
Method Article
Combinando l'analisi di DNA in un'estrazione di greggio virione con l'analisi del RNA dalle foglie infette per scoprire nuovi Virus genomi
In questo articolo
Riepilogo
Qui vi presentiamo un nuovo approccio per identificare virus vegetali con genomi di DNA double-strand. Usiamo metodi standard per estrarre RNA e DNA da foglie infette e svolgere il sequenziamento di nuova generazione. Strumenti bioinformatici assemblare sequenze in contigs, identificano contigs che rappresenta il genoma del virus e assegnano genomi a gruppi tassonomici.
Abstract
Questo approccio metagenoma viene utilizzato per identificare virus vegetali con genomi di DNA circolari e le loro trascrizioni. Spesso virus a DNA vegetale che si verificano in titoli bassi nel loro ospite o non possono essere inoculati meccanicamente a un altro host sono difficili da propagare per conseguire un titolo maggiore di materiale infettivo. Foglie infette sono macinati in un buffer delicato con pH ottimale e composizione ionica consigliato per la maggior parte dei retrovirus di Para di bacilliform di purificazione. L'urea è utilizzata per rompere in sui corpi di inclusione che intrappolano i virions e dissolvere componenti cellulari. Centrifugazione differenziale fornisce ulteriore separazione dei virions da contaminanti di pianta. Quindi proteinasi K trattamento rimuove i capsidi. Quindi il DNA virale è concentrato ed utilizzato per il sequenziamento di nuova generazione (NGS). I dati NGS sono utilizzati per assemblare contigs che vengono sottoposti a NCBI-BLASTn per identificare un sottoinsieme delle sequenze di virus nel dataset generato. In una pipeline parallela, RNA è isolato dalle foglie infette tramite un metodo di estrazione RNA basato su colonna standard. Quindi lo svuotamento ribosoma avviene per arricchire per un sottoinsieme dei trascritti di mRNA e virus. Assemblati sequenze derivate da RNA (RNA-seq) di sequenziamento sono stati sottoposti al NCBI-BLASTn per identificare un sottoinsieme di sequenze di virus in questo set di dati. Nel nostro studio, abbiamo identificato due genomi correlati badnavirus full-length in due DataSet. Questo metodo è preferibile a un altro approccio comune che estrae la popolazione totale di piccole sequenze di RNA per ricostituire la pianta virus sequenze genomic. Questo virus di quest'ultimo metagenomica pipeline recupera relative sequenze che sono retrò-trascrivere gli elementi inseriti nel genoma della pianta. Questo è accoppiato all'analisi biochimiche e molecolare di discernere ulteriormente gli agenti infettivi attivamente. L'approccio documentato in questo studio, recupera sequenze rappresentative di replicare il virus che probabilmente indicare infezione attiva del virus.
Introduzione
Malattie delle piante emergenti guidare i ricercatori a sviluppare nuovi strumenti per l'individuazione degli agenti causali corretti. I primi rapporti di malattie da virus nuovi o ricorrenti si basano su sintomi che si verificano comunemente come mosaico e malformazioni della foglia, vena di compensazione, nanismo, avvizzimento, lesioni, necrosi, o altri sintomi. Lo standard per la segnalazione di un nuovo virus come agente causale di una malattia è per separarla da altri agenti patogeni contaminanti, si propagano in host adatto e riprodurre la malattia inoculando nelle piante sane della specie ospite originale. La limitazione in questo approccio è che molti generi di virus vegetali dipendono da un insetto o altri vettori per la trasmissione a un host adatto o tornare alla originale specie ospiti. In questo caso, la ricerca per il vettore appropriato può essere prolungata, ci possono essere difficoltà a fondare colonie di laboratorio del vettore e ulteriori sforzi sono necessari per elaborare un protocollo per la trasmissione sperimentale. Se le condizioni per gli studi di trasmissione di successo di laboratorio non è possibile, quindi il lavoro cade a corto lo standard per la segnalazione di una nuova malattia di virus. Per virus che si verificano nei loro ospiti naturali a titoli molto bassi, i ricercatori devono identificare host alternativo per la propagazione per mantenere scorte sufficienti infettive per svolgere ricerche. Per specie di virus che infettano soltanto poche piante questo può anche essere un ostacolo per la coltivazione di colture di riserva1.
Negli ultimi anni, gli scienziati stanno impiegando più spesso alto-rendimento NGS e metagenomica approcci per scoprire le sequenze di virus che sono presenti nell'ambiente, che può esistere non correlata ad una malattia nota, ma può essere assegnato a generi e specie tassonomica 2 , 3 , 4. tali approcci alla scoperta e alla categorizzazione dei materiali genetici in un ambiente distinto forniscono un modo per descrivere la diversità di virus in natura o la loro presenza in un certo ecosistema ma non necessariamente confermare un quadro per la definizione agenti causali per una malattia apparente.
Il genere Badnavirus appartiene alla famiglia Caulimoviridae dei virus. Questi virus sono bacilliform in forma con genomi di DNA circolare a doppio filamento di circa 7-9 kb. Tutti i virus si replicano attraverso un intermedio del RNA. Virus esistono come episomi e replicare indipendente della pianta cromosomica del DNA5,6. Gli studi di campo delle popolazioni di virus indicano che queste popolazioni di virus sono geneticamente complesse. Inoltre, le informazioni ottenute attraverso una gamma dei genomi delle piante di sequenziamento ad alta hanno scoperto numerosi esempi di frammenti di genoma badnavirus inserite da eventi illegittimo integration in genomi delle piante. Queste sequenze di badnavirus endogeno non sono necessariamente associate a infezione7,8,9,10,11. Successivamente, l'uso di NGS per identificare nuove badnaviruses come l'agente causale della malattia è complicata dalla diversità della sottopopolazione di genomi episomal così come la presenza di sequenze endogeno12,13.
Mentre non c'è non una pipeline ottima per la scoperta dei genomi di romanzo pararetrovirus, ci sono due approcci comuni per identificare questi virus come agenti causali per la malattia. Un metodo consiste nell'arricchire per piccole sequenze di RNA da foglie infette e poi assemblare queste sequenze per ricostituire il virus genome(s)14,15,16,17. Un altro approccio è l'amplificazione di rotolamento cerchio (RCA) per amplificare i virus a DNA circolare genomi18. Il successo di RCA dipende l'età della foglia e il titolo del virus nel tessuto selezionato. I prodotti RCA sono sottoposti a digestione di limitazione e clonati in plasmidi per direct sequencing19,20,21.
Virus giallo mottle canna (CaYMV) è una badnavirus ed è descritto come la causa eziologica della malattia mottle giallo in canna, anche se solo un frammento di bp 565 del genoma è stato precedentemente isolato da infetto cannas22. Uno studio contemporaneo identificato CaYMV in Alpinia purpurata (fioritura zenzero; CaYMV-Ap)23. L'obiettivo di questo studio era di recuperare sequenze di genoma completo badnavirus da gigli canna infetti. Descriviamo un protocollo per purificare i virus da contaminanti di pianta e quindi isolare il DNA virale da questa preparazione e preparare una libreria di DNA per l'uso in NGS. Questo approccio elimina la necessità di passaggi intermedi di amplificazione molecolare. Isoliamo anche mRNA da piante infette per RNA-seq. NGS, che comprende RNA-seq è stata effettuata utilizzando ogni preparazione di acidi nucleici. Contigs assemblati sono stati trovati a relazionarsi con il taxon Badnavirus in entrambi i set di dati utilizzando il centro nazionale per la biotecnologia e Information (NCBI) strumento di ricerca di base di allineamento locale per gli acidi nucleici (BLASTn). Abbiamo identificato i genomi di due badnavirus specie24.
Protocollo
1. generale Virus purificazione mediante centrifugazione differenziale utilizzando il metodo Standard di Covey et al. 25
- In primo luogo, tagliare 80-100 g di foglie da piante malate e macinare in un frullatore waring a 4 ° C, con 200 mL di tampone di macinazione (0,5 M NaH2PO4, 0,5 M Na2HPO4 (pH 7,2). e 0,5% (p/v) Na2SO3). Indossare un camice da laboratorio e guanti per tutti i passaggi di questa procedura.
- Quindi, trasferire l'omogeneizzato (300 mL) in un becher di 1,0 L. Aggiungere l'omogeneizzato all'interno di una cappa chimica 18 g di urea e 25 mL di 10% detergente non ionico (t-Oct-C6H4- OH9(OCH2CH2)).
Nota: Per questo passaggio è meglio indossare occhiali protettivi e una maschera di respirazione semplice per protezione personale. - Agitare con l'agitatore magnetico brevemente nella cappa e coprire il becher con la pellicola. Quindi trasferire il becher coperto di stagnola in una camera fredda e agitare con l'agitatore magnetico durante la notte a 4 ° C.
- Trasferire l'omogeneizzato per centrifugare bottiglie di rotore (250 mL contenitori) e centrifugare in un rotore ad angolo fisso a 4.000 x g per 10 min a 4 ° C. In una cappa chimica, recuperare il surnatante e filtrare attraverso 4 strati di garza.
- Dividere l'omogeneizzato tra provette in polipropilene 38,5 mL e centrifugare per 2,5 h a 40.000 x g a 4 ° C. In genere, verificare la presenza di una pallina verde nella parte inferiore del tubo e una pallina bianca lungo la lunghezza del tubo. Versare il sovranatante e mantenere entrambi pellet; Posizionare il campione sul ghiaccio.
Nota: Il pellet verde contiene cloroplasti, amido e altri organelli. - Lavorare in una cappa chimica, uso un poliziotto di gomma per separare i pellet. Risospendere il pellet bianco in ogni bottiglia di rotore in 1ml di ddH2O nel corso di 1-2 h mantenendo che le sospensioni durante la notte a 4 ° C per permettere i materiali sciogliere completamente nella soluzione. Centrifugare la sospensione a 6.000 x g e a 4 ° C per 10 min rimuovere i detriti rimanenti.
- Centrifugare la sospensione concentrata a 136.000 x g per 2 ore a 4 ° C a pellet virioni. Risospendere il pellet in 1 mL di tampone (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2).
Nota: È un passaggio facoltativo per il trattamento di virioni con dnasi I (10 µ g/mL) per 10 min a 37 ° C per rimuovere il DNA non-incapsidato, vale a dire, contaminando cloroplasto e DNA mitocondriale. Quindi, inattivare la dnasi che con l'aggiunta di EDTA a 1 mM. - Disturbare i virions con 40 µ l di 2 µ g / µ l di proteinasi K a 37 ° C per 15 min.
- Lavorare all'interno di una cappa chimica per recuperare DNA virione di estrazione biologica. Indossare un camice da laboratorio, guanti e la maschera di protezione durante l'estrazione per la protezione contro potenziali effetti acuti sulla salute. Aggiungere 1 volume di fenolo-cloroformio-alcool isoamilico (49:50:1) al campione e agitare a mano per 20 s. centrifuga a temperatura ambiente per 5 min a 16.000 x g. rimuovere la fase acquosa superiore e il trasferimento ad un nuovo tubo. Ripetere questa estrazione due o più volte. Smaltire la fase organica collocandolo in un vetro di scarto per corretto smaltimento chimico istituzionali26.
- Concentrare il DNA mediante precipitazione dell'etanolo. Utilizzare la concentrazione finale di 0.3 M di acetato di sodio (pH 5.2) e 2,5 volumi di etanolo al 95%. Posizionare il campione a-20 ° C per 30-60 min e centrifugare a 13.000 x g per 10-20 min a pellet il DNA26.
- Lavorando presso un banco di laboratorio, risospendere il pellet di DNA in 1 mL di buffer di TE di 0,1 mM (pH 8.0). Filer la sospensione attraverso una colonna di filtrazione commerciale gel (normalmente utilizzata per reazione a catena della polimerasi (PCR) ripulire) per eliminare i sali e materiale di basso peso molecolare che potrebbe ostacolare NGS.
- Analizzare i campioni con elettroforesi su gel di agarosio all'1% usando il bromuro di etidio macchiatura per visualizzare la qualità delle preparazioni. Valutare la qualità di DNA usando uno spettrofotometro nanodrop.
Nota: Un rapporto di assorbanza del campione a λ 260 e 280 λ tra 1,85 e 2.0 indica in genere che la preparazione è "pulita" di impurità ed è della qualità desiderata. - Analizzare la qualità del DNA (utilizzare 5 pg a 10 ng) utilizzando un chip basato strumento di elettroforesi capillare.
Nota: Output di qualità Mostra picchi puliti, che rappresentano frammenti di DNA distribuiti da dimensioni lungo l'asse x. Altezza del picco indica abbondanza del frammento. Vette frastagliate indicano frammenti parzialmente degradati o contaminanti chimici. Curve rotonde rappresentano una sbavatura del DNA che indica scarsa qualità
2. Biblioteca preparazione usando DNA e amplificazione clonale emulsione (emPCR amplificazione)
Nota: La libreria è in genere preparata da un impianto di NGS che svolge il lavoro orientato al cliente.
- Una soluzione di DNA (> 200 ng) utilizza un nebulizzatore che converte il DNA in frammenti di taglio. Legare le schede commerciali secondo istruzioni27 del manuale.
- Eseguire emPCR amplificazione del campione del DNA secondo istruzioni28,29,30 del produttore. Ripetere il passaggio di lavare tre volte e dopo ogni lavaggio, pallina le perle in un minicentrifuge per 10 s. scartare il supernatante dopo ogni lavaggio.
Nota: La procedura inizia con la preparazione delle perle cattura mediante lavaggio con il tampone di lavaggio commerciale fornito con il kit. emPCR è comunemente usato per l'amplificazione di modello per NGS. - Calore denaturare il DNA o RNA a 95 ° C per 2 min e poi 4 ° C fino all'uso. Uso 200 milioni di molecole di DNA/RNA ai branelli di cattura 5 milioni in un volume finale di 30 µ l. preparano un finto campione a fianco del campione di DNA/RNA e attenersi alla procedura seguente con il campione di acido nucleico, nonché l'esempio finto.
- Eseguire emulsificazione nel Vortex il tubo di emulsione olio per 10 s a velocità massima, poi versare l'intero contenuto (4 mL) in una plastica mescolando il tubo che è compatibile con un omogeneizzatore di piattaforma. Posizionare il tubo di agitazione sulla piattaforma di mescolare l'emulsione a 2.000 rpm per 5 min.
- Dispensare aliquote di 100 µ l di emulsione nelle provette tappo 8-striscia o in una piastra a 96 pozzetti. I tubi di CAP o sigillare la piastra ed eseguire emPCR utilizzo programma consigliato28 del produttore.
Nota: Dopo la PCR è completa, controllare i pozzi per vedere se l'emulsione è intatto e poi procedere. Scartare il pozzo intero se l'emulsione è rotto. - Indossare un camice da laboratorio e lavorare in una cappa chimica per raccogliere le perle del DNA Amplificate (ADB). Vuoto di aspirare l'emulsione dai pozzetti e raccogliere le perle in un tubo da 50 mL. Lavare i pozzetti due volte con 100 µ l di isopropanolo e aspirare il risciacquo per lo stesso tubo 50 mL.
- Vortice i raccolti delle emulsioni e risospendere il ADB con isopropanolo ad un volume finale di 35 mL. A pellet ADB a 930 x g per 5 min, rimuovere il supernatante e aggiungere 10 mL di buffer di valorizzare. Vortice ADB e poi lavare con l'aggiunta di isopropanolo a 40 mL di volume finale centrifuga e scartare il supernatante dopo ogni lavaggio e ripetere la fase di lavaggio due volte.
- Effettuare un lavaggio finale utilizzando etanolo al posto di isopropanolo. Aggiungere miglioramento buffer da 35 mL di volume finale, vortice e pellet le perline a 930 x g per 5 min. eliminare il surnatante ma lasciare 2 mL di buffer di migliorare.
- Trasferire la sospensione ad un tubo del microcentrifuge e centrifugare brevemente per pellet ADB. Dopo aver scartato il supernatante, sciacquare la pallina ADB due volte con 1 mL di tampone d'aumento. Centrifugare e scartare il supernatante dopo ogni lavaggio.
- Per preparare per l'arricchimento di perlina di biblioteca del DNA, aggiungere 1 mL di NaOH N 1 ai talloni. Vortice ADB e poi incubare per 2 min a temperatura ambiente. Centrifugare e scartare il surnatante. Ripetere questo passaggio di lavaggio una volta.
- Aggiungere 1 mL di buffer di ricottura, poi vortice ADB e incubare per 2 min a temperatura ambiente. Brevemente, centrifugare e scartare il surnatante. Ripetere questo passaggio utilizzando nuovamente 100 µ l di tampone di ricottura.
- Per temprare il primer di sequenziamento del DNA, aggiungere 15 µ l di Seq Primer un e 15 µ l di Seq Primer B fornito nel kit. Brevemente mescolare nel vortex e posizionare il tubo del microcentrifuge in un blocco di calore a 65 ° C per 5 min, trasferimento in ghiaccio per 2 min.
- Lavare tre volte con 1,0 mL di tampone di ricottura. Vortexare per 5 s e scartare il supernatante ogni volta.
- Prima di sequenziamento, misurare il numero di perle utilizzando un contatore commerciale della perla. Ci dovrebbe essere almeno 500.000 perline arricchiti.
Nota: Il contatore del branello è uno speciale dispositivo che misura le perle in un tubo del microcentrifuge fornito.
3. generale mRNA isolamento e dsDNA sintesi a partire con infettati Canna lascia che prova mediante RT-PCR per CaYMV utilizzando segnalato diagnostica primer
- Indossare un camice da laboratorio e guanti in lattice per protezione personale in tutti i passaggi successivi. Lavorando presso un banco di laboratorio, raccogliere 12 campioni dalle foglie e immergersi i campioni in azoto liquido. Utilizzare un laminatoio del branello per omogeneizzazione. Utilizzare un kit commerciale che fornisce un metodo standard basata su colonne per isolamento del RNA totale dell'impianto. Aggiungere il buffer di lisi di guanidina-isotiocianato fornito dal kit per il campione di terreno e agitare per 20 s.
- Aggiungere etanolo e mescolare accuratamente, secondo le istruzioni del kit. Aggiungere ogni omogeneato a una colonna di spin che lega il RNA alla membrana. Lavare tre volte ed eluire il RNA in un tubo di recupero24.
- Quantificare il RNA utilizzando uno spettrofotometro per misurare il rapporto di assorbanza a 260 λ e λ 280. verifica il RNA integrità usando l'elettroforesi del gel di agarosio 1% colorato con bromuro di etidio.
Nota: Un rapporto di assorbanza tra 1,85 e 2.0 indica che la preparazione è della qualità desiderata. Trattamento di RNA con dnasi I (10 µ g/mL) per 10 min a 37 ° C. Utilizzare una colonna commerciale spin a concentrare il RNA in RNAsi-libera acqua31. Campioni di RNA piscina prima di procedere. - Utilizzare un kit di rimozione di rRNA di rimuovere piante RNA ribosomiale (rRNA). Aliquotare biglie magnetiche al tubo del microcentrifuge e lavare due volte con RNAsi-libera dell'acqua. Vortice del tubo aliquota per risospendere, mettete il tubo su un supporto magnetico e attendere per liquido cancellare. Scartare il surnatante e sostituire con la soluzione di risospensione di biglie magnetiche. Vortice per risospendere e aggiungere 1 µ l di RNAsi inibitore.
Nota: Tale kit utilizzano oligo-dT associato a biglie magnetiche che ibridano a mRNA. Il metodo utilizza tecnologia di separazione del branello di magnete standard per recuperare le trascrizioni24. - Combinare 500 ng a 1.25 µ g di RNA, acqua RNAsi-libera e buffer di reazione fornito dal kit. Mettere il composto per 10 min a 50 ° C. Togliere dal fuoco e aggiungere biglie magnetiche lavati in acqua libera del RNasi. Vortex brevemente e insieme a temperatura ambiente per 5 min.
- Mettete su un supporto magnetico e attendere che il liquido chiaro. Trasferire il surnatante in una provetta di fresco microcentrifuga. Impostare sul ghiaccio.
- Utilizzare un metodo basato su soluzione capture per arricchimento di esosomi e 200 ng di RNA per preparare la libreria di cDNA.
Nota: La libreria di cDNA di doppio filo in genere viene preparata da un impianto di NGS che svolge il lavoro orientato al cliente. - Frammento del RNA usando una soluzione commerciale per la frammentazione RNA (0,136 g ZnCl2 e 100 mM Tris-HCl pH 7.0). Aggiungere 2 µ l di soluzione a 18 µ l di RNA (Totale 200 ng). Tubi di spin brevemente in microcentrifuga, posizionare i campioni a 70 ° C per 30 s e trasferimento al ghiaccio. Fermare la reazione utilizzando 2 µ l di 0,5 M EDTA pH 8.0 e 28 µ l di 10 mM Tris-HCl a pH 7.5.
- RNA binding a biglie magnetiche mescolando a temperatura ambiente per 10 min. utilizzare un concentratore magnetico per raccogliere le perle e scartare il surnatante. Lavare le perle tre volte con 200 µ l di etanolo al 70%. Elimina ogni lavare e poi asciugare all'aria i branelli pellettati a temperatura ambiente per 3 min. Risospendere in 19 µ l di 10 mM Tris-HCl a pH 7.5.
- Tempri casuale primer di RNA frammentato da riscaldamento a 70 ° C per 10 min e quindi posizionare il tubo sul ghiaccio per 2 min. preparare il primo filo e seconda del cDNA del filo utilizzando un kit di sintesi di cDNA commerciale standard.
- Purificare il cDNA di doppio filo utilizzando un concentratore a biglie magnetiche. Lavare con 800 µ l di etanolo al 70% tre volte. Elimina ogni lavare e asciugare all'aria il pellet a temperatura ambiente per 3 min. risospenda in 16 µ l di 10 mM Tris-HCl a pH 7.5. Utilizzare il concentratore di biglie magnetiche per separare le perle dal cDNA double-stranded, che ora è in soluzione. Rimuovere il cDNA pipettando in una nuova provetta PCR 200 µ l.
- Effettuare riparazione di fine frammento usando Taq polimerasi e una miscela di deossinucleotidi forniti di un kit di preparazione commerciale biblioteca. Il kit commerciale fornisce schede pre-diluiti per aggiungere a ciascuna estremità del cDNA doppio filo utilizzando ligasi commerciale a 25 ° C per 10 min.
4. NGS di DNA biblioteca preparato da preparazione di Virus grezza e dsDNA libreria pronti dal mRNA
- Utilizzare uno strumento di pyrosequencing standard elevato throughput e seguire protocolli consigliati tutti i produttori per generare valori di lettura dirette delle sequenze di DNA. Usare i reagenti di sequenziamento commerciale, tra cui nucleotidi fluorescente contrassegnati.
Nota: Per informazioni, consultare le istruzioni del produttore fornite con lo strumento. - Effettuare analisi di post-sequenziamento utilizzando il software di montaggio di genoma che assembla automaticamente letture per produrre la prima serie di contigs con una lunghezza media di < 700 BP. utilizzare il software di FastQC sul sito iPlant/CyVerse che esegue il controllo di qualità controlli sui dati raw sequenza32. Selezionare sequenze con Phred punteggi ≥ 30 di continuare a ricostruire sequenze più lunghe da più piccola sequenza letture24 utilizzando mapping e il software di amplicon.
Nota: Per informazioni, vedere istruzioni del fabbricante. - Presentare queste contigs assemblato all'analisi di NCBI-BLASTn utilizzando MEGABLAST predefinito modulo così come Viridplantae (TaxID: 33090) e virus (TaxID: 10239) come il organismal che limita i nomi di33. Raccogliere la sottopopolazione di contigs che mostrano l'alta somiglianza segnalata Badnavirus genomi in un report.
- Verificare che i ponteggi riuniti che rappresentano uno o più genomi di virus Full-Length del candidato, producano correttamente nel telaio sequenze che hanno la stessa organizzazione del genoma di badnavirus standard. Per effettuare questa operazione, ingresso il genoma del virus completo lunghezza candidato in un plasmide software di disegno. Quindi di confermare i primi 15 nucleotidi si compone di un tRNAincontrato (TGGTATCAGAGCGAG) che è altamente conservata tra badnaviruses. Individuare il potenziale segnale di poliadenilazione vicino all'estremità 3' del genoma. Annotare l'intero genoma per identificare la presenza di due ORFs piccolo e uno grande ORF codifica una poliproteina. Quindi utilizzare il portale di ExPASy tradurre strumento per identificare i badnavirus ORF1 e ORF2 ORF3 traduzione prodotti34.
Nota: Questo software scientifico è gratuito e sarà generare DNA circolare, identificare tutti aperto lettura cornici e fornisce un'uscita immediata per verificare che la sequenza rappresenta il genoma di DNA circolare di lunghezza completa. - Utilizzare più strumenti di confronto di sequenza, muscolo e CLUSTALW, open source per confrontare il genoma del virus ottenuto da DNA e RNA analisi35,36.
- Cercare nel database di nucleotide NCBI per ottenere le sequenze di genoma completo di 30 badnavirus specie ed esportarli come un documento in formato. fasta. Caricare sequenze a un software che svolge attività di analisi genetica evolutiva delle sequenze insieme le sequenze di genoma del virus ottenute da NGS. Generare più allineamenti di sequenza e alberi di probabilità massima utilizzando muscolo37.
5. qualità valutazione del sequenziamento De Novo dall'amplificazione di PCR del genoma del Virus dalle piante infette
- Le sequenze di genoma recentemente identificati badnavirus Full-Length (formato. fasta) in Primer3, lo strumento gratuito online per derivare PCR primer38di input. Identificare i set di primer che produrrà prodotti sfalsati di 1.000-1.500 bp lungo tutta la lunghezza del genome(s) virus. Inviare le sequenze di una struttura di servizio che sintetizzano e consegnare gli iniettori di PCR.
Nota: L'output identifica le coppie di primer accettabile con temperature di fusione comuni e accettabili e primer precisa luoghi lungo le sequenze introdotte. - Lavora presso un banco di laboratorio e indossa un camice da laboratorio e guanti, isolare 5 µ g di DNA dalle foglie infettate da virus e sano controllo utilizzando un metodo automatizzato che coinvolge particelle standard paramagnetico cellulosa per isolare il DNA da materiale vegetale39 . Congelare il materiale del foglio (20-40 mg) in azoto liquido in un tubo del microcentrifuge e macinare utilizzando un laminatoio del branello. Combinare il campione con il tampone di lisi in una microcentrifuga e aggiungere RNasi A ciascun campione. Vortexare il campione per 10-20 s e brevemente girare il campione per rimuovere particelle solide.
Nota: Cellulosa paramagnetico particelle hanno alta capacità di legare il DNA e isolare alte rese di DNA puro. I metodi di colonna di silice commerciale standard per isolamento del DNA in modo efficiente non estrarre DNA da un'ampia varietà di specie di piante. Di conseguenza, decine di metodi esistono che sono le modifiche di queste procedure per migliorare l'efficienza per le specie vegetali individuali. Il metodo di particella di cellulosa paramagnetico automatizzato è stato scelto perché produce DNA di più e di più alta qualità da più di 25 specie di angiosperme erbacee40. - Utilizzare cartucce di reagente commerciale per isolamento del DNA paramagnetico automatizzato. Aggiungere 300 µ l di acqua gratuita nucleasi a ogni pianta commerciale reagente cartuccia e trasferimento lisato sulla cartuccia stessa. Inserire la cartuccia nel rack cartuccia, mettere un pistone nel pozzo più vicino al tubo di eluizione e inserire il tampone di eluizione nella provetta di eluizione. Caricare le cartucce in macchina l'isolamento di automatizzata degli acidi nucleici ed eseguire la pianta del DNA isolamento protocollo41,42.
- Eseguire PCR per derivare una serie di prodotti di PCR sovrapposti. Utilizzare 5 µM di ogni primer forward e reverse con 35 cicli di amplificazione di PCR. Utilizzare le seguenti condizioni in bicicletta: denaturazione a 95 ° C per 60 s, ricottura a 50 ° C per 45 s e l'estensione a 72 ° C per 1-2 min con estensione finale a 72 ° C per 7-10 min uso una colonna di preconfezionati gel filtrazione per eliminare i sali e materiale di basso peso molecolare come nel passaggio 1.231.
- Calcolare un rapporto molare di 3:1 del prodotto PCR per il vettore a determinare la quantità di prodotto di PCR per legare a 50 ng di plasmide linearizzato pGEM43. Utilizzare un controllo inserimento del DNA per determinare se le legature funzionano in modo efficiente. Eseguire la legatura durante la notte utilizzando T4 DNA ligasi (3 U / µ l) a 4 ° C. Quindi la trasformazione preparati commercialmente JM109 competenti Escherichia coli cellule. Uso di controllo 100 pg di DNA plasmidico uncut come controllo positivo per trasformazione efficiente. Piastra di 100 µ l di cellule trasformate su piastre LB-agar con selezione antibiotica e blu/bianco per recuperare dei plasmidi26. Incubare le piastre per 16-24 h a 37 ° C.
Nota: Il vettore pGEM ha un gene lacZ, che codifica per la β-galattosidasi. Batteri trasformati cresciuti su una piastra contenente ampicillina di 100 µ g/mL, 0,5 mM IPTG, 80 µ g/mL 5-bromo-4-chloro-3-indoyl-β-D-galactopyranosidase (X-gal) diventerà blu a causa di attività della β-galattosidasi. Il plasmide pGEM è linearizzato in un modo che interrompe il gene lacZ . Colonie che contengono gli inserti del prodotto PCR può interferire il gene lacZ e non metabolizza il X-gal. Queste colonie sono bianche. Così colonie con un inserto possono essere differenziati da quelli senza un inserto dal colore della Colonia (bianco e blu)26. - Isolare il DNA da tre colonie utilizzando un kit di isolamento standard basati su colonna plasmide39. Sequenza di tre plasmidi del prodotto di trasformazione. Confrontare ogni sequenza di DNA con i genomi di virus de novo assemblato prodotti da NGS. Utilizzare CLUSTALW per allineare le sequenze e per assicurarsi che essi siano ordinati in modo appropriato.
Risultati
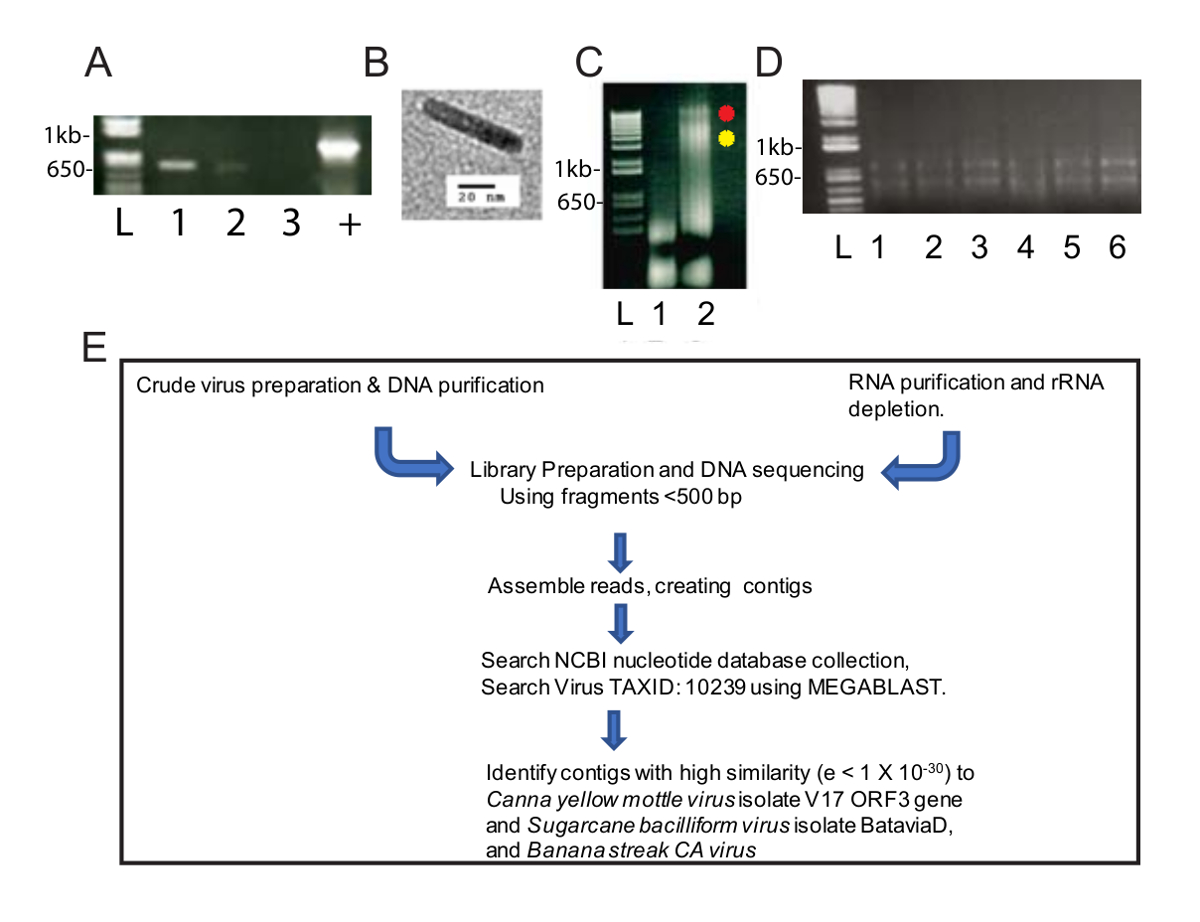
Questo metodo di purificazione del virus modificato fornito un arricchimento del virus DNAs utile per l'identificazione di due specie di virus da NGS e bioinformatica. Dopo l'omogeneizzato è stato centrifugato a 40.000 x g per 2,5 h, c'era una pallina verde nella parte inferiore del tubo e una pallina bianca lungo la lunghezza. La pastiglia verde è stato risospeso in un tubo del microcentrifuge e il pellet bianco è stato risospeso in due provette per microcentrifuga. PCR è stata effettuata utilizzando standard CaYMV diagnostica gli iniettori di PCR, e i prodotti sono stati rilevati in pellet bianco solubilizzate e non la pastiglia verde (Figura 1A). Un campione di preparazione grezza è stato esaminato secondo microscopia elettronica di trasmissione e abbiamo osservato le particelle bacilliform misura 124-133 nm di lunghezza (Figura 1B). Questo è all'interno la lunghezza prevista modale della maggior parte dei badnaviruses. DNA è stata estratta dai pellet bianchi e verdi e risospesi separatamente. Figura 1C, abbiamo caricato 5 µ l di DNA estratto dal verde e pellet bianco campione (1,6 µ g di DNA per la frazione verde) e 3,1 µ g di DNA per la frazione di bianco da 0,8% agarosio elettroforesi su gel e analizzato il DNA seguendo il bromuro di etidio la macchiatura. La frazione verde contenuto basso peso molecolare del DNA, mentre la frazione bianca prodotto due bande di più alto peso molecolare del DNA, così come il più basso peso molecolare del DNA (Figura 1C). Il gel ha presentato nella Figura 1C è stato eseguito per 40 min a 100 V e lo striscio in vicolo 3 suggerisce che la tensione di gel dovrebbe essere abbassata per produrre bande più chiare. Questi dati suggeriscono che la pallina bianca è stata arricchita per i virions. La concentrazione di DNA (0,6 µ g/mL) estratta dal campione bianco era basso, ma adeguata per NGS, che richiede un minimo di 10 ng di DNA per procedere. DNAs frammentati sono stati utilizzati per preparare una libreria per NGS.
In parallelo, il RNA è stato estratto dalle piante infette canna (Figura 1D) per high throughput RNA-seq. Un flusso di lavoro standard è stato effettuato per la preparazione di biblioteca, NGS, creando contigs e identificare sequenze di genoma virale (Figura 1E). Sono stati confrontati i risultati di uscita da usando il DNA e RNA come materiali di partenza.
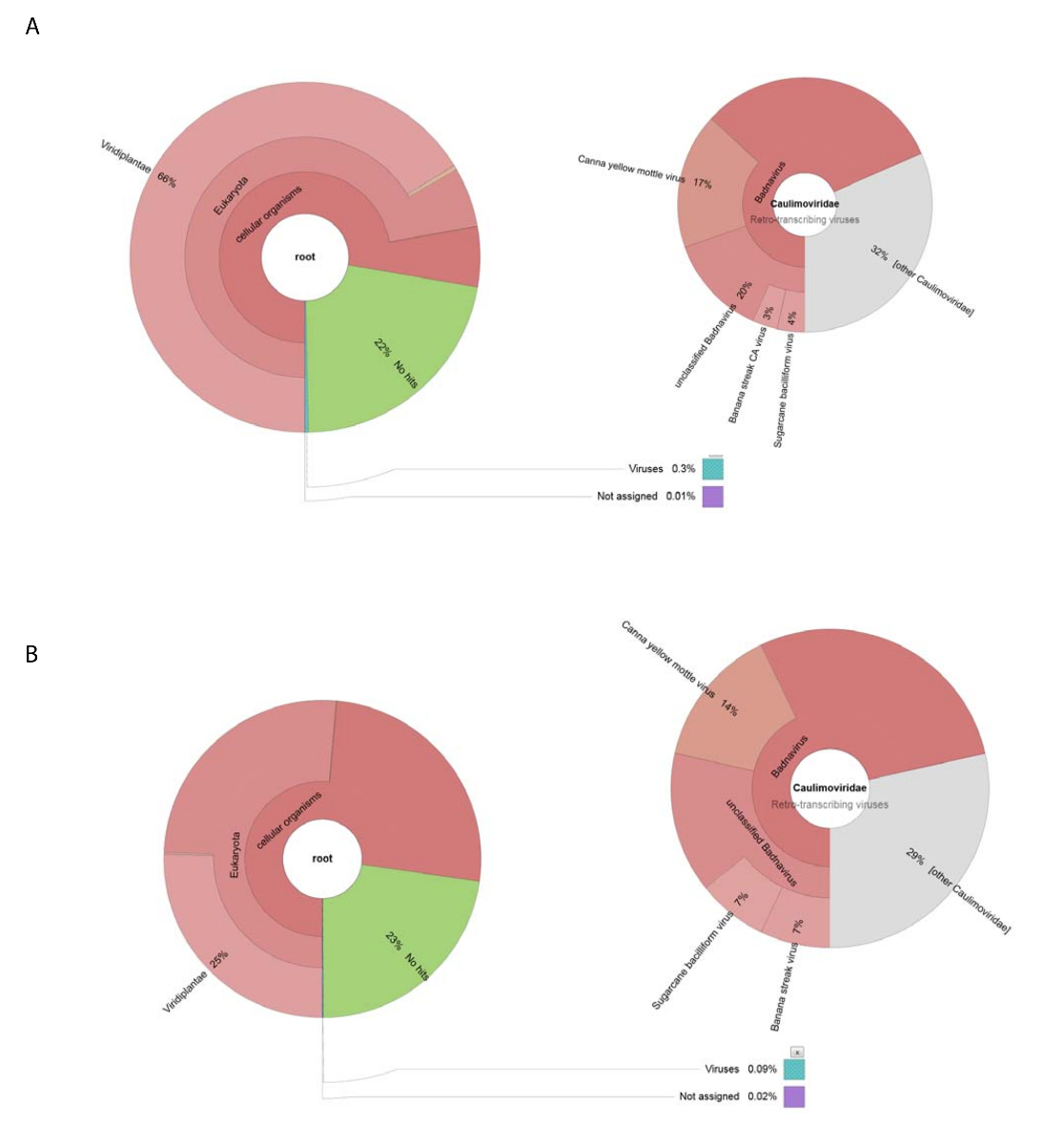
Abbiamo ottenuto 188.626 crudo DNA letture da NGS usando il DNA isolato dalla preparazione di virus grezza. Letture sono state assemblate in 13.269 contigs e BLASTn è stato utilizzato per la ricerca il dataset NCBI delle sequenze nucleotidiche (utilizzando Viridplantae TaxID: 33090 e Virus TaxID: 10239 come gli organismi limitanti) (Figura 1E). I risultati di NCBI-BLASTn ha rivelato che erano 93% del de novo assemblati contigs sequenze cellulari, 22% erano sconosciuti, e 0,3% erano virus contigs (Figura 2A). La maggior parte dei contigs categorizzato come sequenze di cellulari sono stati identificati come mitocondriale o cloroplasto DNA. All'interno del dataset del virus contigs, 32% di contigs i virus sono stati collegati ai membri del Caulimoviridae (che non erano Badnavirus sequenze) e 58% di questi sono stati collegati con Badnavirus. Di contigs il virus, 29% erano molto simili (e < 1 x 10-30) al CaYMV isolare V17 ORF3 gene (EF189148.1), virus del bacilliform di canna da zucchero isolare Batavia D, completa del genoma (FJ439817.1), e Banana streak CA virus completo (genoma KJ013511). All'interno di questa popolazione, c'erano contigs lungo che ha assomigliato a due genomi di integrale.
Alto-rendimento RNA-seq prodotta 153.488 pulite singole sequenze letture con una media lunghezza di < 500 b.p. Contig Assemblea di leggere questo ridotto a 8.243 contigs. Questi sono stati sottoposti a NCBI-BLASTn (utilizzando Viridplantae TaxID: 33090 e Virus TaxID: 10239 come gli organismi limitanti) e le uscite collocate il 76% del contigs in una categoria di sequenze cellulari di impianto, 23% erano sconosciute, e 0,1% sono stati categorizzati come virus contigs ( Figura 2 B). esame più attento della popolazione della popolazione 0,1% di virus contigs determinato che il 68% di questi sono stati assegnati a Caulimoviridae (Figura 2B). Tre grandi contigs all'interno di questa popolazione sono stati identificati con alta somiglianza (e < 1 X 10-30) al CaYMV isolare V17 ORF3 gene (EF189148.1), virus del bacilliform di canna da zucchero isolare Batavia D, completa del genoma (FJ439817.1) e Banana streak Virus di CA completa del genoma (KJ013511). Esaminando i tre contigs, unimmo manualmente due di questi per produrre un genoma di virus full-length.
Abbiamo confrontato il virus genoma lunghezza contigs prodotta mediante sequenziamento del DNA e RNA come impalcatura reciproca per confermare la presenza di due genomi di virus full-length. Un genoma del virus full-length di 6.966 bp è stato provvisoriamente chiamato virus giallo mottle associato Canna 1 (CaYMAV-1) (Figura 3A). Il secondo genoma era 7.385 bp e una variante del CaYMV infettare Alpinia purpurata (CaYMV-Ap01) (Figura 3A).
Infine, gli iniettori di PCR che sono stati progettati per clone ~ 1.000 bp frammento di ogni virus, sono stati utilizzati per rilevare differenzialmente entrambi genomi in una popolazione di 227 piante di canna che rappresenta nove varietà commerciali. In molti casi singoli impianti sono stati infettati con entrambi i virus. Forniamo un esempio di rilevazione di RT-PCR di CaYMAV-1 e CaYMV-Ap01 nelle 12 piante. Tre di questi erano positivi solo per CaYMV-Ap01 e nove erano positive per entrambi i virus (Figura 3B).

Figura 1 : Preparazioni di acido nucleico del virus e NGS workflow. Agarosio (A) (1,0%) elettroforesi di frammenti PCR bp 565 di genomi di CaYMV. Due prodotti di PCR sono stati rilevati in campioni preparati dal pellet bianco (corsie 1, 2) ma non nel campione pastiglia verde (corsia 3). Controllo positivo (+) rappresenta un prodotto PCR amplificato dalla pianta infetta del DNA che è stato isolato utilizzando un metodo automatizzato che coinvolgono particelle standard cellulosa paramagnetico. Lane L contiene la scala di DNA utilizzata come standard per misurare le dimensioni delle bande di DNA lineare nelle corsie di campione. (B) esempio di particella virale visualizzata da microscopia elettronica di trasmissione nel pellet bianco recuperati da frazionamento grezzo di foglie di canna infetti. (C) agarosio (0,8%) gel elettroforesi di DNA recuperato dal verde (corsia 1) e pellet bianchi (lane 2) quello testato positivo mediante PCR in pannello A. I puntini rossi e gialli accanto a corsia 2 identificano due bande di DNA ad elevato peso molecolare che si verificano nella frazione di bianco. (D) agarosio (1%) elettroforesi del gel di RNA totale recuperato dalla purificazione di RNA basata su colonne. Lane L contiene la scala di DNA utilizzata come standard per misurare le dimensioni delle bande lineari nelle corsie di campione. Lane 1-6 contiene RNA isolato dalle foglie di canna infetti che sono stati riuniti per un singolo campione per ribo-svuotamento e pipeline schematica di RNA-seq. (E) di acido nucleico isolamenti, preparazione di biblioteca, sequenziamento, contig assieme e il genoma del virus scoperta. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2 : Grafici di krona visualizzando le categorie tassonomiche di contigs. (A) il grafico a sinistra mostra l'abbondanza e la distribuzione tassonomica di contigs assemblati dalla preparazione grezza virus. Il grafico a destro raffigura le proporzioni del virus contigs connesso con la famiglia Caulimoviridae , Badnavirus genere e tre specie strettamente imparentate. (B) il pannello a sinistra mostra l'abbondanza di contigs derivate da RNA-seq basato sulla loro distribuzione tassonomica. Sulla destra si trova il grafico raffigurante l'abbondanza di contigs all'interno della popolazione del virus contigs connesso con la famiglia Caulimoviridae , Badnavirus genere e tre specie strettamente imparentate. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3 . Caratterizzazione dei genomi di CaYMAV-1 e CaYMV-Ap01. (A) rappresentazione schematica di mottle Canna giallo associare virus 1 (CaYMAV) e virus giallo mottle Canna simile al genoma isolato da Alpinia purpurata (CaYMV-Ap01). Posizioni del nucleotide 1-10 è identificato come l'inizio del genoma e contiene un tipico della maggior parte dei genomi di badnavirus sito anticodonemet tRNA. Le posizioni di stop e start per traduzione di open reading frame (ORF) 1 e 2 sono adiacenti. Queste proteine hanno sconosciuto funzioni. ORF3 è una poliproteina contenente dito di zinco (ZnF), proteasi (Pro), della trascrittasi inversa (RT) e domini RNAsi H. Una sequenza di segnale di poli (a) 3' è conservata per entrambi genomi di virus. (B) l'analisi di RT-PCR è stata effettuata utilizzando RNA isolato dalle foglie infetti da virus e gli iniettori che rilevano CaYMAV e CaYMV-Ap01. Nella stessa popolazione di 12 impianti, tre sono stati infettati con CaYMV-Ap01 solo, mentre i restanti sono stati infettati con CaYMAV e CaYMV-Ap01. (+) indica il controllo positivo e (-) indica il controllo negativo. Questa figura è riprodotto/modificati da Wijayasekara et al. 24 con permesso. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Negli ultimi anni una varietà di metodi sono stati impiegati per studiare la biodiversità di virus vegetali in ambienti naturali che comprendono arricchimento per particelle simili al virus (VLP) o virus specifici RNA o DNA2,3,44, 45,46 . Questi metodi sono seguiti da analisi NGS e bioinformatica. L'obiettivo di questo studio era quello di trovare l'agente causale di una malattia comune in una pianta coltivata. La malattia è stata segnalata per essere il risultato di un virus sconosciuto che ha senza involucro bacilliform particelle, e per cui solo un frammento di bp 565 è stato clonato47. Questa informazione è stata sufficiente per precedenti ricercatori ipoteticamente assegnare il virus al genere Badnavirus all'interno della famiglia Caulimoviridae. Mentre i rapporti anteriori supposto che canna mottle malattia in Gigli di canna era il risultato di un singolo badnavirus, utilizzando l'approccio di metagenomica delineato in questo studio, abbiamo determinato che la malattia è stata causata da due badnavirus provvisorio specie24. Così, la forza di un approccio metagenoma per scoprire l'agente causale di una malattia è che ora possiamo identificare situazioni dove ci possono essere più di una causa.
Il nostro approccio combinando dati di sequenziamento del DNA e RNA è approfondita e dimostra anche che i risultati utilizzando due approcci ha prodotto risultati coerenti e ha confermato la presenza di due virus correlati. Abbiamo impiegato una procedura modificata per isolamento di caulimoviruses e prodotto un campione che è stato arricchito per gli acidi nucleici di virus associato e che sono stati protetti all'interno del capside del virus. Un laboratorio di servizio è stato incaricato di effettuare il sequenziamento del DNA. Il concetto essenziale per il sequenziamento de novo è che quel DNA polimerasi incorpora il fluorescente etichettato nucleotidi in un filamento di DNA modello durante cicli sequenziali della sintesi del DNA. Il contigs assemblati seguita da NGS sono state presentate in un workflow di bioinformatic producendo contigs pochi che sono stati identificati come virus contigs. Ulteriore conferma di virus due genomi10,24,48,49,50 è stata ottenuta tramite analisi bioinformatica del RNA-seq dati ottenuti dalle preparazioni di RNA ribo-vuotati. Un risultato interessante era di imparare che le popolazioni delle sequenze recupero mediante sequenziamento del DNA e RNA fornito distribuzioni simili degli acidi nucleici virali e non virali. Per il sequenziamento del DNA e RNA, < 0,5% delle sequenze erano di origine del virus. All'interno della popolazione del virus sequenze 78-82% apparteneva alla famiglia Caulimoviridae. Confrontando i contigs virus assemblato dal sequenziamento del DNA e RNA, abbiamo confermato che i due genomi assemblati si è verificato in entrambi i set di dati.
Una preoccupazione di usare solo sequenziamento del DNA per identificare i nuovi genomi di virus è che il genoma di badnavirus è un DNA circolare aperto. Abbiamo ipotizzato che sequenze sovrapposte discontinuità nel genoma potrebbero presentare ostacoli per l'assemblaggio del genoma da contigs. L'esame iniziale dei risultati del sequenziamento del DNA ha rivelato due genomi di virus simili. Abbiamo supposto che questi genomi sia rappresentavano la diversità genetica di una specie che non è stata studiata, o rappresentate due specie co-infettare lo stesso impianto di24. Di conseguenza, l'analisi bioinformatica collettiva dei set di dati ottenuti da NGS DNA e RNA 16s, attivata la conferma della presenza di due genomi di integrale.
C'è un altro rapporto che si è sviluppato un metodo alternativo per l'estrazione VLP e acidi nucleici da omogenati di pianta per gli studi di metagenomica, basati su procedure per recuperare il DNA dal virus del mosaico del cavolfiore (CaMV; un caulimovirus)3. Questo approccio identificato romanzo RNA e virus a DNA sequenze nelle piante non coltivate. I passaggi derivati dalla procedura di isolamento caulimovirus utilizzata in questo studio per individuare l'agente causale di una malattia delle piante coltivate sono a differenza i passaggi derivati per l'estrazione VLP da piante naturalmente infettati24. Il successo di entrambi i metodi modificati suggerisce che la procedura di quadro per l'isolamento di caulimovirus può essere un prezioso punto di partenza per studi metagenomica di virus vegetali in generale.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Ricerca è stata finanziata dal centro dell'Oklahoma per l'avanzamento della scienza e tecnologia applicata ricerca programma fase II AR 132-053-2; e dal dipartimento di Oklahoma di colture speciali agricoltura ricerca programma di sovvenzione. Si ringrazia il Dr. HongJin Hwang e la struttura di nucleo di bioinformatica OSU che è stata sostenuta da sovvenzioni da NSF (EOS-0132534) e NIH (2P20RR016478-04, 1P20RR16478-02 e 5P20RR15564-03).
Materiali
| Name | Company | Catalog Number | Comments |
| NaH2PO4 | Sigma-Aldrich St. Louis MO | S5976 | Grinding buffer for virus purification |
| Na2HPO4 | Sigma-Aldrich | S0751 | Grinding buffer for virus purification |
| Na2SO3 | Thermo-Fisher Waltham, MA | 28790 | Grinding buffer for virus purification |
| urea | Thermo-Fisher | PB169-212 | Homogenate extraction |
| Triton X-100 | Sigma-Aldrich | X-100 | Homogenate extraction |
| Cheesecloth | VWR Radnor, PA | 21910-107 | Filter homogenate |
| Tris | Thermo-Fisher | BP152-5 | Pellet resuspension& DNA resuspension buffers |
| MgCl2 | Spectrum, Gardena, CA | M1035 | Pellet resuspension buffer |
| EDTA | Spectrum | E1045 | Stops enzyme reactions |
| Proteinase K | Thermo-Fisher | 25530 | DNA resuspension buffer |
| phenol:chloroform:isoamylalcohol | Sigma-Aldrich | P2069 | Dissolve virion proteins |
| DNAse I | Promega | M6101 | Degrade cellular DNA from extracts |
| 95% ethanol | Sigma-Aldrich | 6B-100 | Virus DNA precipitation |
| Laboratory blender | VWR | 58984-030 | Grind leaf samples |
| Floor model ultracentrifuge &Ti70 rotor | Beckman Coulter, Irving TX | A94471 | Separation of cellular extracts |
| Floor model centrifuge and JA-14 rotor | Beckman Coulter | 369001 | Separation of cellular extracts |
| Magnetic stir plate | VWR | 75876-022 | Mixing urea into samples overnight |
| Rubber policeman | VWR | 470104-462 | Dissolve virus pellet |
| 2100 bioanalyzer Instrument | Agilent Genomics, Santa Clare, CA | G2939BA | Sensitive detection of DNA and RNA quality and quantity |
| 2100 Bioanalyzer RNA-Picochip | 5067-1513 | Microfluidics chip used to move, stain and measure RNA quality in a 2100 Bioanalyzer | |
| 2100 Bioanalyzer DNA-High Sensitive chip | 5067-4626 | Microfluidics chip used to move, stain and measure DNA quality in a 2100 Bioanalyzer | |
| Nanodrop spectrophotometer | Thermo-Fisher | ND-2000 | Analysis of DNA/RNA quality at intermediate steps of procedures |
| Plant total RNA isolation kit | Sigma-Aldrich | STRN50-1KT | Isolate RNA for RNA-seq |
| RNase-free water | VWR | 10128-514 | Resuspension of DNA and RNA for NGS |
| RNA concentrator spin column | Zymo Research, Irvine, CA | R1013 | Prepare RNA for RNA-seq |
| rRNA removal kit | Illumina, San Diego, CA | MRZPL116 | Prepare RNA for RNA-seq |
| DynaMag-2 Magnet | ThermoFisher | 12321D | Prepare RNA for RNA-seq |
| RNA enrichment system | Roche | 7277300001 | Prepare RNA for RNA-seq |
| Agarose | Thermo-Fisher | 16500100 | Gel analysis of DNA/RNA quality at intermediate steps of procedures |
| Ethidium bromide | Thermo-Fisher | 15585011 | Agarose gel staining |
| pGEM-T +JM109 competent cells | Promega, Madison, WI | A3610 | Clone genome fragments |
| pFU Taq polymerase | Promega | M7741 | PCR amplify virus genome |
| dNTPs | Promega | U1511 | PCR amplify virus genome |
| PCR oligonucleotides | IDT, Coralvill, IA | Custom order | PCR amplify virus genome |
| Miniprep DNA purification kit | Promega | A1330 | Plasmid DNA purification prior to sequencing |
| PCR clean-up kit | Promega | A9281 | Prepare PCR products for cloning |
| pDRAW32 software | ACAClone | Computer analysis of circular DNA and motifs | |
| MEGA6.0 software | MEGA | Molecular evolutionary genetics analysis | |
| Primer 3.0 | Simgene.com | ||
| Quant-iT™ RiboGreen™ RNA Assay Kit | Thermo-Fisher | R11490 | Fluorometric determination of RNA quantity |
| GS Junior™ pyrosequencing System | Roche | 5526337001 | Sequencing platform |
| GS Junior Titanium EmPCR Kit (Lib-A) | Roche | 5996520001 | Reagents for emulsion PCR |
| GS Jr EmPCR Bead Recovery Reagents | Roche | 5996490001 | Reagents for emulsion PCR |
| GS Junior EmPCR Reagents (Lib-A) | Roche | 5996538001 | Reagents for emulsion PCR |
| GS Jr EmPCR Oil & Breaking Kit | Roche | 5996511001 | Reagents for emulsion PCR |
| GS Jr Titanium Sequenicing kit* | Roche | 5996554001 | Includes sequencing reagents, enzymes, buffers, and packing beads |
| GS Jr. Titanium Picotiter Plate Kit | Roche | 5996619001 | Sequencing plate with associated reagents and gaskets |
| IKA Turrax mixer | 3646000 | Special mixer used with Turrax Tubes | |
| IKA Turrax Tube (specialized mixer) | 20003213 | Specialized mixing tubes with internal rotor for creating emulsions | |
| GS Nebulizers Kit | Roche | 5160570001 | Nucleic acid size fractionator for use during library preparations |
| GS Junior emPCR Bead Counter | Roche | 05 996 635 001 | Library bead counter |
| GS Junior Bead Deposition Device | Roche | 05 996 473 001 | Holder for Picotiter plate during centrifugation |
| Counterweight & Adaptor for the Bead Deposition Devices | Roche | 05 889 103 001 | Used to balance deposition device with picotiter plate centrifugation |
| GS Junior Software | Roche | 05 996 643 001 | Software suite for controlling the instrument, collecting and analyzing data |
| GS Junior Sequencer Control v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Run Processor v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS De Novo Assembler v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Reference Mapper v. 3.0 | Roche | (Included in item 05 996 643 001 above) | |
| GS Amplicon Variant Analyzer v. 3.0 | Roche | (Included in item 05 996 643 001 above) |
Riferimenti
- Dijkstra, J., Jager, C. P. Practical Plant Virology : Protocols and Exercises. , Springer-Verlag. Berlin Heidelberg. 1 edn (1998).
- Roossinck, M. J. Plant virus metagenomics: biodiversity and ecology. Annu Rev Genet. 46, 359-369 (2012).
- Melcher, U., et al. Evidence for novel viruses by analysis of nucleic acids in virus-like particle fractions from Ambrosia psilostachya. J Virol Methods. 152 (1-2), 49-55 (2008).
- Stobbe, A. H., Schneider, W. L., Hoyt, P. R., Melcher, U. Screening metagenomic data for viruses using the e-probe diagnostic nucleic Acid assay. Phytopathology. 104 (10), 1125-1129 (2014).
- Borah, B. K., et al. Bacilliform DNA-containing plant viruses in the tropics: commonalities within a genetically diverse group. Mol Plant Pathol. 14 (8), 759-771 (2013).
- Bousalem, M., Douzery, E. J., Seal, S. E. Taxonomy, molecular phylogeny and evolution of plant reverse transcribing viruses (family Caulimoviridae) inferred from full-length genome and reverse transcriptase sequences. Arch Virol. 153 (6), 1085-1102 (2008).
- Geering, A. D., et al. Banana contains a diverse array of endogenous badnaviruses. J Gen Virol. 86, Pt 2 511-520 (2005).
- Kunii, M., et al. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: implications for integration and evolution. BMC Genomics. 5, 80(2004).
- Laney, A. G., Hassan, M., Tzanetakis, I. E. An integrated badnavirus is prevalent in Figure germplasm. Phytopathology. 102 (12), 1182-1189 (2012).
- Gambley, C. F., Geering, A. D., Steele, V., Thomas, J. E. Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch Virol. 153 (8), 1599-1604 (2008).
- Gayral, P., et al. A single Banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J Virol. 82 (13), 6697-6710 (2008).
- Lyttle, D. J., Orlovich, D. A., Guy, P. L. Detection and analysis of endogenous badnaviruses in the New Zealand flora. AoB Plants. 2011, 008(2011).
- Le Provost, G., Iskra-Caruana, M. L., Acina, I., Teycheney, P. Y. Improved detection of episomal Banana streak viruses by multiplex immunocapture PCR. J Virol Methods. 137 (1), 7-13 (2006).
- Singh, K., Talla, A., Qiu, W. Small RNA profiling of virus-infected grapevines: evidences for virus infection-associated and variety-specific miRNAs. Funct Integr Genomics. 12 (4), 659-669 (2012).
- Alfson, K. J., Beadles, M. W., Griffiths, A. A new approach to determining whole viral genomic sequences including termini using a single deep sequencing run. J Virol Methods. 208, 1-5 (2014).
- Kreuze, J. F., et al. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology. 388 (1), 1-7 (2009).
- Zheng, Y., et al. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology. 500, 130-138 (2017).
- James, A. P., Geijskes, R. J., Dale, J. L., Harding, R. M. Molecular characterisation of six badnavirus species associated with leaf streak disease of banana in East Africa. Annals of Applied Biology. 158 (3), 346-353 (2011).
- Baranwal, V. K., Sharma, S. K., Khurana, D., Verma, R. Sequence analysis of shorter than genome length episomal Banana streak OL virus like sequences isolated from banana in India. Virus Genes. 48 (1), 120-127 (2014).
- Sukal, A., Kidanemariam, D., Dale, J., James, A., Harding, R. Characterization of badnaviruses infecting Dioscorea spp. in the Pacific reveals two putative novel species and the first report of dioscorea bacilliform RT virus 2. Virus Res. 238, 29-34 (2017).
- BÖmer, M., Turaki, A. A., Silva, G., Kumar, P. L., Seal, S. E. A sequence-independent strategy for amplification and characterisation of episomal badnavirus sequences reveals three previously uncharacterised yam badnaviruses. Viruses. 8 (7), (2016).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Canna yellow mottle virus detected in Canna in Florida. Plant Health Progress. , August 2-4 (2004).
- Zhang, J., et al. Characterization of Canna yellow mottle virus in a new host, Alpinia purpurata, in Hawaii. Phytopathology. 107 (6), 791-799 (2017).
- Wijayasekara, D., et al. Molecular characterization of two badnavirus genomes associated with Canna yellow mottle disease. Virus Res. 243, 19-24 (2018).
- Covey, S. N., Noad, R. J., al-Kaff, N. S., Turner, D. S. Caulimovirus isolation and DNA extraction. Methods Mol Biol. 81, 53-63 (1998).
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: A laboratory manual. 2nd edn. , Cold Spring Harbor Press. (1989).
- Radford, A. D., et al. Application of next-generation sequencing technologies in virology. J Gen Virol. 93, Pt 9 1853-1868 (2012).
- Kanagal-Shamanna, R. Emulsion PCR: Techniques and Applications. Methods Mol Biol. 1392, 33-42 (2016).
- Getts, D. R., et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflammation. 9, 246(2012).
- van Dijk, E. L., Jaszczyszyn, Y., Thermes, C. Library preparation methods for next-generation sequencing: tone down the bias. Exp Cell Res. 322 (1), 12-20 (2014).
- Gel filtration principles and methods. GE Healthcare. , (2010).
- Goff, S., et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Frontiers in Plant Science. 2, (2011).
- Lin, Z., et al. Next-generation sequencing and bioinformatic approaches to detect and analyze influenza virus in ferrets. J Infect Dev Ctries. 8 (4), 498-509 (2014).
- Artimo, P., et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, Web Server issue 597-603 (2012).
- Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5, 113(2004).
- Hung, J. H., Weng, Z. Sequence Alignment and Homology Search with BLAST and ClustalW. Cold Spring Harb Protoc. 2016 (11), (2016).
- Sohpal, V. K., Dey, A., Singh, A. MEGA biocentric software for sequence and phylogenetic analysis: a review. Int J Bioinform Res Appl. 6 (3), 230-240 (2010).
- Untergasser, A., et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 40 (15), 115(2012).
- Dhaliwa, A. DNA extraction and purification. Mater Methods. 3, 191(2013).
- Moeller, J. R., Moehn, N. R., Waller, D. M., Givnish, T. J. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Moeller, J. R., et al. Paramagnetic cellulose DNA isolation improves DNA yield and quality among diverse plant taxa. Appl. Plant Sci. 2 (10), (2014).
- Grooms, K. Review: Improved DNA Yield and Quality from Diverse Plant Taxa. , (2015).
- Nishimori, A., et al. In vitro and in vivo antivirus activity of an anti-programmed death-ligand 1 (PD-L1) rat-bovine chimeric antibody against bovine leukemia virus infection. PLoS One. 12 (4), 0174916(2017).
- Rojas, M. R., Gilbertson, R. L. Plant Virus Evolution. Roossinck, M. J. 1, Springer-Verlag. 27-51 (2008).
- Roossinck, M. J. The big unknown: plant virus biodiversity. Curr Opin Virol. 1 (1), 63-67 (2011).
- Roossinck, M. J., Martin, D. P., Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology. 105 (6), 716-727 (2015).
- Momol, M. T., Lockhart, B. E. L., Dankers, H., Adkins, S. Plant Health Progress. , Online (2004).
- Eni, A., Hughes, J. D., Asiedu, R., Rey, M. Sequence diversity among badnavirus isolates infecting yam (Dioscorea spp.). Archives of Virology. 153 (12), Ghana, Togo, Benin and Nigeria. 2263-2272 (2008).
- Harper, G., et al. The diversity of Banana streak virus isolates in Uganda. Arch Virol. 150 (12), 2407-2420 (2005).
- Muller, E., Sackey, S. Molecular variability analysis of five new complete cacao swollen shoot virus genomic sequences. Arch Virol. 150 (1), 53-66 (2005).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati