
Method Article
Medindo a ligação de nucleotídeos a proteínas de membrana intactas e funcionais em tempo real
Neste Artigo
Resumo
Este protocolo apresenta um método para medir a ligação de adenina nucleotídica a receptores em tempo real em ambiente celular. A ligação é medida como transferência de energia de ressonância de Förster (FRET) entre derivados de nucleotídeos trinitrofenila e proteínas marcadas com um aminoácido fluorescente não canônico.
Resumo
Desenvolvemos um método para medir a ligação de nucleotídeos de adenina a receptores transmembrana intactos e funcionais em um ambiente celular ou de membrana. Este método combina a expressão de proteínas marcadas com o aminoácido fluorescente não canônico ANAP, e FRET entre ANAP e derivados nucleotídicos fluorescentes (trinitrofenil). Apresentamos exemplos de ligação de nucleotídeos a canais iônicos KATP marcados com ANAP, medidos em membranas plasmáticas sem teto e remendos de membrana excisados de dentro para fora sob pinça de tensão. Este último permite medições simultâneas da ligação do ligante e da corrente do canal, uma leitura direta da função da proteína. O tratamento e a análise dos dados são discutidos extensivamente, juntamente com possíveis armadilhas e artefatos. Este método fornece informações mecanicistas ricas sobre o gating ligante-dependente de canais deK ATP e pode ser facilmente adaptado ao estudo de outras proteínas nucleotídeos-reguladas ou qualquer receptor para o qual um ligante fluorescente adequado pode ser identificado.
Introdução
Várias classes importantes de proteínas são diretamente reguladas pela ligação do ligante. Estes variam de enzimas solúveis a proteínas embutidas na membrana, incluindo receptores tirosina quinases, receptores acoplados à proteína G (GPCRs) e canais iônicos. Os GPCRs e canais respondem por ~34% e ~15% de todos os alvos atuais de drogas, respectivamente 1,2. Portanto, há um considerável interesse bioquímico, bem como médico no desenvolvimento de métodos que forneçam insights mecanísticos sobre as interações ligante-receptor. Métodos tradicionais para medir a ligação de ligantes, incluindo estudos de marcação por fotoafinidade e ligação a radioligantes, requerem grandes quantidades de proteína parcialmente purificada e são tipicamente realizados sob condições não fisiológicas e escalas de tempo. Um método ideal exigiria apenas pequenas quantidades de proteína, poderia ser realizado em proteínas intactas expressas em um ambiente celular ou de membrana, poderia ser monitorado em tempo real e seria compatível com leituras diretas da função proteica.
A transferência de energia de ressonância de Förster (FRET) é um método que detecta a proximidade entre duas moléculas marcadas fluorescentemente3. O FRET ocorre quando um fluoróforo de doador excitado transfere energia de forma não radiativa para uma molécula receptora (normalmente outro fluoróforo). A transferência de energia resulta na supressão da emissão de fluorescência do doador e sensibilização da emissão do receptor (se o receptor for um fluoróforo). A eficiência da transferência depende da6ª potência da distância entre o doador e o receptor. Além disso, o doador e o receptor devem estar próximos (geralmente menos de 10 nm) para que o FRET ocorra. Como tal, o FRET pode ser explorado para medir a ligação direta entre um receptor de proteína fluorescentemente marcado e um ligante fluorescente.
Várias proteínas diferentes são reguladas ou ativadas pela ligação de nucleotídeos de adenina intracelulares ou extracelulares (ATP, ADP, AMP, cAMP). Muitas proteínas transportadoras requerem hidrólise de ATP para seu ciclo de reação, incluindo transportadores de de ligação a ATP e ATPases do tipo P, como a bomba Na+/K+ 4,5. Os canais de K+ sensíveis ao ATP (KATP), o regulador de condutância transmembrana da fibrose cística (CFTR) e os canais regulados por nucleotídeos cíclicos são canais iônicos que são limitados pela ligação de nucleotídeos de adenina intracelular, tornando-os extremamente sensíveis a mudanças no metabolismo celular e transdução de sinal 6,7,8. Os receptores P2X e P2Y purinérgicos respondem a alterações no ATP extracelular, que pode ser liberado como neurotransmissor ou como resultado de dano tecidual9. Desenvolvemos um ensaio baseado em FRET para a medição da ligação de adenina nucleotídica a proteínas de membrana em tempo real. Aplicamos previamente este método para estudar a ligação de nucleotídeos a canaisde K ATP 10,11.
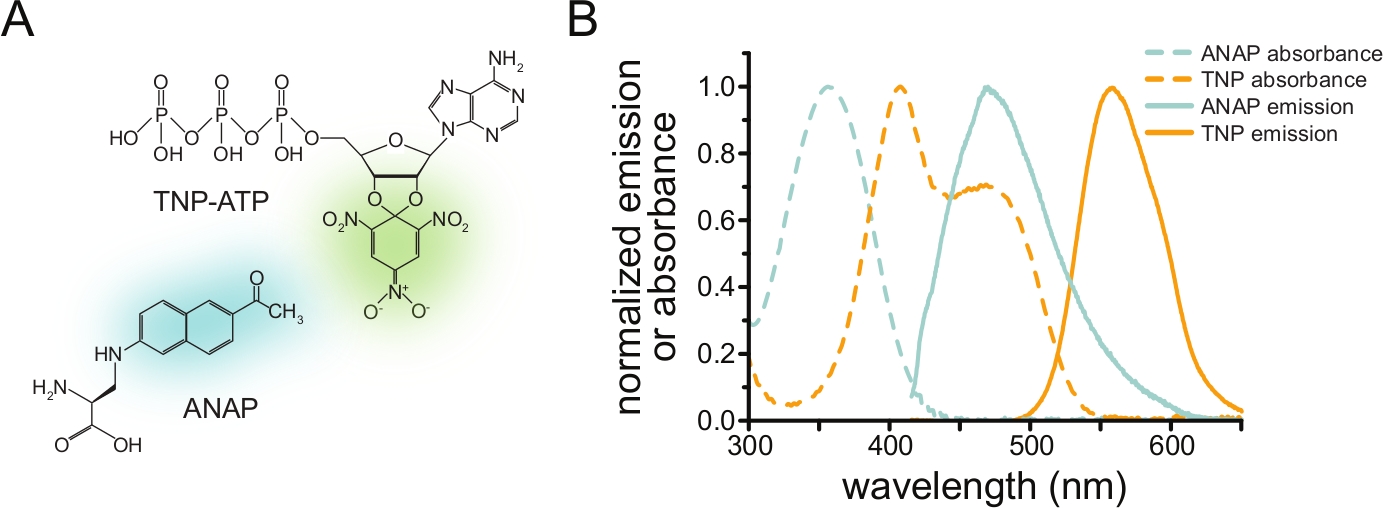
Para medir a ligação de nucleotídeos via FRET, uma proteína de interesse deve primeiro ser marcada com um fluoróforo. A etiqueta fluorescente deve ser inserida especificamente na proteína de interesse, de modo que esteja perto o suficiente do local de ligação do ligante para que o FRET ocorra, com especial cuidado para garantir que a etiqueta não afete a estrutura e a função gerais da proteína. Para isso, empregamos uma técnica desenvolvida por Chatterjee et al., usando supressão de stop-codon âmbar para inserir um aminoácido fluorescente não canônico (l-3-(6-acetilnaftaleno-2-ilamino)-2-aminopropiônico; ANAP) no local desejado12. Medimos a ligação de nucleotídeos como FRET entre a proteína marcada com ANAP e derivados nucleotídicos trinitrofenil (TNP) fluorescentes (Figura 1A). O espectro de emissão da ANAP sobrepõe-se ao espectro de absorbância dos nucleotídeos de TNP, condição necessária para que ocorra o FRET (Figura 1B). Aqui descrevemos dois tipos diferentes de experimento de ligação. No primeiro, a ligação de nucleotídeos ao lado intracelular dos canais deK ATP marcados com ANAP é medida em células que foram destelhadas por sonicação, deixando fragmentos aderidos da membrana plasmática sobre uma lamínula de cobertura de vidro10,11,13,14.
No segundo método, a ligação de nucleotídeos a canais KATP marcados com ANAP é medida em um patch de membrana sob pinça de tensão, permitindo a medição simultânea de correntes iônicas e fluorescência. Combinando essas duas abordagens experimentais, mudanças na ligação podem ser diretamente correlacionadas com mudanças na função do canal11. Resultados típicos, armadilhas potenciais e análise de dados são discutidos.
Protocolo
1. Preparação das lamínulas
NOTA: Essas etapas devem ocorrer em uma capa estéril de cultura de tecidos. As quantidades são dadas para a preparação de 10 pratos.
- Coloque dez lâminas de vidro de borossilicato autoclavadas de 30 mm individualmente em dez placas estéreis de 35 mm não tratadas e enxágue uma vez com 2 mL de água destilada estéril.
- Diluir 1 mL de solução de poli-L-lisina a 0,1% p/v em água destilada estéril até um volume total de 10 mL (concentração final de 0,01% p/v). Misture bem, em seguida, pipetar 1 mL em cada tampa e incubar à temperatura ambiente por 20 min.
- Aspirar a poli-L-lisina e lavar duas vezes cada lamínula com pelo menos 2 mL de água destilada estéril. Deixe até secar completamente, ou seja, pelo menos 3 h.
2. Semeadura de células HEK-293T
NOTA: Essas etapas devem ocorrer em uma capa de cultura de tecidos. As células HEK-293T foram escolhidas por seu baixo background atual e facilidade de crescimento em cultura. Este protocolo pode ser adaptado a outros tipos celulares.
- Enxaguar um frasco T75 confluente 80-90% de células HEK-293T uma vez com 12 mL de solução salina tamponada com fosfato (PBS) antes de incubar com 2 mL de tripsina por 2-5 min, ou até que as células estejam totalmente destacadas e quase completamente dissociadas.
- Ressuspender as células adicionando 10 mL de meio Dulbecco's Modified Eagle Medium (DMEM) suplementado com 10% de soro fetal bovino, 100 U/mL de penicilina e 100 μg/mL de estreptomicina. Pipetar suavemente contra o fundo do balão para quebrar os aglomerados de células restantes.
- Adicionar 2 mL de DMEM suplementado ao número desejado de placas de 35 mm contendo lamínulas revestidas. Adicionar 100 μL de células ressuspensas a cada prato. Incubar durante a noite a 37 °C.
3. Transfecção
NOTA: Essas etapas devem ocorrer em uma capa de cultura de tecidos. São dadas quantidades para a transfecção de 10 pratos. Para a incorporação ANAP sítio-específica, o códon de DNA na posição destinada à marcação deve ser substituído pelo códon de parada âmbar (TAG). Este construto é co-transfectado com dois plasmídeos: pANAP e peRF1-E55D12,15. A pANAP codifica várias cópias de um par de tRNA/tRNA sintetase específico da ANAP. Na presença de ANAP, a transfecção desse plasmídeo produz RNAt carregado com ANAP que reconhece o códon âmbar. peRF1-E55D codifica um fator de liberação ribossomal negativo dominante que aumenta o rendimento da proteína marcada com ANAP de comprimento total.
- Preparar um tubo de 1,5 mL com 10 μg de pANAP, 10 μg de peRF1-E55D e DNA para a construção destinada à marcação com ANAP. Levar a um volume final de 500 μL com DMEM não suplementado.
- Num tubo separado, preparar 3 μL de reagente de transfecção à base de lípidos (ver Tabela de Materiais) para cada 1 μg de ADN e levar a um volume final de 500 μL com DMEM não suplementado.
- Combinar as misturas de DNA e reagentes de transfecção em um único tubo e incubar por 20 min à temperatura ambiente.
- Adicionar 400 μL do estoque de 1 mM ANAP (sal de trifluoroacetato em NaOH 30 mM) a 20 mL de DMEM suplementado para uma concentração final de 20 μM ANAP. Substitua a mídia antiga das células chapeadas por 2 mL da mídia contendo ANAP por prato.
- Pipetar 10% da mistura de transfecção de DNA em cada prato. Incubar a 33 °C durante 2-4 dias antes das experiências. A incubação a 33 °C retarda a divisão celular e aumenta o rendimento de proteína por célula16.
4. Experimentos com membranas sem telhado
- Use um par de pinças para quebrar uma tampa com células transfectadas em fragmentos menores.
- Siga um dos procedimentos abaixo para destedilhar as células.
- Se estiver usando lamínulas pré-revestidas, enxágue um fragmento com PBS e, em seguida, coloque-o no fundo de uma placa de 35 mm contendo 2 mL de PBS. Sonicar brevemente usando um sonicador de sonda (50 W, 20%-40% de amplitude, sonda de 3 mm) posicionado 3-5 mm acima da amostra para destelhar as células e deixar para trás fragmentos aderentes da membrana plasmática (Figura 2A,C).
NOTA: A potência, a duração e a altura da sonda do sonicador acima da amostra podem ser variadas para obter um alto rendimento de membranas destelhadas sem desnudar completamente a lamínula. - Se não estiver usando lamínulas de cobertura pré-revestidas, enxágue um fragmento de lamínula de cobertura com PBS e, em seguida, mergulhe em um tubo contendo 0,1% p/v de poli-L-lisina por ~30 s antes de sonicar brevemente (como na etapa 4.2.1) para destelhar as células e deixar para trás fragmentos de membrana plasmática destelhados/parcialmente destelhados (Figura 2A,C,D). Demonstrou-se que exposições breves à poli-L-lisina melhoram a aderência à lamínula13.
- Se estiver usando lamínulas pré-revestidas, enxágue um fragmento com PBS e, em seguida, coloque-o no fundo de uma placa de 35 mm contendo 2 mL de PBS. Sonicar brevemente usando um sonicador de sonda (50 W, 20%-40% de amplitude, sonda de 3 mm) posicionado 3-5 mm acima da amostra para destelhar as células e deixar para trás fragmentos aderentes da membrana plasmática (Figura 2A,C).
- Coloque o fragmento sonicado em uma placa de 35 mm com fundo de vidro contendo 2 mL de solução de banho e monte em um microscópio invertido equipado com uma objetiva de imersão em água de 60x NA. A porta da câmera do microscópio é conectada a um espectrógrafo em série com uma câmera CCD de alta sensibilidade. Perfundir a câmara de banho (0,5 – 1 mL/min) com tampão utilizando bomba peristáltica. A composição do tampão irá variar dependendo da proteína em estudo.
NOTA: Se o usuário não tiver acesso a um objetivo com uma longa distância de trabalho, pode ser impossível focar nos fragmentos de membrana destelhados devido à altura extra do deslizamento da tampa. Uma alternativa é semear células diretamente em pratos com fundos de vidro de poli-L-lisina (veja Tabela de Materiais para um exemplo). Isso também reduzirá possíveis aberrações na imagem associadas ao foco através de dois pedaços de vidro. Essas aberrações não afetam a forma dos espectros adquiridos. - Identificar fragmentos de membrana sem telhado expressando o canal marcado com ANAP procurando fluorescência do canal (Figura 2C,D).
NOTA: Recomenda-se o uso de uma etiqueta fluorescente adicional (onde o espectro de emissão é distinguível do espectro de emissão ANAP) para ajudar a identificar membranas sem telhado contendo a proteína de interesse. Os experimentos na Figura 2C,D foram realizados em canais marcados com ANAP com etiquetas de proteína fluorescente C-terminal. - Acople parcialmente a máscara do espectrômetro (aumente ~10%) entre a porta da câmera no microscópio e o espectrógrafo. A sombra da máscara aparecerá na imagem da câmera. Alinhe a membrana sem teto com a máscara do espectrômetro, ajustando o estágio do microscópio. Adquira uma imagem de campo brilhante e fluorescência da membrana sem telhado. Estes serão usados para selecionar uma região de interesse para análise.
- Aproximar a ponta do sistema de perfusão de microvolume da membrana destelhada.
NOTA: Para reduzir a fluorescência de fundo, a saída do sistema de perfusão foi substituída por uma ponta personalizada feita de vidro borossilicato. - Para obter imagens dos espectros de fluorescência, excite a membrana com um LED de 385 nm através de um filtro de excitação passa-banda de 390/18 nm e um dicroico de borda de 416 nm. Coletar a luz emitida através de um filtro de emissão passa-longa de 400 nm (Figura 2B).
- Acione a máscara do espectrômetro e certifique-se de que a luz emitida seja passada. Acione as grades do espectrômetro (300 ranhuras/mm). Com as grades instaladas, a luz difratada pelo espectrômetro será projetada no chip da câmera CCD para produzir imagens espectrais (Figura 3A). Essas imagens retêm informações espaciais na dimensão y . A dimensão x é substituída pelo comprimento de onda.
- Opcionalmente, se a proteína de interesse for marcada com uma proteína fluorescente, adquira uma imagem espectral da proteína fluorescente usando o conjunto de filtros apropriado.
- Tome uma ou mais exposições de 0,1-10 s no início do experimento enquanto perfunde a solução tampão livre de nucleotídeo. Estes serão utilizados para corrigir e normalizar os dados ao longo do resto da experiência (ver secção 5 abaixo).
NOTA: A escolha do tempo de exposição dependerá do nível de expressão alcançado, do brilho do fluoróforo e da ótica. O tempo de exposição deve ser escolhido para maximizar o sinal e minimizar a taxa de clareamento observada. O intervalo de tempo de exposição indicado no ponto 4.10 é adequado para medições de ligação ao equilíbrio, mas pode ser útil para medir alterações cinéticas mais lentas10. A capacidade de usar tempos de exposição curtos para rastrear cinética mais rápida será limitada pelos níveis de expressão de proteínas e fotoclareamento, em vez de hardware. - Aplicar uma faixa de concentrações de TNP-ATP (geralmente preparado em solução de banho) para estabelecer uma curva de concentração-resposta. Perfundir cada solução durante pelo menos 1 min para garantir que seja atingido um estado estacionário e lavar cada concentração com solução de banho durante, pelo menos, 1 min.
NOTA: É importante garantir que o sistema de perfusão possa atingir rapidamente o equilíbrio (Figura 2E) e atingir a concentração local correta de TNP-ATP (Figura 2F). - Tomar uma exposição (com a mesma duração utilizada na etapa 4.10) em cada concentração e no final de cada washout.
5. Análise espectral
NOTA: Estas instruções são escritas para uso com o código de análise "pcf.m", que pode ser encontrado no GitHub. https://github.com/mpuljung/spectra-analysis10. Código adicional e alternativo pode ser encontrado em https://github.com/smusher/KATP_paper_201911. Descrevemos aqui as operações realizadas pelo software para que o usuário possa criar seu próprio código ou optar por analisar os dados manualmente.
- Inicie o programa de análise digitando o nome do programa ("pcf") na linha de comando.
- Quando uma caixa de diálogo de arquivo/pasta aberta for aberta com o prompt: "Selecionar arquivos para ROI", selecione os nomes de arquivo associados às imagens de campo brilhante e fluorescência da membrana destelhada. Um prompt aparecerá na linha de comando para digitar o nome do arquivo de saída.
- Digite o nome do arquivo e pressione enter.
- Quando o software exibir as imagens de campo brilhante e fluorescência, selecione uma região de interesse (ROI) na imagem espectral correspondente à localização do fragmento de membrana sem telhado ou do patch excisado (ver secção 6) seguindo as instruções do software. Selecione uma região de fundo na mesma imagem espectral (representando a mesma faixa de comprimento de onda da ROI) correspondente a uma seção de deslizamento de cobertura ou placa sem membrana conectada (Figura 3A). O software solicitará que você clique na parte superior do ROI e pressione Enter, clique na parte inferior do ROI e pressione Enter e, em seguida, repita esse processo para a região de fundo.
- Quando uma caixa de diálogo de arquivo/pasta aberta for aberta com o prompt: "Selecionar arquivo para o espectro FP", selecione o nome do arquivo associado ao espectro de proteína fluorescente (FP) (etapa opcional 4.9). Se nenhum espectro FP foi adquirido, selecione um arquivo de espectro diferente. O espectro FP serve como controle de qualidade para distinguir entre proteína marcada e fluorescência de fundo.
- Quando uma caixa de diálogo de arquivo/pasta aberta for aberta com o prompt: "Selecionar arquivos para análise", selecione todos os arquivos correspondentes aos espectros ANAP (das etapas 4.10 a 4.12), incluindo os arquivos necessários para a correção da água sanitária.
- Quando uma caixa de diálogo de arquivo/pasta aberta for aberta com o prompt: "Selecionar arquivos para coleta de branqueamento", selecione o subconjunto de arquivos da etapa 5.6 correspondente aos espectros iniciais adquiridos em solução livre de nucleotídeos no início do experimento ou espectros adquiridos durante as lavagens em solução livre de nucleotídeo a serem usados para correção (das etapas 4.10 a 4.12).
- Média de linha de cada imagem para produzir espectros, ou seja, média da intensidade de todos os pixels na dimensão y de uma ROI ou região de fundo em cada comprimento de onda. (Figura 3B). Subtraia o espectro de fundo médio resultante do espectro médio adquirido da ROI para remover a fluorescência e a fluorescência de fundo do TNP-ATP não ligado (Figura 3C). Essas etapas são executadas automaticamente pelo software.
- Determine a intensidade da ANAP para cada exposição calculando a média da intensidade de uma janela de 5 nm centrada em torno do pico ANAP dos espectros subtraídos (tipicamente ~470 nm, mas pode variar dependendo do microambiente local do resíduo ANAP).
NOTA: A Figura 3D mostra 6 espectros obtidos a partir de exposições consecutivas de 10 s de um fragmento de membrana sem telhado expressando canais marcados com ANAP. O inset mostra a intensidade média do pico de cada espectro. O software irá encontrar automaticamente o comprimento de onda de pico no primeiro espectro adquirido e usar esse valor em todo o espectro. A intensidade será calculada automaticamente pelo software. - Normalizar as intensidades de PANA para cada experimento dividindo a intensidade de ANAP de uma dada exposição (F) pela intensidade de ANAP da primeira exposição da série temporal, que foi tomada na etapa 4.10 (Fmax). Novamente, o software executa esses cálculos automaticamente.
- Execute as etapas abaixo para obter dados.
- Para corrigir o fotoclareamento ANAP, primeiro ajuste um único decaimento exponencial, (F/Fmax) = A*exp(-t/τ)+(1-A), onde t é o tempo de exposição cumulativo, τ é a constante de tempo e A é a amplitude) para as etapas intermediárias de lavagem entre as aplicações de TNP-ATP ou para múltiplas exposições iniciais tomadas antes da lavagem com TNP-ATP (Figura 3D, inserção).
NOTA: O software exibirá esse ajuste e solicitará que ele seja aceito ou rejeitado. Se o ajuste for rejeitado, outra oportunidade será fornecida para selecionar arquivos para correção de clareamento. - Divida os espectros ANAP normalizados (na etapa 5.10) pelo valor previsto do ajuste exponencial da etapa 5.11.1 em cada ponto de tempo (Figura 3E).
NOTA: Para o exemplo mostrado, a fluorescência de pico normalizada observada em 50 s é de 0,65 e a fluorescência prevista a partir do ajuste exponencial é de 0,64. Para corrigir o clareamento, divida o valor observado (0,65, Figura 3E inset, círculo vazio) pelo valor previsto (0,64, Figura 3E inset, linha tracejada) para produzir o valor corrigido (~1, Figura 3E inset, círculo colorido). Se a correção do clareamento for adequada, a intensidade da PANA de todas as exposições adquiridas na ausência de nucleotídeos deve ser aproximadamente igual (Figura 3E). Esses cálculos são realizados automaticamente pelo software. - Obter a saída como uma imagem plotando os dados e uma planilha com abas contendo os espectros brutos, espectros subtraídos, espectros corrigidos para fotobranqueamento e os dados de pico para cada arquivo para que análises adicionais possam ser realizadas.
- Para corrigir o fotoclareamento ANAP, primeiro ajuste um único decaimento exponencial, (F/Fmax) = A*exp(-t/τ)+(1-A), onde t é o tempo de exposição cumulativo, τ é a constante de tempo e A é a amplitude) para as etapas intermediárias de lavagem entre as aplicações de TNP-ATP ou para múltiplas exposições iniciais tomadas antes da lavagem com TNP-ATP (Figura 3D, inserção).
6. Experimentos de fluorometria patch-clamp
- Puxe pipetas de remendo de capilares de vidro borossilicato de paredes espessas para uma resistência de 1,5 MΩ a 2,5 MΩ quando preenchidas com solução de pipeta. A composição da solução da pipeta varia de acordo com a proteína em estudo.
- Transfira uma lamínula com células transfectadas para uma placa de 35 mm com fundo de vidro contendo 2 mL de solução de banho e monte em um microscópio invertido equipado com uma objetiva de imersão em água de 60x de alto NA. Perfundir a câmara de banho (0,5 – 1 mL/min) com solução de banho com bomba peristáltica. Quanto à solução de pipeta, a solução de banho irá variar dependendo da proteína em estudo.
- Identificar uma célula que expresse canais marcados com ANAP procurando fluorescência na membrana celular.
- Encha uma pipeta de remendo com solução de pipeta. Aplique uma pressão positiva suave na pipeta e coloque na câmara de banho. Pressione a pipeta contra a membrana da célula e aplique uma sucção suave para obter uma vedação GΩ (Figura 4A).
- Excise o adesivo movendo rapidamente o suporte da pipeta para longe da célula (Figura 4A).
NOTA: A excisão do adesivo desta forma deve formar um adesivo de dentro para fora, com os domínios citosólicos da proteína expostos ao sistema de perfusão. Se a localização do sítio de ligação de nucleotídeos em estudo não for citosólica, será necessário usar adesivos externos ou gravações de células inteiras para realizar experimentos de PCF. - Aproximar a ponta da pipeta do remendo da ponta do sistema de perfusão e verificar se o remendo está dentro da fenda da máscara do espectrômetro (Figura 4A).
- Aplicar TNP-ATP e espectros de imagem como nas etapas 4.10-4.12, enquanto simultaneamente registra a resposta da corrente iônica à aplicação de nucleotídeos.
OBS: O vidro da pipeta pode introduzir aberrações espaciais e reflexos nas imagens adquiridas. No entanto, essas aberrações não afetarão a forma dos espectros adquiridos e a luz de excitação refletida é facilmente separada da fluorescência usando o espectrógrafo ou um filtro de emissão passa-longa. - Analise os espectros. Os espectros obtidos a partir de manchas excisadas podem exibir subtração excessiva da fluorescência não ligada do TNP-ATP devido à exclusão do TNP-ATP do vidro da pipeta do remendo (Figura 4C-E). Essa subtração excessiva não afeta o espectro de emissão da ANAP e, portanto, pode ser ignorada.
NOTA: Como o sinal de fluorescência em manchas excisadas será menor do que em membranas sem telhado, é importante usar um tempo de exposição que forneça sinal a ruído alto o suficiente sem clarear a ANAP muito rapidamente.
Resultados
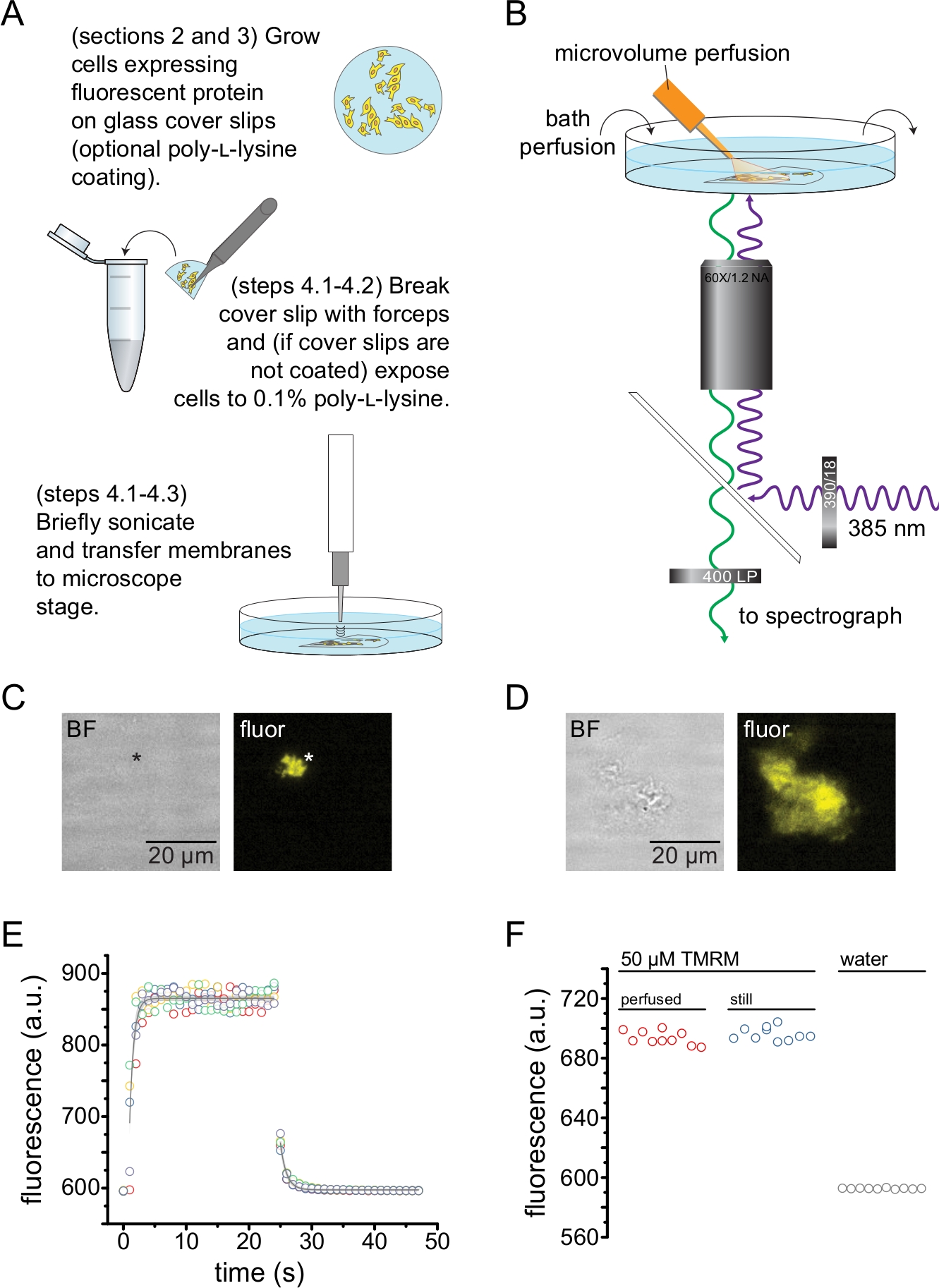
A Figura 2 mostra o arranjo experimental básico para medir a ligação de nucleotídeos a proteínas fluorescentes em fragmentos de membrana sem telhado obtidos por sonicação (Figura 2A,B). Duas abordagens diferentes foram usadas para obter membranas destelhadas, cultivando diretamente as células em lamínulas de cobertura revestidas com poli-L-lisina ou cultivando células em vidro não tratado e expondo-as brevemente à poli-L-lisina (0,1% em água) antes do destelhado. A Figura 2C mostra um fragmento de membrana típico sem telhado de uma célula HEK-293T expressando canaisde K ATP marcados com proteína fluorescente laranja (OFP). As membranas sem teto eram virtualmente invisíveis em imagens de campo claro e foram identificadas pela fluorescência de proteínas de membrana marcadas ou por contracoloração com um corante de membrana como octadecil rodamina B13. Além das membranas destelhadas, a sonicação das células HEK-293T também produziu fragmentos celulares parcialmente destelhados (Figura 2D)10,17. Esses fragmentos eram visíveis em campo claro. Isso pode ser o resultado de membranas plasmáticas babadas que são pouco aderentes ao vidro da cobertura. Alternativamente, esses fragmentos podem conter vesículas e membranas de organelas intracelulares. Como tal, é preferível adquirir imagens apenas de membranas "verdadeiras" sem teto, uma vez que a proteína-alvo marcada associada às membranas intracelulares pode refletir estágios intermediários de processamento e montagem pós-traducional. O cultivo de células em vidro revestido com poli-L-lisina é recomendado, pois isso resultou em um maior rendimento de membranas "verdadeiras" sem telhado após a sonicação.
Um sistema de perfusão de microvolumes foi aplicado a nucleotídeos fluorescentes para minimizar as quantidades necessárias em um experimento típico (Figura 2B). A ponta de vidro revestida de poliimida fornecida foi substituída por uma ponta de vidro borossilicato puxada à mão em nossa configuração de perfusão, o que reduziu o fundo de fluorescência. Para minimizar o acúmulo de nucleotídeos ao redor das membranas não cobertas que estão sendo fotografadas, toda a câmara de banho foi lentamente perfundida com tampão. Dessa forma, desejamos medir a taxa de troca de solução do nosso sistema de perfusão de microvolume e verificar se conseguimos atingir a concentração de ligante pretendida em nossa região de interesse, ou seja, que o ligante do nosso sistema de perfusão não foi diluído diretamente no meio de banho antes de atingir a membrana destelhada. Para controlar essas possibilidades, foi medida a lavagem e o wash-out de uma solução de 50 μM de tetrametilrodamina-5-maleimida (TMRM) de nosso sistema de perfusão de microvolume direcionados à superfície de uma placa de fundo de vidro de cobertura perfundida com água (Figura 2E). A cinética de troca de solução foi reprodutível e bem descrita por um único decaimento exponencial com constantes de tempo menores que 1 s tanto para o wash-in quanto para o wash-out. Esses tempos de troca de solução limitam nossa capacidade de medir a cinética de ligação e desvinculação de ligantes em nossa configuração atual. Para verificar se conseguimos atingir a concentração desejada do ligante na superfície da lamínula, comparamos a intensidade de fluorescência de 50 μM de TMRM entregue à lamínula pelo nosso sistema de perfusão de microvolume com 50 μM de TMRM em banho de parada (Figura 2F). Nenhuma diferença na intensidade foi observada, verificando-se que concentrações adequadas do ligante na superfície do deslizamento de cobertura com nosso sistema de perfusão de microvolume podem ser alcançadas, mesmo quando o banho é perfundido.
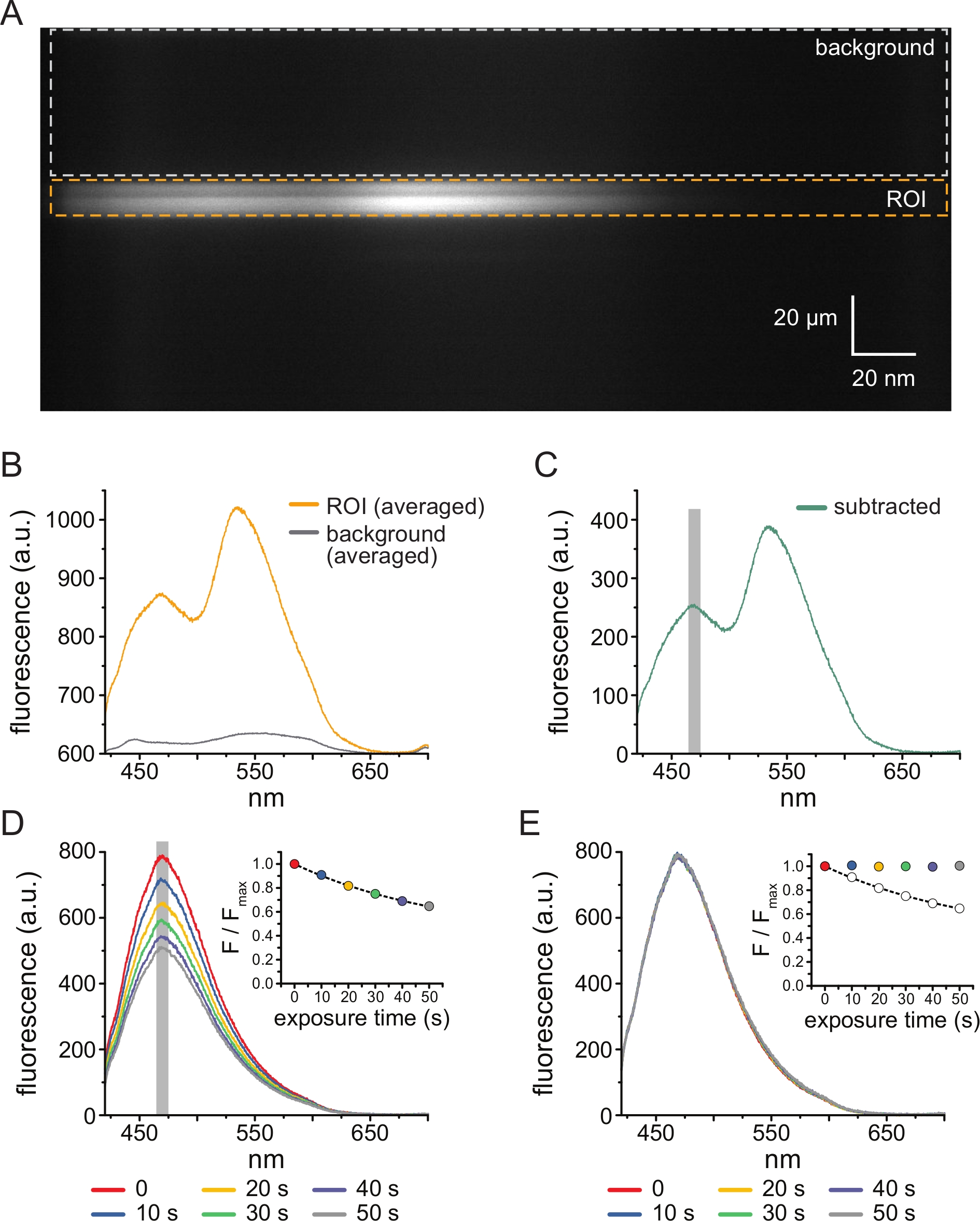
A Figura 3A mostra uma imagem espectral obtida de canais de K ATP marcados com ANAP em uma membrana sem telhado de uma célula HEK-239T exposta a 5 μM TNP-ATP. Para a obtenção dessas imagens, a luz emitida pela membrana destelhada foi direcionada através de um espectrômetro em série com uma câmera CCD. A fluorescência emitida foi difratada das grades e projetada no chip da câmera, produzindo espectros. As imagens resultantes retêm informações espaciais na dimensão y, mas a dimensão x foi substituída pelo comprimento de onda. A região de interesse (ROI), correspondente à membrana destelhada, é delineada em laranja. Duas regiões de alta intensidade são evidentes na imagem, correspondendo ao pico de emissão de ANAP e TNP-ATP. Isso foi melhor apreciado no espectro de comprimento de onda por comprimento de onda (em toda a ROI) mostrado na Figura 3B. O pico ~470 nm corresponde à ANAP incorporada ao KATP; o pico ~535 nm corresponde ao TNP-ATP. Para corrigir a fluorescência de fundo e a excitação direta do TNP-ATP em solução, uma região de fundo (Figura 3A, cinza) foi selecionada de cada imagem. A média do espectro de fundo é mostrada na Figura 3B. O espectro final foi obtido subtraindo-se o espectro de fundo médio do espectro de ROI médio (Figura 3C).
A ANAP é propensa a artefatos fotoclareadores. A Figura 3D mostra a redução do pico de fluorescência da ANAP após múltiplas exposições. O pico de fluorescência de várias exposições na ausência de TNP-ATP (ou de lavagens entre concentrações de TNP-ATP) foi ajustado a um decaimento exponencial único e isso foi usado para corrigir artefatos de fotoclareamento (Figura 3E). Recomenda-se a realização de experimentos de concentração-resposta de baixas a altas e altas a baixas concentrações de nucleotídeos. Se a correção do clareamento não introduzir artefatos adicionais, os resultados devem ser comparáveis11.
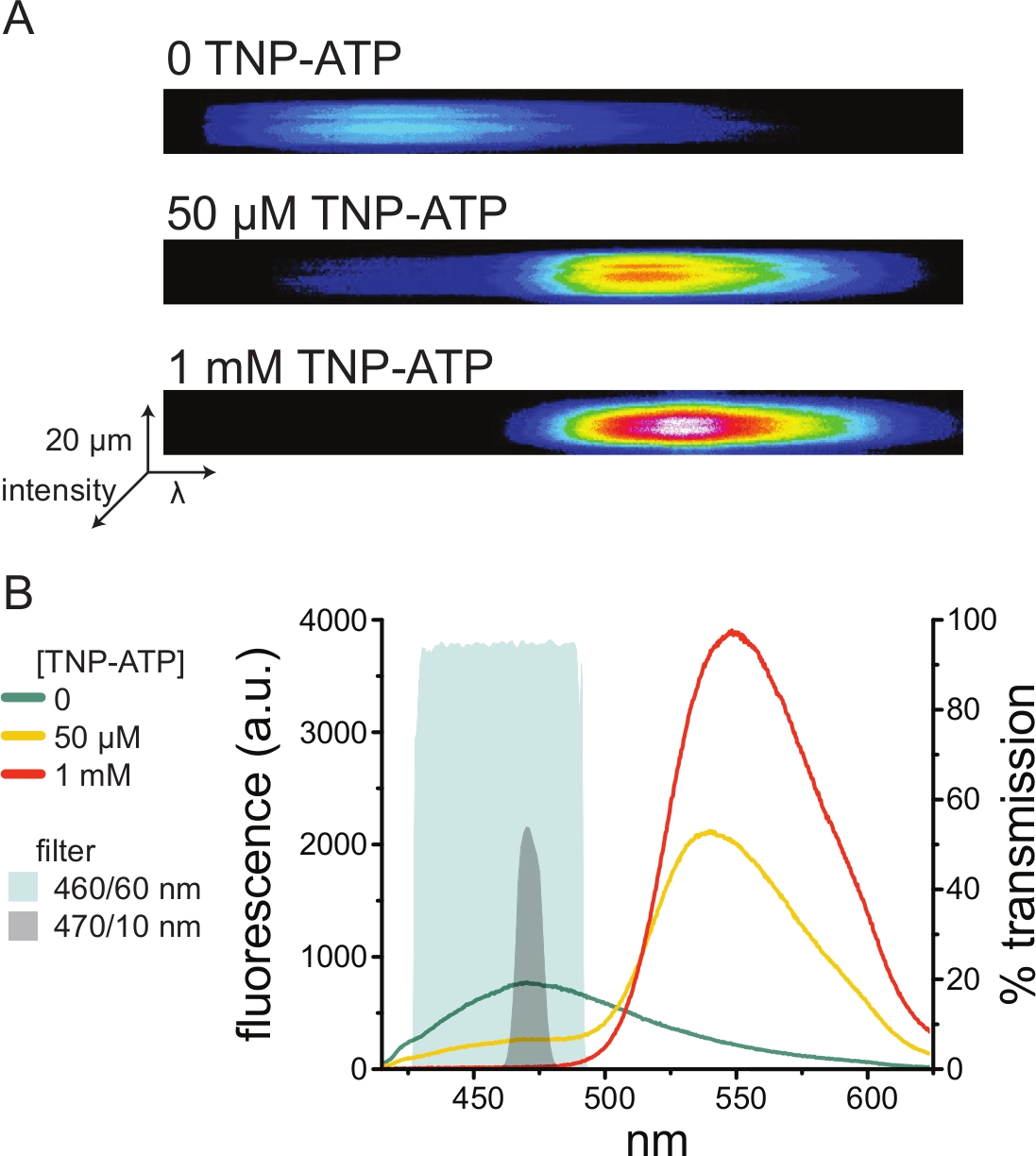
A Figura 5A mostra imagens espectrais representativas de uma membrana sem telhado obtidas de uma célula expressando canais de K ATP marcados com ANAP na ausência e presença de TNP-ATP. Os espectros corrigidos são mostrados na Figura 5B. Observando os espectros de emissão, houve uma clara separação entre a emissão de fluorescência do doador e do receptor. Como foi observada alguma ligação inespecífica do TNP-ATP a membranas plasmáticas virgens de células HEK-293T não transfectadas, recomenda-se quantificar o FRET como uma redução na fluorescência do doador (ANAP)10,11. Esse pico foi específico para o receptor marcado.
Para ligantes que induzem uma mudança conformacional em seu receptor, estudos de ligação isoladamente não fornecem informações diretas e mecanisticamente significativas sobre o processo de ligação do ligante18. A relação concentração-resposta para ligação do ligante depende não apenas da afinidade intrínseca de ligação, mas também da mudança conformacional induzida pela ligação do ligante, e da propensão inerente do receptor a mudar de conformação na ausência do ligante. Para entender melhor os processos que sublinham as interações ligante-receptor, as medidas de ligação podem ser emparelhadas com experimentos que fornecem uma leitura da função da proteína. Para este fim, os canais iônicos são um sistema modelo ideal, pois suas correntes podem ser medidas com resolução de tempo sub-ms até o nível de molécula única usando pinça de tensão. Historicamente, medições pareadas de corrente e fluorescência têm fornecido informações significativas sobre a abertura e fechamento (gating) de canais iônicos dependentes de tensão e ligantes 19,20,21. Experimentos têm sido conduzidos para medir simultaneamente correntes iônicas e nucleotídeos cíclicos fluorescentes ligando-se a vários canais nucleotídeos cíclicosregulados 22,23,24. Esses estudos empregaram um ligante que aumentou seu rendimento quântico após a ligação. A fluorescência do ligante não ligado no volume da solução próximo ao remendo pode ser subtraída por imagem dos remendos por microscopia confocal22,23. Em nossos estudos, a ligação foi medida usando a redução da fluorescência da ANAP. Como este sinal é específico para o canal e o FRET entre ANAP e TNP-ATP é fortemente dependente da distância (metade máxima em ~43 Å), a contaminação do nosso sinal por nucleotídeos não especificamente ligados e não ligados foi evitada.
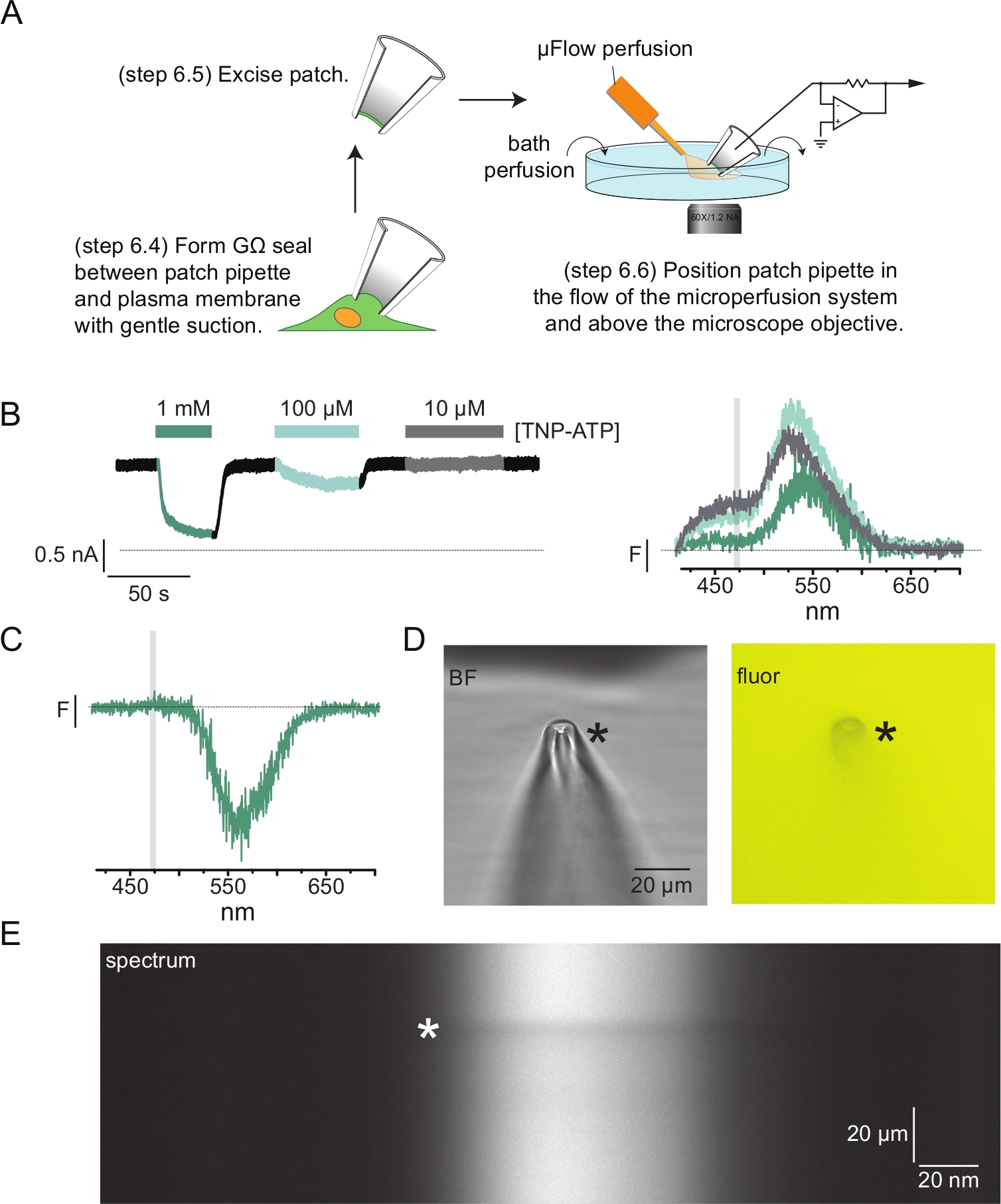
A Figura 4A mostra um experimento típico de fluorometria patch-clamp (PCF). Um selo de alta resistência (GΩ) foi formado entre uma pipeta de vidro borossilicato preenchida com solução salina (conectada a um amplificador de pinça de tensão) e uma célula expressando KATP marcado com ANAP. Após a formação do selo, a pipeta foi afastada da célula, permitindo o acesso aos sítios de ligação de nucleotídeos intracelulares. A pipeta foi então posicionada sobre a objetiva do microscópio, centrada na fenda da máscara do espectrômetro e a saída do sistema de perfusão de microvolume (modificado com uma ponta de vidro borossilicato) foi aproximada da pipeta (Figura 4D). A tensão foi controlada e as correntes foram medidas a partir dos canais no patch. Correntes e espectros representativos de canais KATP marcados com ANAP são mostrados na Figura 4B, codificados por cores para corresponder os espectros às correntes. Os espectros de emissão foram corrigidos para fundo e clareamento e para membranas sem telhado.

Figura 1: ANAP e TNP-ATP formam um par FRET adequado. (A) Estruturas da ANAP e TNP-ATP. As metades fluorescentes são destacadas. (B) Espectros de absorbância e emissão de fluorescência de ANAP e TNP-ATP. A sobreposição entre a emissão ANAP e a absorbância TNP-ATP é necessária para o FRET. Adaptado de Puljung e colaboradores (publicado sob a Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)10. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Medição da ligação de nucleotídeos em membranas plasmáticas sem teto. (A) Esquema para a preparação de membranas plasmáticas sem teto a partir de células aderentes que expressam uma proteína fluorescente de membrana. São fornecidas instruções para as células cultivadas em lamínulas revestidas com poli-L-lisina ou não tratadas. (B) Instalação experimental para medição da ligação de nucleotídeos em membranas sem teto. (C) Campo brilhante e imagens fluorescentes de uma membrana plasmática completamente descoberta derivada de uma célula que expressa canais KATP marcados com proteína fluorescente laranja (OFP). O asterisco marca a posição da membrana, que é quase invisível na imagem de campo brilhante. O OFP foi excitado com um LED largo de 565 nm através de um filtro passa-banda de 531/40 nm e a luz dicroica de borda de 562 nm foi coletada através de um filtro passa-banda de 593/40 nm. (D) Campo brilhante e imagens fluorescentes de um fragmento de membrana parcialmente destelhado derivado de uma célula que expressa canais KATP marcados com proteína fluorescente laranja (OFP). (E) Curso de tempo de troca de solução adquirido usando a configuração descrita em B. Cinco réplicas técnicas são mostradas. O sistema de perfusão de microvolume foi carregado com 50 μM de tetrametilrodamina-5-maleimida (TMRM). O banho foi perfundido com água a uma taxa de ~0,5 mL/min. Os dados dos cursos de tempo wash-on (fluorescência crescente) e wash-out (fluorescência decrescente) foram ajustados com um único decaimento exponencial da forma F = A*exp(-x/τ) + y0. A constante de tempo (τ) para wash-in foi de ~0,6 s. A constante de tempo para wash-out foi de ~1,0 s. O TMRM foi excitado com um LED largo de 565 nm através de um filtro passa-banda de 540/25 nm e a luz dicroica de borda de 565 nm foi coletada através de um filtro passa-banda de 605/55 nm. (F) Comparação da intensidade de fluorescência de uma solução de 50 μM de TMRM aplicada utilizando o sistema de perfusão de microvolume como em B e de um banho de alambique contendo 50 μM de TMRM. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Subtração de fundo e correção do clareamento. (A) Imagem espectral (informação espacial na dimensão y, comprimento de onda na dimensão x) de uma membrana plasmática sem teto de uma célula expressando canais KATP marcados com ANAP. 5 μM TNP-ATP foi aplicado usando o arranjo descrito na Figura 2B. A caixa laranja denota a região de interesse (ROI), correspondente à membrana destelhada. A caixa cinza indica a região de fundo usada para corrigir o espectro. (B) Espectros de emissão derivados das médias comprimento de onda por comprimento de onda das regiões ROI e de fundo em A. (C) Espectro derivado subtraindo o espectro de fundo médio do espectro de ROI médio em B. A janela de 5 nm ao redor do pico ANAP usada para determinar a intensidade média é mostrada como uma área sombreada em cinza. (D) Espectros adquiridos de seis exposições consecutivas de 10 s de uma membrana plasmática sem teto de uma célula expressando canais deK ATP marcados com ANAP. Observe o decremento na fluorescência resultante do fotoclareamento. O inset mostra o ajuste de fluorescência de pico normalizado com um único decaimento exponencial da forma F/Fmax = A*exp(-t/τ) + (1-A). Os símbolos na inserção são codificados por cores para corresponder aos espectros. (E) Os mesmos espectros de D corrigidos para fotoclareamento. O inset mostra o pico normalizado de fluorescência de D como círculos abertos, com o pico de fluorescência corrigido mostrado usando círculos preenchidos. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Medidas simultâneas de ligação de nucleotídeos e correntes de canal usando fluorometria patch-clamp (PCF). (A) Esquema mostrando o arranjo experimental para medição de ligação de nucleotídeos e correntes iônicas. (B) Exemplo de correntes (esquerda) e espectros (direita) adquiridos de um patch de membrana excisado de uma célula que expressa canais KATP marcados com ANAP. As correntes foram registradas a um potencial de retenção de -60 mV, digitalizadas a 20 kHz e filtradas a 5 kHz. A área sombreada em cinza corresponde à faixa de comprimento de onda a partir da qual a intensidade da ANAP foi quantificada. Adaptado de Usher e colaboradores (publicado sob a Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (C) Espectro adquirido a partir de um adesivo de membrana excisado de uma célula que expressa canais KATP marcados com ANAP expostos a 1 mM TNP-ATP. Observe o pico negativo correspondente à faixa de comprimento de onda sobre a qual a fluorescência TNP-ATP é observada. A área sombreada em cinza denota a faixa de comprimento de onda usada para quantificar a fluorescência ANAP como em B. Adaptado de Usher et al., publicado sob a Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (D) Campo brilhante e imagens fluorescentes de uma pipeta de remendo exposta a 1 mM TNP-ATP. O asterisco marca a ponta da pipeta. (E) Imagem espectral da mesma pipeta de remendo em 1 mM TNP-ATP. O asterisco marca a posição da pipeta. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Ligação do TNP-ATP aos canais KATP marcados com ANAP. (A) Imagens espectrais de uma membrana plasmática sem teto de uma célula expressando canais de K ATP marcados com ANAP na ausência de TNP-ATP ou na presença de 50 μM ou 1 mM TNP-ATP. As intensidades são mostradas como um mapa de calor. (B) Espectros médios de comprimento de onda a partir das imagens em A mostrando a extinção da fluorescência ANAP por TNP-ATP. As áreas sombreadas representam dois filtros passa-banda diferentes que podem ser usados para medir a têmpera ANAP se um espectrômetro não estiver disponível. Clique aqui para ver uma versão maior desta figura.
{kind=link}

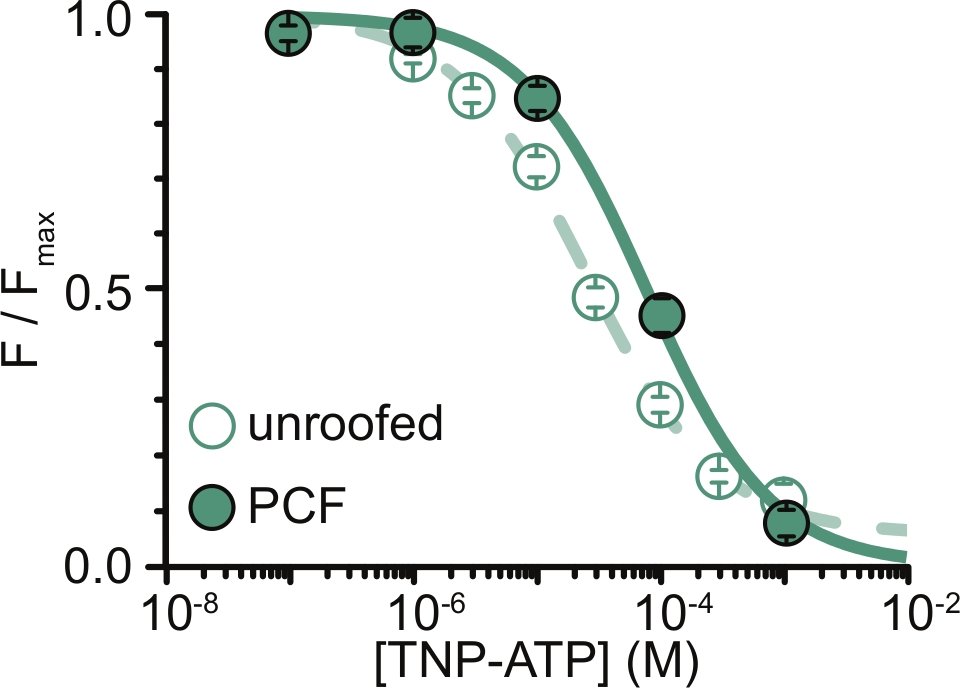
Figura 6: Têmpera dos canais de K ATP marcados com ANAP porTNP-ATP em membranas destelhadas e PCF. Sobreposição de dados de Usher e colaboradores (publicados sob a Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. Os dados foram ajustados à equação de Hill: F / F max = E max + (1 – E max) / (1+10(EC50 –[TNP-ATP])*h). F é a fluorescência medida, F max é a fluorescência máxima na ausência de nucleotídeo, Emax é a têmpera máxima em concentrações de nucleotídeos saturantes e h é a inclinação de Hill. EC50, (a concentração de nucleotídeos na qual a têmpera é metade máxima) e [TNP-ATP] são valores logarítmicos. Membranas sem teto: CE50 = -4,59 (25,7 μM), h = 0,82, Emax = 0,93. PCF: CE50 = -4,11 (77,6 μM), h = 0,87, Emax = 1,00. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Desenvolvemos um método para medir a ligação de adenina nucleotídica em tempo real a proteínas intactas da membrana. Nosso método baseia-se em várias outras técnicas estabelecidas, incluindo a marcação de proteínas com ANAP usando supressão de códon âmbar stop 12, destelhamento celular14 e fluorometria voltage-clamp/PCF 19,20,21,22,23,24,25 . A síntese dessas abordagens permite a mensuração da ligação de nucleotídeos com alta resolução espacial e temporal. De fato, em nosso trabalho anterior, conseguimos distinguir entre diferentes sítios de ligação no mesmo complexo proteico usando essa abordagem10,11. É importante ressaltar que essa técnica pode ser aplicada diretamente a pequenas quantidades de proteína em um ambiente celular em condições que preservem a função proteica. O uso de nosso método de ligação em conjunto com a leitura eletrofisiológica direta de correntes de canais iônicos nos permite obter informações ricas sobre os fundamentos moleculares do fechamento de canais11.
Como os espectrômetros são equipamentos de laboratório fora do padrão, a intensidade da ANAP também pode ser monitorada em relativo isolamento usando filtros passa-faixa. A Figura 5B mostra as propriedades espectrais de dois desses filtros. O filtro passa-banda de 470/10 nm filtra efetivamente o sinal de fluorescência do TNP-ATP e se sobrepõe bem com a fluorescência ANAP de pico. No entanto, o pico de transmitância deste filtro é de apenas cerca de 50%, o que pode dificultar a obtenção de bons sinais de membranas escuras (ou em remendos de membrana excisados sob pinça de tensão). Outra opção é um filtro passa-banda de 460/60 nm. Há um pouco mais de sobreposição entre o filtro de 460/60 nm e o pé do pico de emissão de TNP-ATP em comparação com o filtro de 470/10 nm. No entanto, a passagem de banda de 460/60 nm tem uma transmitância de 90-95% em uma ampla faixa do pico ANAP, o que seria esperado para aumentar o sinal de emissão de fluorescência.
A ANAP é um fluoróforo ambientalmente sensível 12,26,27. O pico de emissão e o rendimento quântico variam dependendo do local de incorporação na proteína de interesse, podendo mudar à medida que a proteína muda de conformação. Tais mudanças seriam imediatamente evidentes a partir dos espectros de emissão, mas não seriam tão óbvias quando a intensidade ANAP é medida usando filtros. Em qualquer caso, controles apropriados são necessários para demonstrar que o sinal de fluorescência não varia devido a mudanças no ambiente local ao redor da ANAP subsequentes à ligação de nucleotídeos. Experimentos de controle com nucleotídeos não marcados podem ajudar a verificar que quaisquer mudanças na intensidade da ANAP são o resultado de FRET entre os nucleotídeos ANAP e TNP. Os nucleotídeos TNP podem ligar-se não especificamente às membranas derivadas de células não transfectadas (seja à membrana plasmática ou às proteínas da membrana nativa)10. Quantificamos a ligação como um decremento na fluorescência do doador, pois este sinal é específico do canal marcado. No entanto, recomendamos a realização de experimentos de controle adicionais para cada par agonista/receptor, por exemplo, mutando o sítio de ligação de nucleotídeos, se conhecido, para verificar se a mudança na fluorescência do doador é realmente o resultado da ligação direta ao receptor marcado11. Finalmente, recomenda-se trabalhar com construções que contenham uma etiqueta de proteína fluorescente além do rótulo ANAP. Isso ajuda a diferenciar a fluorescência do receptor marcado do fundo/autofluorescência. A fluorescência de fundo pode ser distinguida da ANAP pelo pico e forma dos espectros de emissão10, mas tais determinações podem ser muito difíceis quando apenas conjuntos de filtros são usados. Além disso, células e membranas sem teto expressando receptores fluorescentes podem ser identificadas usando o tag de proteína fluorescente sem ter que excitar ANAP e correr o risco de fotoclareamento excessivo.
Em muitos de nossos registros de PCF, observamos um forte pico negativo em nossos espectros em altas concentrações de TNP-ATP (Figura 4C). Esse pico negativo é um artefato do nosso protocolo de subtração de fundo. A Figura 4D mostra imagens fluorescentes e de campo claro de uma pipeta de remendo exposta a 1 mM TNP-ATP. Uma sombra na ponta da pipeta é evidente, resultante da exclusão do TNP-ATP do volume das paredes da pipeta, que é mais evidente dentro do plano de foco. A imagem espectral da Figura 4E mostra uma faixa escura, correspondente a essa sombra. Quando uma região acima ou abaixo dessa faixa escura é usada para subtração de fundo, ela produz um pico negativo. É importante ressaltar que esse pico ocorreu em uma faixa de comprimento de onda correspondente à emissão de TNP-ATP e não afetou nossas medidas de extinção da ANAP.
A principal limitação de nossos experimentos foi a obtenção da expressão adequada na membrana plasmática de construtos marcados com ANAP para medir fluorescência. Era geralmente mais fácil adquirir espectros de alta qualidade a partir de membranas sem telhado do que em PCF, devido ao seu tamanho maior e à nossa capacidade de escanear rapidamente um prato inteiro de membranas destelhadas, ao contrário do PCF, onde os adesivos só podem ser obtidos um de cada vez. Em nossos experimentos, os dados de membranas sem teto e experimentos de PCF foram semelhantes, mas não equivalentes (Figura 6)11. No entanto, não há nenhuma razão a priori para que esta seja uma observação universal, pois as proteínas em uma pipeta de remendo podem estar em um estado funcional diferente daquelas em membranas sem teto.
Aqui, tentativas têm sido feitas para maximizar a expressão de nossos construtos marcados com ANAP, em particular reduzindo a temperatura de cultura celular para 33 °C10,11,16. Em nossa experiência, a tentativa de identificar locais na proteína em que a PANA seria uma substituição conservadora não resultou consistentemente em construtos que expressassem bem. Tivemos mais sucesso na varredura sistemática de regiões inteiras de proteínas para sítios de incorporação de ANAP e triagem de candidatos para expressão de superfície10. O sistema de marcação ANAP também funciona em ovócitos de Xenopus laevis, o que permite a excisão de remendos de membrana muito maiores, aumentando o sinal para o ruído26,27,28.
Enquanto níveis maiores de expressão devem resultar em sinais mais brilhantes, o número mínimo de canais necessários para medir a fluorescência depende de vários fatores, incluindo o brilho do fluoróforo, o grau de fotoclareamento, a intensidade da luz de excitação e o plano de foco. Teoricamente, as estimativas poderiam ser feitas correlacionando-se a intensidade da fluorescência e a corrente do canal, como já demonstradoanteriormente28,29. No entanto, a confiabilidade de tais estimativas requer algum conhecimento da condutância de canal único e da probabilidade aberta do canal. Além dos fatores listados acima, o sinal de fluorescência também será afetado por canais associados a vesículas ou seções da membrana plasmática presas ao vidro da pipeta que não estão sob pinça de tensão.
Este método é prontamente adaptado ao estudo de outros canais iônicos nucleotídeos-sensíveis. A CFTR é estruturalmente semelhante à subunidade do receptor acessório de sulfonilureia do KATP30,31. Assim como o KATP, O CFTR é controlado pela ligação de nucleotídeos, tornando-se um alvo futuro óbvio do nosso método7. Os receptores P2X purinérgicos são canais iônicos ligados por ATP extracelular9. O TNP-ATP atua como antagonista dos receptores P2X32,33. Portanto, não será útil para estudar a ativação do P2X, embora possa ser usado em ensaios de competição com agonistas do P2X. Alternativamente, outros derivados fluorescentes de ATP com sobreposição espectral suficiente com emissão ANAP podem ser usados para estudar a ativação. Alexa-647-ATP é um agonista P2X fluorescente34. O R0 calculado entre Alexa-647 e ANAP é ~85 Å, o que significa que a ligação direta ao P2X deve resultar em supressão substancial do ANAP incorporado ao canal. No entanto, um R0 tão longo também resultará na extinção de Alexa-647-ATP ligado a subunidades vizinhas e aumenta a probabilidade de que a ligação de nucleotídeos não específicos resulte em FRET. Como o sítio de ligação do ligante nos receptores P2X é extracelular, as medidas de ligação seriam realizadas em células intactas, em pinça de voltagem de célula inteira ou em adesivos de membrana externos. Nosso método também pode ser estendido para estudar a ligação e ativação de transportadores e bombas eletrogênicas e não-eletrogênicas que dependem de ATP para seu ciclo de reação, bem como receptores P2Y acoplados à proteína G. Finalmente, embora tenhamos desenvolvido este método para medir a ligação de adenina nucleotídica (TNP-ATP, TNP-ADP, TNP-AMP), a mesma abordagem pode ser usada para estudar a ligação a praticamente qualquer receptor para o qual um ligante fluorescente adequado tenha sido identificado.
Divulgações
Os autores declaram não haver conflitos de interesse.
Agradecimentos
Agradecemos a Raul Terron Exposito pela excelente assistência técnica. Este trabalho foi financiado pelo Conselho de Pesquisa em Biotecnologia e Ciências Biológicas (BB/R002517/1; MCP e FMA) e Wellcome Trust (203731/Z/16/A; SGU)
Materiais
| Name | Company | Catalog Number | Comments |
| T75 tissue-culture treated flask | StarLab | CC7682-4875 | |
| 0.1% w/v poly-L-lysine | Sigma-Aldrich | P8920 | |
| 30 mm borosilicate cover glass slips | VWR | 631-0174 | |
| 35 mm non-treated sterile dishes | CytoOne | CC7672-3340 | |
| 35 mm cover glass bottom dish | WPI | FD35-PDL-100 | |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 31966021 | |
| Foetal bovine serum (FBS) | Gibco | 10500-064 | |
| Penicillin/Streptomycin | Gibco | 15140-122 | |
| TrypLE select (tryosin) | Gibco | 12563-011 | Trypsin/EDTA reagent |
| Phosphate buffered saline (PBS) | Gibco | 14040-091 | |
| UltraPure distilled water | Invitrogen | 10977-035 | |
| HEK293T cells | ATTC | CRL-3216 | Used between passages 5-30 |
| ANAP-TFA | AsisChem | ASIS-0014 | Reconstituted in 30 mM NaOH to a final concentration of 1 mM |
| pANAP expression plasmid | Addgene | Plasmid #48696 | Encodes tRNA/tRNA synthetase pair for expression of ANAP-tagged protein |
| peRF1-E55D | Chin Lab (MRC Laboratory of Molecular Biology, Cambridge, UK) | Jason Chin: DOI: 10.1021/ja5069728 | Encodes dominant-negative eukaryotic ribosomal release factor |
| TransIT-LT1 | Mirus Bio | MIR 2300 | Lipopolyplex transfection reagent |
| Thick-walled borosilicate glass capillaries | Harvard Apparatus | GC150F-15 | |
| Tetramethylrhodamine-5-maleimide | Sigma-Aldrich | 94506 | |
| TNP-ATP | Jena Bioscience | NU-221L | Delivered at 10 mM in water |
| Nikon Eclipse TE2000-U inverted microscope microscope | Nikon | ||
| 60x water immersion objective (1.4 NA) | Nikon | MRD07602 | |
| 4-Wavelength High-Power LED Head | ThorLabs | LED4D245 | 385/490/565/625 nm LEDs |
| Four-Channel LED Driver | ThorLabs | DC4100 | |
| 390/18 nm band-pass excitation filter | ThorLabs | MF390-18 | For ANAP excitation |
| 400 nm long-pass emission filter | ThorLabs | FEL0400 | For imaging ANAP spectra |
| 416 nm edge dichroic | ThorLabs | MD416 | For imaging ANAP spectra |
| 460/60 nm band-pass emission filter | ThorLabs | MF460-60 | Suggested wide band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 470/10 nm band-pass emission filter | ThorLabs | FB470-10 | Suggested narrow band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 531/40 band-pass excitation filter | Brightline | FF01-531/40-25 | For orange fluorescent protein (OFP) excitation |
| 540/25 nm band-pass excitation filter | Chroma | D540/25X | For tetramethylrhodamine-5-maleimide (TMRM) excitation |
| 562 nm edge dichroic | Semrock | FF562-Di03 | For imaging OFP fluorescence |
| 565 nm edge dichroic | Chroma | 565DC | For imaging TMRM fluorescence |
| 593/40 nm band-pass excitation filter | Brightline | FF01-387/11-25 | For imaging OFP fluorescence |
| 605/55 nm band-pass emission filter | Chroma | D605/55M | For imaging TMRM fluorescence |
| IsoPlane-160 Imaging Spectrometer | Princeton Instruments | IsoPlane-160 | |
| PIXIS 400BR_eXcelon Camera | Princeton Instruments | PIXIS: 400BR_eXcelon | |
| Axopatch 200B amplifier | Molecular Devices | Axopatch 200B-2 | |
| Digidata 1440A digitizer | Molecular Devices | Digidata 1440A | |
| Probe sonicator | Sonics & Materials | VC-50 | For unroofing |
| REGLO digital peristaltic pump | Ismatec | ISM 832 | For bath perfusion |
| Microvolume perfusion system | ALA Scientific Instruments | ALA μFlow-8 | For TNP-ATP perfusion |
| pClamp 10.6.2 | Molecular Devices | Recording and analysing currents | |
| Lightfield 5.20.1507 | Princeton Instruments | Acquisition software for images and spectra | |
| Matlab | Mathworks | For data analysis | |
| Python 3.8.1 | Python Software Foundation | For data analysis |
Referências
- Garcia, M. L., Kaczorowski, G. J. Ion channels find a pathway for therapeutic success. Proceedings of the National Academy of Sciences of the United States of America. 113 (20), 5472-5474 (2016).
- Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B., Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. 16 (12), 829-842 (2017).
- Lakowicz, J. R. Principles of fluorescence spectroscopy. 3rd edn. , Springer. (2006).
- Higgins, C. F., Linton, K. J. The ATP switch model for ABC transporters. Nature Structural & Molecular Biology. 11 (10), 918-926 (2004).
- Toyoshima, C., Cornelius, F. New crystal structures of PII-type ATPases: excitement continues. Current Opinion in Structural Biology. 23 (4), 507-514 (2013).
- Craven, K. B., Zagotta, W. N. CNG and HCN channels: two peas, one pod. Annual Review of Physiology. 68, 375-401 (2006).
- Csanady, L., Vergani, P., Gadsby, D. C. Strict coupling between CFTR's catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proceedings of the National Academy of Sciences of the United States of America. 107 (3), 1241-1246 (2010).
- Vedovato, N., Ashcroft, F. M., Puljung, M. C. The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophysical Journal. 109 (12), 2452-2460 (2015).
- Burnstock, G. Introduction to the Special Issue on Purinergic Receptors. Advances in Experimental Medicine and Biology. 1051, 1-6 (2017).
- Puljung, M., Vedovato, N., Usher, S., Ashcroft, F. Activation mechanism of ATP-sensitive K(+) channels explored with real-time nucleotide binding. Elife. 8, 41103(2019).
- Usher, S. G., Ashcroft, F. M., Puljung, M. C. Nucleotide inhibition of the pancreatic ATP-sensitive K+ channel explored with patch-clamp fluorometry. Elife. 9, 52775(2020).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. Journal of the American Chemical Society. 135 (34), 12540-12543 (2013).
- Gordon, S. E., Senning, E. N., Aman, T. K., Zagotta, W. N. Transition metal ion FRET to measure short-range distances at the intracellular surface of the plasma membrane. Journal of General Physiology. 147 (2), 189-200 (2016).
- Heuser, J. The production of 'cell cortices' for light and electron microscopy. Traffic. 1 (7), 545-552 (2000).
- Schmied, W. H., Elsasser, S. J., Uttamapinant, C., Chin, J. W. Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. Journal of the American Chemical Society. 136 (44), 15577-15583 (2014).
- Lin, C. Y., et al. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PloS One. 10 (4), 0123562(2015).
- Usukura, J., et al. Use of the unroofing technique for atomic force microscopic imaging of the intra-cellular cytoskeleton under aqueous conditions. Journal of Electron Microscopy. 61 (5), 321-326 (2012).
- Colquhoun, D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. British Journal of Pharmacology. 125 (5), 924-947 (1998).
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Zheng, J., Zagotta, W. N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 28 (2), 369-374 (2000).
- Zheng, J., Zagotta, W. N. Patch-clamp fluorometry recording of conformational rearrangements of ion channels. Science's STKE. 2003 (176), 7(2003).
- Biskup, C., et al. Relating ligand binding to activation gating in CNGA2 channels. Nature. 446 (7134), 440-443 (2007).
- Kusch, J., et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron. 67 (1), 75-85 (2010).
- Wu, S., et al. State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophysical Journal. 100 (5), 1226-1232 (2011).
- Cha, A., Bezanilla, F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 19 (5), 1127-1140 (1997).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proceedings of the National Academy of Sciences of the United States of America. 110 (20), 8272-8277 (2013).
- Kalstrup, T., Blunck, R. S4-S5 linker movement during activation and inactivation in voltage-gated K(+) channels. Proceedings of the National Academy of Sciences of the United States of America. 115 (29), 6751-6759 (2018).
- Dai, G., Aman, T. K., DiMaio, F., Zagotta, W. N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nature Structural & Molecular Biology. 26 (8), 686-694 (2019).
- Liu, C., et al. Patch-clamp fluorometry-based channel counting to determine HCN channel conductance. Journal of General Physiology. 148 (1), 65-76 (2016).
- Hwang, T. C., et al. Structural mechanisms of CFTR function and dysfunction. Journal of General Physiology. 150 (4), 539-570 (2018).
- Puljung, M. C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. Journal of General Physiology. 150 (5), 653-669 (2018).
- Kasuya, G., et al. Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nature Communications. 8 (1), 876(2017).
- Virginio, C., Robertson, G., Surprenant, A., North, R. A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Molecular Pharmacology. 53 (6), 969-973 (1998).
- Bhargava, Y., Nicke, A., Rettinger, J. Validation of Alexa-647-ATP as a powerful tool to study P2X receptor ligand binding and desensitization. Biochemical and Biophysical Research Communications. 438 (2), 295-300 (2013).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados