É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Caracterização funcional de Carboxylesterases em casa resistente a inseticida moscas, Musca Domestica
Neste Artigo
Resumo
Aqui, apresentamos um protocolo para produzir casa voar carboxylesterase proteínas em vitro com um sistema de expressão baculovirus mediada por células de inseto e mais tarde funcionalmente caracterizar seus papéis em metabolisante permetrina, desse modo, conferindo piretroide resistência por estudos baseados em células MTT ensaio e in vitro metabólica.
Resumo
Mediada por Carboxylesterase metabolismo é pensado para jogar um papel importante na resistência de inseticida em vários tipos de insetos. Vários carboxylesterase de genes foram encontrados acima-está regulada em tensão de voar a casa resistente, Considerando que seus papéis em conferir resistência inseticida permaneceram para ser explorado. Aqui, nós projetamos um protocolo para a caracterização funcional de carboxylesterases. Três experimentos de exemplo são apresentados: (1) expressão e isolamento de proteínas carboxylesterase através de um inseto mediada por baculovirus Spodoptera frugiperda (Sf9) sistema de expressão de células; (2) um MTT baseada em célula (3-[4, 5-dimethykthiazol-2-yl] -2, 5-diphenyltetrazolium brometo) ensaio de citotoxicidade para medir a tolerância das células de inseto à permetrina diferentes tratamentos; e (3) em vitro estudos metabólicos para explorar os recursos metabólicos do carboxylesterases em direção a permetrina. O gene carboxylesterase MdαE7 foi clonada de uma casa resistente voar estirpe ALHF e usado para construir um baculovirus recombinante para Sf9 infecção de células. As realidades de célula contra permetrina diferentes tratamentos foram medidas com o ensaio de MTT. A tolerância de célula reforçada do grupo experimental (células de baculovirus infectado MdαE7-recombinante) comparado com os dos grupos controle (CAT-recombinante baculovirus infectados células e células de baculovirus infectados de GFP-recombinante) a permetrina tratamentos sugeriram que os recursos do MdαE7 em metabolizar inseticidas, protegendo assim as células de danos químicos. Além disso, carboxylesterase proteínas foram expressas em células de inseto Sf9 e isoladas para realizar um estudo metabólico em vitro . Nossos resultados indicaram uma significativa em vitro eficiência metabólica de MdαE7 em direção a permetrina, indicando diretamente o envolvimento do carboxylesterases em metabolizar inseticidas e assim conferir resistência inseticida em casa voa.
Introdução
Resistência de inseticida é atualmente um grande problema de casa fly controle mundial1,2. Os esforços para determinar o mecanismo de resistência de inseticida facilita o melhor entendimento desta questão e, assim, fornecer novas estratégias para efetivamente prevenir ou minimizar a propagação de resistência desenvolvimento3. Carboxylesterases, como uma das enzimas de desintoxicação principais, têm atraído muita atenção por seus papéis em sequestro e metabolizar inseticidas em vários insetos4,5,6. Nosso estudo anterior identificou vários carboxylesterases em moscas domésticas e seus níveis de expressão não foram apenas constitutivamente acima-está regulada na cepa resistente do ALHF, mas também podem ser induzido para maiores níveis em resposta aos tratamentos de permetrina7 . No entanto, as caracterizações funcionais destes genes carboxylesterase em metabolizar inseticidas continuam a ser explorado.
Desde o primeiro relatório no início da década de 19808, um sistema de expressão de gene estrangeira mediada por baculovirus tem sido amplamente empregado devido a sua eficiência de produção de alta proteína e proteína eukaryotic processamento recursos9. Este sistema binário é composto de dois elementos essenciais: o baculovirus recombinante construído entregando estrangeiros genes em células hospedeiras e a expressão em larga escala de proteínas interessadas por células infectadas por baculovirus recombinante. Nas últimas décadas, o sistema de expressão baculovirus mediada por células tem sido amplamente utilizado para produzir milhares de proteínas recombinantes, variando de enzimas citosólica a proteínas de membrana-limite no inseto e10células de mamífero. Nosso estudo anterior com êxito manifestou várias enzimas CYP450 em células de inseto Sf9 com este sistema de11. Neste estudo, temos construído um baculovirus recombinante-carboxylesterase para infectar células de inseto Sf9, explorou a tolerância de célula à permetrina diferentes tratamentos e em grande escala expressa carboxylesterase proteínas em vitro para funcional exploração. Em vez de investigar várias misturas de Isozima carboxylesterase de insetos homogenates adoptado pela anteriores estudos12,13, este sistema de expressão de células de inseto mediada por baculovirus permite a expressão específica e isolamento de proteínas específicas para melhor caracterização das suas propriedades bioquímicas e estruturais.
O ensaio baseado em sal de tetrazólio (MTT) é um método colorimétrico de alta produtividade, desenvolvido e otimizado para medir a viabilidade celular. Este ensaio baseia-se o mecanismo que apenas as células vivas são capazes de metabolizar o reagente MTT amarela a um precipitado de formazan cor roxo escuro, que pode ser analisado colorimetricamente depois dissolvido em solventes orgânicos14, 15. Mais precisos, mas demorados métodos, tais como a exclusão de Trypan azul e a timidina titulação ensaio16,17, vários foram desenvolvidos nos últimos anos. No entanto, o ensaio de MTT baseada em célula ainda atualmente é reconhecido como o método mais rápido e facilmente operado para detectar rapidamente a viabilidade celular. Aqui, usamos o ensaio de MTT para explorar a tolerância celular contra tratamentos insecticidas. A tolerância melhorada de células quando infectadas com baculovirus recombinante carboxylesterase fortemente suporta as funções metabólicas de carboxylesterases aos inseticidas, que por sua vez, sugere sua participação na resistência de inseticida.
Além disso, um ensaio metabólico em vitro foi também realizado neste estudo. Em comparação com ensaios de carboxylesterase geral que usam substratos comuns tais como acetato de α-napthyl (α-at) e β-naftil acetato (β-NA) para refletir a actividades hidrolíticas de carboxylesterases, o estudo metabólico em vitro é considerado como uma maneira exata para medir diretamente as atividades de carboxylesterases em direção a inseticidas18. Este método tem sido empregado com sucesso em vários tipos de insetos para caracterizar vários citocromo P450s em associação com inseticida resistência11,19,20. No entanto, este método ainda não foi aplicado em estudos carboxylesterase. Com a disponibilidade de carboxylesterase proteínas produzidas pelo sistema de expressão mediada por baculovirus, podemos realizar um em vitro estudo metabólico de carboxylesterases em direção a permetrina, que mais pode fornecer fortes evidências do envolvimento de carboxylesterases em conferir resistência piretroide em casa voa.
Protocolo
1. expressão e isolamento de proteínas do alvo com um sistema de expressão de células de inseto mediada por Baculovirus
- Direcionalmente clone sem corte-terminados produtos PCR de proteínas do alvo de moscas domésticas.
- Projetar primers PCR de proteína verde fluorescente (GFP) e o gene MdαE7 casa voar com base em suas sequências e os requisitos especiais do vetor escolhido (tabela 1).
- Usar uma DNA-polimerase termoestável, correcção e primers da etapa 1.1.1 para realizar uma reação de PCR de 150 µ l (30 µ l de tampão de reação, 3 µ l de dNTPs 10 mM, 1,5 µ l de DNA polimerase, 6 µ l de DNA de casa modelo voar, 7,5 µ l de primer para a frente 7,5 µ l de primer reverso, com água até um volume final de 150 µ l). Aqueça a reação de PCR a 98 ° C por 30 s, seguido por 35 ciclos de 98 ° C por 10 s, 53 ° C por 30 s e 72 ° C por 60 s e em seguida uma extensão final a 72 ° C por 2 min).
- Execute um gel de agarose 1% com 150 µ l de produto do PCR.
- Impostos especiais de consumo os fragmentos de DNA alvo do agarose gel com um bisturi afiado e limpo: 1317 bp para MdαE7 e 858 bp para as boas práticas agrícolas. Purifica o DNA usando gel comercialmente disponível kit de extração seguindo o protocolo do fabricante (Tabela de materiais). Dissolva o DNA purificado em 15 µ l de água destilada.
- Execute um gel de agarose 1% com 1 µ l de DNA purificado para verificar a integridade. Use outro 1 µ l de DNA purificado para medir concentrações com o espectrofotómetro.
- Construir um plasmídeo de entrada para as proteínas alvo
- Configure a reação de clonagem. Mistura purificada recentemente produtos DNA da etapa 1.1.3 e comercialmente disponível entrada vetores contendo attL-sites (Tabela de materiais) em uma relação molar de 1:1 (0,5-2 µ l de produto fresco de PCR em concentrações de 70-200 ng / µ l: vetor de 0,5 µ l). Em seguida, adicione 1 µ l de solução salina comercialmente disponível (1,2 M NaCl2 e 0,06 M MgCl2) e adicionar água até um volume final de 6 µ l. Misture delicadamente e incubar a temperatura ambiente durante 1 h.
- Transferir 4 µ l de clonagem de produtos de reacção na etapa 1.2.1 em 50 µ l de quimicamente competentes de Escherichia coli células. Incubar no gelo por 30 min. calor-choque as células por 30 s em banho de água de 42 ° C, sem agitação.
- Coloque o tubo de volta no gelo para outro µ l de 2 min. Adicione 250 de temperatura S.O.C médio (2% triptona extrato de levedura 0,5% 10 mM NaCl; 2,5 mM KCl; 10 mM MgCl2; 10 mM de MgSO4; glicose 20 mM). Incube a 37 ° C, durante 1 h, agitando suavemente.
- Espalhe 50-200 µ l de cultura bacteriana na etapa 1.2.3 nas placas seletivas LB (1 g de triptona 1 g de NaCl 0,5 g de extrato de levedura; 1,5 g de ágar-ágar dissolvido em 100 mL de água destilada. Autoclave e adicionar 0,1% de canamicina 50mg/mL). Incube as placas LB para 16 h a 37 ° C para o crescimento da colônia.
- Escolha 5 a 10 colônias e re-suspendê-las individualmente em 5 µ l de água destilada.
- Realizar PCR adicionando 5 µ l de tampão de reação, 0,5 µ l de dNTPs 10 mM, 0,25 µ l de DNA polimerase, 1 µ l de suspensa colônias, 1,25 µ l de 10 µM M13 forward da primeira demão, 1,25 µ l de 10 µM inversa da primeira demão do gene alvo e da água para o volume final de 25 µ l. aquecer o PCR reação a 98 ° C por 30 s, seguido por 35 ciclos de 98° C por 10 s, 53 ° C por 30 s e 72 ° C por 60 s e em seguida uma extensão final a 72 ° C por 2 min (tabela 1).
- Re-cultura 3 µ l de suspensa colônias da etapa 1.2.5 em mídia TB (100 mL de tampão fosfato contendo 0,17 M KH2PO4 e 0,72 M K2HPO4 com 450 mL do meio de base de caldo de triptona, 12 g de fermento e 2 mL de 6 g glicerol, esterilizado, contendo 0,1% canamicina de 50 mg/mL) por 16 h. extrair DNA de plasmídeo ultra-pura seguindo o protocolo do fabricante.
- Uso de 200 ng / µ l plasmídeo ultra-pura da etapa 1.2.7 para Sanger comercial sequenciando com primers para diante e reversos de M13. Verificar a inserção correta do gene alvo no vetor de acordo com o mapa de sequência fornecido pelo fabricante.
- Armazenar as amostras de DNA de plasmídeo ultra-pura verificada a sequência de MdαE7 e GFP em-20 ° C.
Nota: Estas são a entrada plasmídeo DNA de MdαE7 e GFP.
- Construa um baculovirus recombinante, realizando a reação de recombinação (LR) Lambda.
- Respectivamente, misture 1 µ l de plasmídeo de entrada ng / µ l 300 DNA de GFP, MdαE7 da etapa 1.2.7 ou entrada do plasmídeo DNA do gene do acetyltransferase de cloranfenicol (CAT) fornecido pelo kit de transfecção celular com 5 µ l de comercialmente disponíveis (contendo sites attL) C-termo DNA linear (contendo sites attR) em 250 µ l tubos de PCR. Adicione tampão TE para fazer um volume total de 8 µ l.
Nota: As reações de GFP e CAT foram usadas como grupos de controle. - Adicione 2 µ l de comercialmente disponível mistura enzima recombinação (LR) a Lambda (Tabela de materiais) em cada mistura de passo 1.3.1 para catalisar a reação de LR. Delicadamente, misturar e incubar a 25 ° C durante a noite (≈16 h).
Nota: A reação de LR facilita a recombinação de um attL contendo entrada plasmídeo com uma attcontendo R C-termo DNA linear para gerar um attB-contendo vírus expressando. Esta etapa produz o baculovirus recombinantes para cada proteína do alvo. - Diluir 1 µ l de produtos de reacção LR da etapa 1.3.2 20 X. Realize PCR usando um Polyhedrin para a frente da primeira demão e V5 reverso primer (tabela 1). Use 5 µ l de produtos do PCR para executar um gel de agarose a 1%, para verificar a qualidade. A Figura 1 mostra um exemplo de produto de reação o LR do gene MdαE7.
- Respectivamente, misture 1 µ l de plasmídeo de entrada ng / µ l 300 DNA de GFP, MdαE7 da etapa 1.2.7 ou entrada do plasmídeo DNA do gene do acetyltransferase de cloranfenicol (CAT) fornecido pelo kit de transfecção celular com 5 µ l de comercialmente disponíveis (contendo sites attL) C-termo DNA linear (contendo sites attR) em 250 µ l tubos de PCR. Adicione tampão TE para fazer um volume total de 8 µ l.

Figura 1: resultados do exemplo de reação MdαE7 LR por análise PCR. Diluir 2 µ l de reação LR 200-fold e usar 2 µ l da diluição juntamente com Polyhedrin cartilha para diante e reverso primer V5 para executar um 25 µ l do PCR. Use 5 µ l de produtos do PCR para executar o gel de agarose 1% para verificar a qualidade da reação de LR. um
- Transfect inseto Sf9 de células
- Insetos células Sf9 de cultura com 5 mL de meio de crescimento de célula completa (meio livre de soro com 10% de soro fetal bovino (FBS)) no T25 trataram frascos em 27 ˚ c.
- Remover o sobrenadante e irrigue células conectado à parte inferior com 3 mL de meio de cultura fresco célula completa. Transferi 0,5 mL de células re-suspensas num balão de novo T25 tratados. Adicione 4,5 mL de meio de crescimento completo. Incube a 27 ° C, por 3-4 dias até a próxima transferência.
- Semente de 2 mL de inseto de crescimento log-fase Sf9 células de cultura (≈3.0-5,0 × 106 células) uniformemente na cultura de pilha bem. Permitir que células anexar pelo menos 3 h à temperatura de capuz.
- Verificar a fixação da célula ao observar com um microscópio de fase invertido em 250 X. Remover o meio de cultura celular e substitua por 2 mL do meio de inseto da Grace.
- Preparar a solução de mistura A transfeccao (8 µ l de reagente de infecção de célula comercialmente disponíveis (Tabela de materiais) com 100 µ l de meio inseto de Grace) e solução de mistura B transfeccao de cada gene alvo (9 µ l dos produtos de reação LR da etapa 1.3 com 100 µ l de meio inseto da Grace unsupplemented) em 1,5 mL centrifugar tubos, respectivamente.
- Misture suavemente mistura de transfeccao A e B juntos tocando os tubos. Incube a temperatura ambiente por 35 min no capô.
- Uniformemente, adicione a mistura da etapa 1.4.6 gota a gota sobre as células semeadas da etapa 1.4.4. Selar poços com fitas e incubar a 27 ° C durante a noite.
- Substitua 2 mL do meio de inseto da Grace com 2 mL do meio de crescimento completo. Adicione 100 µM ganciclovir em cada poço para selecionar negativamente contra não-recombinante baculovirus. Selar poços com fita e incubar a 27 ° C, durante 72 h.
- Coletar 72 h pós infecção meio de cultura celular de cada poço e transferir para tubos de centrífuga de 1,5 mL. Centrifugar a 1.500 x g por 5 min a 4 ° C para remover células ou fragmentos grandes.
- Transferi o sobrenadante para novos tubos de centrífuga de 1,5 mL. Armazená-los em 4 ° C, com proteção da luz.
Nota: Estas são as soluções de estoque de vírus P1 para cada gene alvo. - Amplifica o estoque de viral baixa-título P1 (1 × 105-1 × 106 pfu/mL) para uma ação viral foram P2 (5 × 107-1 × 108 pfu/mL).
- Semente de 2 mL de inseto de crescimento log-fase Sf9 células de cultura (≈3.0-5,0 × 106 células) uniformemente na cultura de pilha bem. Permitir que células anexar pelo menos 3 h à temperatura de capuz.
- Inocule a 5 µ l de estoque de vírus P1 obtido na etapa 1.4.10 na célula semeada bem de passo 1.4.12, respectivamente. Em seguida, adicione 100 µM ganciclovir para cada poço. Selar poços e incubar a 27 ° C, durante 72 h.
- Colete P2 soluções estoque de vírus de infecção de post de 72 h. Armazenar a 4 ° C, com proteção da luz.
Nota: Estas são as soluções de estoque de vírus P2 para cada gene alvo. - (Opcional) Repita os passos 1.4.12-1.4.14 com 5 µ l de estoque de vírus P2 para coletar P3 soluções estoque de vírus.
Nota: Estas são as soluções de estoque de vírus P3 para cada gene alvo. - Armazene todos os baculovirus construídos em 4 ° C, com proteção da luz.
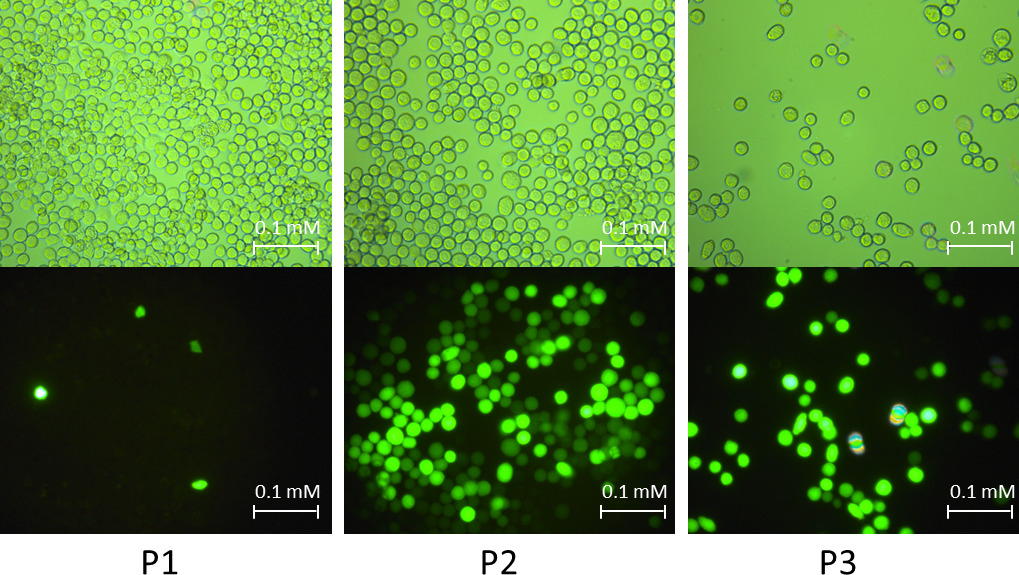
Nota: O gene GFP e gato serviram como grupo de controle. Figura 2 mostrou os sinais das células quando infectado pelo baculovirus recombinante-GFP em fases diferentes de amplificação.

Figura 2: exemplo de sinais infectados de Sf9 células em estágios de amplificação de baculovirus diferente. A figura mostra células de baculovirus recombinante-GFP sob luz natural e fluorescentes luz. Na fase de infecção P1, a taxa de infecção é baixa. Na fase de infecção P2, a taxa de infecção foi aprimorada significativamente. Na fase de infecção P3, quase todas as células apresentou sintomas de afastamento da placa de cultura de células, aumenta de diâmetro da célula, bem como a cessação do crescimento celular. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Expressão em larga escala de proteínas do alvo em células de inseto Sf9
- 10 mL de log crescimento inseto Sf9 células em fase com meio de crescimento completo T25 não tratados frascos de cultura.
- Adicione 200 µ l de solução-mãe vírus P2 (obtida na etapa 1.4.14) para infectar culturas celulares na etapa 1.5.1 para 72 h.
- Colete 72 h pós infecção meio de cultura celular. Centrifugar a 1.500 x g por 5 min a 4 ° C.
- Descarte o sobrenadante e lavar pelotas duas vezes com 1 mL de tampão PBS de 0,1 M (pH 7,5).
- Dissolva totalmente pelotas de célula com 1 mL de extração de proteínas de célula inseto & lise (Tabela de materiais). Adicione 10% de glicerol na lise celular. Imediatamente, armazenar a-80 ° C.
- Repita as etapas 1.5.1-1.5.5 com P2 vírus solução estoque de proteínas CAT para servir como grupo controle.
Nota: Repita o passo 1.5 em triplicado para as preparações de proteína diferentes.
2. um ensaio de citotoxicidade MTT baseada em célula
- 5 mL de log crescimento (1,5-2,5 × 106 células/mL) inseto Sf9 células em fase com meio de crescimento completo T25 não tratados frascos de cultura.
- Adicione 25 µ l de solução-mãe vírus P1 (obtida na etapa 1.4.14) para infectar culturas celulares na etapa 2.1 para 48 h com agitação suave em 27 ˚ c.
- Prepare 100 mM permetrina soluções-padrão estoque em acetonitrila. Use o acetonitrilo para diluir a 50 mM, 25 mM, 12,5 mM e 6,25 mM adicionando 500 µ l, 250 µ l, 125 µ l e 62,5 µ l permetrina solução em um volume total de 1.000 µ l, respectivamente.
- Uniformemente as sementes 200 µ l de culturas de células infectadas da etapa 2.2 suplementado com 300 µ l de meio completo crescimento em placa de 24 em uma densidade de 2 × 105 células/mL.
- Adicione 4 µ l das soluções-padrão permetrina (6,25 mM, 12,5 mM, 25 mM e 50 mM) em cada poço tornar-se uma concentração final de µM 50, 100 µM, 200 µM e 400 µM, respectivamente. Apenas adicione 4 µ l de acetonitrilo em poços para cálculos de viabilidade celular (Figura 3).
- Placas com fitas de vedação e incubar a 27 ° C, durante 48 h com proteção da luz.
- Preparar o reagente MTT (Thiazolyl azul tetrazólio brometo de) 5mg/mL dissolvido em buffer (2,0 g de NaCl, 0,05 g de KCl, 0,36 g de NaH2PO4∙2H2O, 0,28 g de NaH2PO4, 0,05 g de KH2PO4 dissolvido em 200 mL de água destilada água, pH 7,5).
- Remova o meio de cultura celular de passo 2.6 após incubação de 48 h sem tocar a camada celular conectado à parte inferior. Adicione 200 µ l de reagente MTT na etapa 2.7 em cada poço. Incube a 37 ° C por 4 h até formazan roxo escuro precipitados formaram em cada poço (Figura 3).
- Incube a 37 ° C por 4 h até formazan roxo escuro precipita-se forma em cada poço. Adicione 500 µ l de DMSO para dissolver totalmente precipitados (Figura 3).

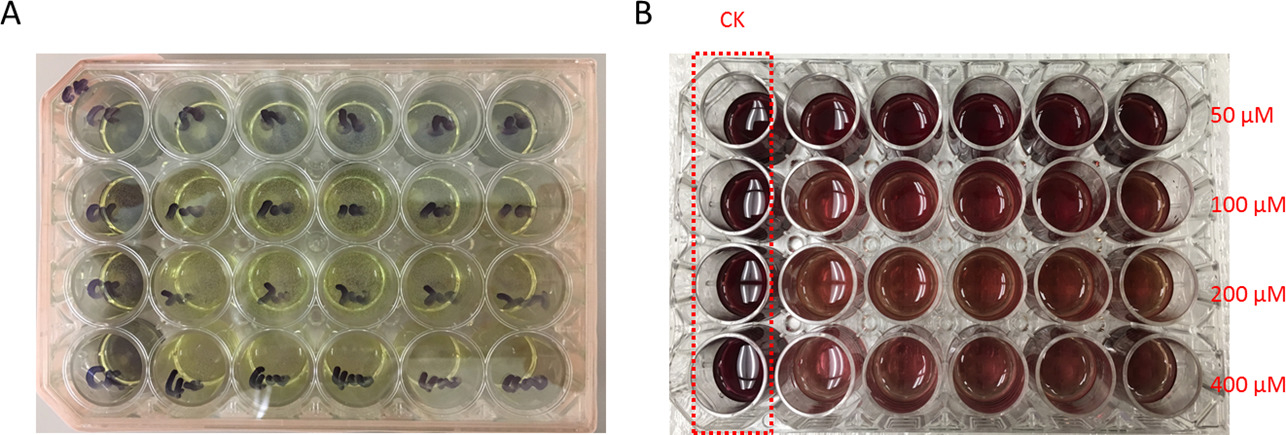
Figura 3: exemplo de resultados MTT. Após 48 h pós infecção com solução-mãe de vírus P1 da solução de baculovirus recombinante CAT, uniformemente semente 500 µ l células expressando o gene CAT em uma placa de cultura de células 24 poços. Respectivamente, adicione 4 µ l permetrina em uma dose diferente (6,25 mM, 12,5 mM, 25 mM e 50 mM) em cada linha para fazer a concentração final de 50 µM e 100 µM, 200 µM 400 µM. A linha de controle foi tratada com acetonitrilo apenas e marcada como CK no prato. (A) após 48 h de incubação a 27 ° C, elimine o meio de cultura de células na camada superior e substitua com 200 µ l de reagentes MTT coloridos amarelos. Então incube a 37 ° C por 48 h. roxo escuro colorida redução precipita-se forma em cada bem, indicando que as células de sobrevivência são capazes de metabolizar a cor amarela reagentes MTT no precipitado de cor roxo escuro. (B) 500 µ l de solvente DMSO foi adicionado em cada bem para dissolver o precipitado formado. O valor de absorvância de cada bem foi detectado pelo espectrofotômetro a 540 nm. Como aumenta a concentração de permetrina, a cor gradualmente irá mudar de vermelho escuro para a luz vermelha, sugerindo que as realidades de célula foram gradualmente diminuiu. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Transferi 200 µ l de solução dissolvida em placa de 96 poços. Medir os valores de absorvância a 540 nm, utilizando um leitor de microplacas (Tabela de materiais).
- Calcule a viabilidade celular, comparando os valores de absorvância das células de permetrina tratada com aqueles de apenas células de acetonitrilo Tratado.
- Para o grupo de controle, repita todos os passos com P2 solução stock de vírus de chloramphenicol acetyltransferase (CAT) baculovirus recombinante obtido na etapa 1.4.
- Repita 4 vezes para preparações de vírus diferentes.
3. in Vitro ensaio metabólico
- Prepare 100 mM permetrina soluções-padrão estoque em acetonitrila. Use o acetonitrilo para diluir a 50 mM, 25 mM, 12,5 mM e 6,25 mM. Detecta a área do pico correspondente sob cada concentração usando HPLC. Criar e calcular a curva padrão de permetrina baseada a área do pico com concentrações diferentes de permetrina.
- Medir as concentrações de proteína na etapa 1.5 com Bradford métodos21.
- Prepare-se 700 µ l de reação metabólica com padrão de permetrina 40 µM e 1 mg de proteínas obtido na etapa 1.5 dissolvida no buffer de 0.2 M Tris-HCl (pH 7,4). Incube a 30 ° C, durante 2 h com agitação suave. Protege da luz.
- Saciar a reação adicionando 700 µ l de acetonitrilo gelado. Incube a 30 ˚ c por mais 30 minutos com agitação suave. Protege da luz.
- Centrifugue a mistura de reação a 16.000 x g por 2 min à temperatura ambiente. Recolha o sobrenadante e filtrar através de uma membrana de 0,45 µm. Transferi a filtragem em frascos de vidro castanho ultraclean para análise HPLC.
- Executar a HPLC sob as condições cromatográficas ideais (fase móvel r: acetonitrilo de 90% e 10% de água; Fase móvel b: 5% acetonitrila ajustado ao pH 2.3 com ácido fosfórico 85%). Gradiente eluir com uma taxa de fluxo de 1 mL/min e medida no comprimento de onda de 232 nm.
- Calcule a porcentagem de depleção de permetrina, comparando com reações sem proteína amostra adicionada.
- Use a proteína CAT obtida na etapa 1.5.6 para servir como o controle.
- Repeti todos os passos com preparações de proteína diferentes na etapa 1.5.7.
Resultados
A viabilidade celular em direção a permetrina diferentes tratamentos (ensaio de MTT)
A citotoxidade de permetrina foi examinada em MdαE7-recombinante baculovirus infectado Sf9 células (grupo experimental) e CAT-recombinante baculovirus (fornecido pelo kit de baculovirus infectado) infectados células (grupos de controle). As tolerâncias de célula reforçada de permetrina em MdαE7 expressando células fortemente apoiar as fun...
Discussão
Nas últimas décadas, sistemas de expressão heteróloga têm sido amplamente utilizados para expressar e isolar grandes quantidades de proteínas, permitindo assim a determinação bioquímica e funcional e caracterização de enzimas em vitro. Até à data, vários sistemas de modelo diferente, incluindo Escherichia colie Pichia pastoris, Sacccharomyces cerevisiae, Spodoptera frugiperda foram adaptados para a expressão da proteína recombinante e a escolha da sistema i...
Materiais
| Name | Company | Catalog Number | Comments |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs inc. | M0491L | |
| QIAquick Gel Extraction Kit | QIAGEN | 28704 | |
| pENTR/D-TOPO Cloning Kit, with One Shot TOP10 Chemically Competent E. coli | Invitrogen by life technology | K240020 | S.O.C medium and universal M13 sequence primers were included in this kit. |
| PureLink HiPure Plasmid Miniprep Kit | Invitrogen by life technology | K210002 | |
| Gateway LR Clonase II Enzyme mix for BaculoDirectTM Kits | Invitrogen by life technology | 11791-023 | |
| BaculoDirect C-Term Linear DNA Transfection Kit | Invitrogen by life technology | 12562-019 | Cellfectin transfection reagent and ganciclovir were included in this kit |
| pENTR-CAT plasmid | Invitrogen by life technology | Included in BaculoDirect C-Term Linear DNA Transfection Kit, concentration: 0.5 ug/uL | |
| Heat inactivated Fetal Bovine Serum, Certified | Gibco by Life Technologies | 10082-139 | |
| Sf9 cells in Sf-900 III SFM | Gibco by Life Technologies | 12659017 | |
| Insect Cell-PE LB Insect Cell Protein Extraction & Lysis Buffer | G Biosciences by A Geno Technology Inc | 786-411 | |
| Sf-900 III SFM (1×) Serum Free Medium Complete | Gibco by Life Technologies | 12658-019 | |
| Grace's Insect Medium, unsupplemented | Gibco by Life Technologies | 11595030 | |
| Permethrin (isomers) analytical standard | SUPELCO by Solutions WithinTM | 442748 | |
| Methanol (analytical graded) | Sigma-Aldrich | 67-56-1 | |
| Acetonitrile (analytical graded) | Sigma-Aldrich | 75-05-8 | |
| GHP Acrodisc 25 mm Syringe Filters with 0.45 μm GHP Membrane (HPLC Certified) | Pall Life Sciences | 21890388 | |
| Alliance Waters 2695 HPLC System | Waters | ||
| T100 Thermal Cycle | Bio-Rad Laboratories Inc. | 1861096 | |
| Nanodrop 2000/2000c Spectrophotometers | ThermoFisher Scientific | ND2000CLAPTOP | |
| Cytation 5 Cell Imaging Multi-Mode Reader | BioTek |
Referências
- Scott, J. G., et al. Insecticide resistance in house flies from the United States: Resistance levels and frequency of pyrethroid resistance alleles. Pesticide Biochemistry and Physiology. 107 (3), 377-384 (2013).
- Li, M., et al. A whole transcriptomal linkage analysis of gene co-regulation in insecticide resistant house flies, Musca domestica. BMC Genomics. 14, 803 (2013).
- Liu, N. Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annual Review of Entomology. 60, 537-559 (2015).
- Grigoraki, L., et al. Transcriptome profiling and genetic study reveal amplified carboxylesterase genes implicated in temephos resistance, in the Asian tiger mosquito Aedes albopictus. e0003771. 9, e0003771 (2015).
- Grigoraki, L., et al. Carboxylesterase gene amplifications associated with insecticide resistance in Aedes albopictus: Geographical distribution and evolutionary origin. PLOS Neglected Tropical Diseases. 11, e0005533 (2017).
- Wheelock, C., Shan, G., Ottea, J. Overview of carboxylesterases and their role in the metabolism of insecticides. Journal of Pesticide Science. 30, 75-83 (2005).
- Feng, X., Li, M., Liu, N. Carboxylesterase genes in pyrethroid resistant house flies, Musca domestica. Insect Biochemistry and Molecular Biology. 92, 30-39 (2018).
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 65 (1-2), 55-63 (1983).
- Jarvis, D. L. Baculovirus-insect cell expression systems. Methods in Enzymology. 463, 191-222 (2009).
- Berger, I., Fitzgerald, D. J., Richmond, T. J. Baculovirus expression system for heterologous multiprotein complexes. Nature Biotechnology. 22 (12), 1583 (2004).
- Gong, Y., Li, T., Feng, Y., Liu, N. The function of two P450s, CYP9M10 and CYP6AA7, in the permethrin resistance of Culex quinquefasciatus. Scientific Reports. 7 (1), 587 (2017).
- Cao, C. W., Zhang, J., Gao, X. W., Liang, P., Guo, H. L. Overexpression of carboxylesterase gene associated with organophosphorous insecticide resistance in cotton aphids, Aphis gossypii (Glover). Pesticide Biochemistry and Physiology. 90 (3), 175-180 (2008).
- Zhang, L., Gao, X., Liang, P. Beta-cypermethrin resistance associated with high carboxylesterase activities in a strain of house fly, Musca domestica (Diptera: Muscidae). Pesticide Biochemistry and Physiology. 89, 65-72 (2007).
- Van Meerloo, J., Kaspers, G. J., Cloos, J. Cell sensitivity assays: the MTT assay. Cancer cell culture. , 237-245 (2011).
- Stockert, J. C., Blázquez-Castro, A., Cañete, M., Horobin, R. W., Villanueva, &. #. 1. 9. 3. ;. MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets. Acta Histochemica. 114 (8), 785-796 (2012).
- Strober, W. Trypan blue exclusion test of cell viability. Current Protocols in Immunology. , (2001).
- Riss, T. L., Moravec, R. A., Niles, A. L., Duellman, S., Benink, H. A., Worzella, T. J., Minor, L. Cell viability assays. Assay Guidance Manual. , (2013).
- Wheelock, C. E., Shan, G., Ottea, J. Overview of carboxylesterases and their role in the metabolism of insecticides. Journal of Pesticide Science. 30 (2), 75-83 (2005).
- Li, X., Schuler, M. A., Berenbaum, M. R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annual Review of Entomology. 52, 231-253 (2007).
- Nakamura, Y., et al. The in vitro metabolism of a pyrethroid insecticide, permethrin, and its hydrolysis products in rats. Toxicology. 235 (3), 176-184 (2007).
- Kruger, N. J. The Bradford method for protein quantitation. The protein protocols handbook. , 15-21 (2002).
- Macauley-Patrick, S., Fazenda, M. L., McNeil, B., Harvey, L. M. Heterologous protein production using the Pichia pastoris expression system. Yeast. 22 (4), 249-270 (2005).
- Berger, I., Fitzgerald, D. J., Richmond, T. J. Baculovirus expression system for heterologous multiprotein complexes. Nature Biotechnology. 22 (12), 1583 (2004).
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology. 72 (2), 211 (2006).
- Bulter, T., et al. Functional expression of a fungal laccase in Saccharomyces cerevisiae by directed evolution. Applied Microbiology and Biotechnology. 69 (2), 987-995 (2003).
- Stepanenko, A. A., Dmitrenko, V. V. Pitfalls of the MTT assay: Direct and off-target effects of inhibitors can result in over/underestimation of cell viability. Gene. 574 (2), 193-203 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados