |
| PCR oligonucleotides | IDT, Coralvill, IA | Custom order | PCR amplify virus genome |
| Miniprep DNA purification kit | Promega | A1330 | Plasmid DNA purification prior to sequencing |
| PCR clean-up kit | Promega | A9281 | Prepare PCR products for cloning |
| pDRAW32 software | ACAClone | Computer analysis of circular DNA and motifs | |
| MEGA6.0 software | MEGA | Molecular evolutionary genetics analysis | |
| Primer 3.0 | Simgene.com | ||
| Quant-iT™ RiboGreen™ RNA Assay Kit | Thermo-Fisher | R11490 | Fluorometric determination of RNA quantity |
| GS Junior™ pyrosequencing System | Roche | 5526337001 | Sequencing platform |
| GS Junior Titanium EmPCR Kit (Lib-A) | Roche | 5996520001 | Reagents for emulsion PCR |
| GS Jr EmPCR Bead Recovery Reagents | Roche | 5996490001 | Reagents for emulsion PCR |
| GS Junior EmPCR Reagents (Lib-A) | Roche | 5996538001 | Reagents for emulsion PCR |
| GS Jr EmPCR Oil & Breaking Kit | Roche | 5996511001 | Reagents for emulsion PCR |
| GS Jr Titanium Sequenicing kit* | Roche | 5996554001 | Includes sequencing reagents, enzymes, buffers, and packing beads |
| GS Jr. Titanium Picotiter Plate Kit | Roche | 5996619001 | Sequencing plate with associated reagents and gaskets |
| IKA Turrax mixer | 3646000 | Special mixer used with Turrax Tubes | |
| IKA Turrax Tube (specialized mixer) | 20003213 | Specialized mixing tubes with internal rotor for creating emulsions | |
| GS Nebulizers Kit | Roche | 5160570001 | Nucleic acid size fractionator for use during library preparations |
| GS Junior emPCR Bead Counter | Roche | 05 996 635 001 | Library bead counter |
| GS Junior Bead Deposition Device | Roche | 05 996 473 001 | Holder for Picotiter plate during centrifugation |
| Counterweight & Adaptor for the Bead Deposition Devices | Roche | 05 889 103 001 | Used to balance deposition device with picotiter plate centrifugation |
| GS Junior Software | Roche | 05 996 643 001 | Software suite for controlling the instrument, collecting and analyzing data |
| GS Junior Sequencer Control v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Run Processor v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS De Novo Assembler v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Reference Mapper v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Amplicon Variant Analyzer v. 3.0 | Roche | (Included in item 05 996 643 001 above) |
Referências
- Dijkstra, J., Jager, C. P. Practical Plant Virology : Protocols and Exercises. , Springer-Verlag. Berlin Heidelberg. 1 edn (1998).
- Roossinck, M. J. Plant virus metagenomics: biodiversity and ecology. Annu Rev Genet. 46, 359-369 (2012).
- Melcher, U., et al. Evidence for novel viruses by analysis of nucleic acids in virus-like particle fractions from Ambrosia psilostachya. J Virol Methods. 152 (1-2), 49-55 (2008).
- Stobbe, A. H., Schneider, W. L., Hoyt, P. R., Melcher, U. Screening metagenomic data for viruses using the e-probe diagnostic nucleic Acid assay. Phytopathology. 104 (10), 1125-1129 (2014).
- Borah, B. K., et al. Bacilliform DNA-containing plant viruses in the tropics: commonalities within a genetically diverse group. Mol Plant Pathol. 14 (8), 759-771 (2013).
- Bousalem, M., Douzery, E. J., Seal, S. E. Taxonomy, molecular phylogeny and evolution of plant reverse transcribing viruses (family Caulimoviridae) inferred from full-length genome and reverse transcriptase sequences. Arch Virol. 153 (6), 1085-1102 (2008).
- Geering, A. D., et al. Banana contains a diverse array of endogenous badnaviruses. J Gen Virol. 86, Pt 2 511-520 (2005).
- Kunii, M., et al. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics. 5, 80(2004).
- Laney, A. G., Hassan, M., Tzanetakis, I. E. An integrated badnavirus is prevalent in Figure germplasm. Phytopathology. 102 (12), 1182-1189 (2012).
- Gambley, C. F., Geering, A. D., Steele, V., Thomas, J. E. Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch Virol. 153 (8), 1599-1604 (2008).
- Gayral, P., et al. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J Virol. 82 (13), 6697-6710 (2008).
- Lyttle, D. J., Orlovich, D. A., Guy, P. L. Detection and analysis of endogenous badnaviruses in the New Zealand flora. AoB Plants. 2011, 008(2011).
- Le Provost, G., Iskra-Caruana, M. L., Acina, I., Teycheney, P. Y. Improved detection of episomal Banana streak viruses by multiplex immunocapture PCR. J Virol Methods. 137 (1), 7-13 (2006).
- Singh, K., Talla, A., Qiu, W. Small RNA profiling of virus-infected grapevines: evidences for virus infection-associated and variety-specific miRNAs. Funct Integr Genomics. 12 (4), 659-669 (2012).
- Alfson, K. J., Beadles, M. W., Griffiths, A. A new approach to determining whole viral genomic sequences including termini using a single deep sequencing run. J Virol Methods. 208, 1-5 (2014).
- Kreuze, J. F., et al. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology. 388 (1), 1-7 (2009).
- Zheng, Y., et al. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology. 500, 130-138 (2017).
- James, A. P., Geijskes, R. J., Dale, J. L., Harding, R. M. Molecular characterisation of six badnavirus species associated with leaf streak disease of banana in East Africa. Annals of Applied Biology. 158 (3), 346-353 (2011).
- Baranwal, V. K., Sharma, S. K., Khurana, D., Verma, R. Sequence analysis of shorter than genome length episomal Banana streak OL virus like sequences isolated from banana in India. Virus Genes. 48 (1), 120-127 (2014).
- Sukal, A., Kidanemariam, D., Dale, J., James, A., Harding, R. Characterization of badnaviruses infecting Dioscorea spp. in the Pacific reveals two putative novel species and the first report of dioscorea bacilliform RT virus 2. Virus Res. 238, 29-34 (2017).
- BÖmer, M., Turaki, A. A., Silva, G., Kumar, P. L., Seal, S. E. A sequence-independent strategy for amplification and characterisation of episomal badnavirus sequences reveals three previously uncharacterised yam badnaviruses. Viruses. 8 (7), (2016).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Canna yellow mottle virus detected in Canna in Florida. Plant Health Progress. , August 2-4 (2004).
- Zhang, J., et al. Characterization of Canna yellow mottle virus in a new host, Alpinia purpurata, in Hawaii. Phytopathology. 107 (6), 791-799 (2017).
- Wijayasekara, D., et al. Molecular characterization of two badnavirus genomes associated with Canna yellow mottle disease. Virus Res. 243, 19-24 (2018).
- Covey, S. N., Noad, R. J., al-Kaff, N. S., Turner, D. S. Caulimovirus isolation and DNA extraction. Methods Mol Biol. 81, 53-63 (1998).
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: A laboratory manual. 2nd edn. , Cold Spring Harbor Press. (1989).
- Radford, A. D., et al. Application of next-generation sequencing technologies in virology. J Gen Virol. 93, Pt 9 1853-1868 (2012).
- Kanagal-Shamanna, R. Emulsion PCR: Techniques and Applications. Methods Mol Biol. 1392, 33-42 (2016).
- Getts, D. R., et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflammation. 9, 246(2012).
- van Dijk, E. L., Jaszczyszyn, Y., Thermes, C. Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res. 322 (1), 12-20 (2014).
- Gel filtration principles and methods. GE Healthcare. , (2010).
- Goff, S., et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Frontiers in Plant Science. 2, (2011).
- Lin, Z., et al. Next-generation sequencing and bioinformatic approaches to detect and analyze influenza virus in ferrets. J Infect Dev Ctries. 8 (4), 498-509 (2014).
- Artimo, P., et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, Web Server issue 597-603 (2012).
- Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5, 113(2004).
- Hung, J. H., Weng, Z. Sequence Alignment and Homology Search with BLAST and ClustalW. Cold Spring Harb Protoc. 2016 (11), (2016).
- Sohpal, V. K., Dey, A., Singh, A. MEGA biocentric software for sequence and phylogenetic analysis: a review. Int J Bioinform Res Appl. 6 (3), 230-240 (2010).
- Untergasser, A., et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 40 (15), 115(2012).
- Dhaliwa, A. DNA extraction and purification. Mater Methods. 3, 191(2013).
- Moeller, J. R., Moehn, N. R., Waller, D. M., Givnish, T. J. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Moeller, J. R., et al. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Grooms, K. Review: Improved DNA Yield and Quality from Diverse Plant Taxa. , (2015).
- Nishimori, A., et al. In vitro and in vivo antivirus activity of an anti-programmed death-ligand 1 (PD-L1) rat-bovine chimeric antibody against bovine leukemia virus infection. PLoS One. 12 (4), 0174916(2017).
- Rojas, M. R., Gilbertson, R. L. Plant Virus Evolution. Roossinck, M. J. 1, Springer-Verlag. 27-51 (2008).
- Roossinck, M. J. The big unknown: plant virus biodiversity. Curr Opin Virol. 1 (1), 63-67 (2011).
- Roossinck, M. J., Martin, D. P., Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology. 105 (6), 716-727 (2015).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Plant Health Progress. , Online (2004).
- Eni, A., Hughes, J. D., Asiedu, R., Rey, M. Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.). Archives of Virology. 153 (12), Ghana, Togo, Benin and Nigeria. 2263-2272 (2008).
- Harper, G., et al. The diversity of Banana streak virus isolates in Uganda. Arch Virol. 150 (12), 2407-2420 (2005).
- Muller, E., Sackey, S. Molecular variability analysis of five new complete cacao swollen shoot virus genomic sequences. Arch Virol. 150 (1), 53-66 (2005).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados