
Method Article
Misurare il legame nucleotidico a proteine di membrana intatte e funzionali in tempo reale
In questo articolo
Riepilogo
Questo protocollo presenta un metodo per misurare il legame dei nucleotidi adenina ai recettori in tempo reale in un ambiente cellulare. Il legame è misurato come trasferimento di energia di risonanza di Förster (FRET) tra derivati del nucleotide trinitrofenile e proteine marcate con un amminoacido fluorescente non canonico.
Abstract
Abbiamo sviluppato un metodo per misurare il legame dei nucleotidi adenina ai recettori transmembrana intatti e funzionali in un ambiente cellulare o di membrana. Questo metodo combina l'espressione di proteine marcate con l'amminoacido fluorescente non canonico ANAP e FRET tra ANAP e derivati nucleotidici fluorescenti (trinitrofenili). Presentiamo esempi di legame nucleotidico a canali ionici KATP marcati con ANAP misurati in membrane plasmatiche senza tetto e patch di membrana asportate e inside-out sotto morsetto di tensione. Quest'ultimo consente misurazioni simultanee del legame del ligando e della corrente del canale, una lettura diretta della funzione proteica. Il trattamento e l'analisi dei dati sono ampiamente discussi, insieme a potenziali insidie e artefatti. Questo metodo fornisce ricche intuizioni meccanicistiche sul gating ligando-dipendente dei canali KATP e può essere facilmente adattato allo studio di altre proteine regolate da nucleotidi o di qualsiasi recettore per il quale può essere identificato un ligando fluorescente adatto.
Introduzione
Diverse importanti classi di proteine sono direttamente regolate dal legame del ligando. Questi vanno dagli enzimi solubili alle proteine incorporate nella membrana, tra cui il recettore tirosin-chinasi, i recettori accoppiati alle proteine G (GPCR) e i canali ionici. I GPCR e i canali rappresentano ~ 34% e ~ 15% di tutti gli attuali bersagli farmacologici, rispettivamente 1,2. Pertanto, vi è un notevole interesse biochimico e medico nello sviluppo di metodi che forniscono intuizioni meccanicistiche sulle interazioni ligando-recettore. I metodi tradizionali per misurare il legame del ligando, tra cui l'etichettatura della fotoaffinità e gli studi sul legame del radioligando, richiedono grandi quantità di proteine parzialmente purificate e sono tipicamente eseguiti in condizioni e scale temporali non fisiologiche. Un metodo ideale richiederebbe solo piccole quantità di proteine, potrebbe essere eseguito su proteine intatte espresse in un ambiente cellulare o di membrana, potrebbe essere monitorato in tempo reale e sarebbe compatibile con letture dirette della funzione proteica.
Il trasferimento di energia di risonanza di Förster (FRET) è un metodo che rileva la vicinanza tra due molecole marcate con fluorescenza3. La FRET si verifica quando un fluoroforo donatore eccitato trasferisce energia in modo non radiativo a una molecola accettore (tipicamente un altro fluoroforo). Il trasferimento di energia provoca l'estinzione dell'emissione di fluorescenza del donatore e la sensibilizzazione dell'emissione dell'accettore (se l'accettore è un fluoroforo). L'efficienza di trasferimento dipende dalla 6apotenza della distanza tra il donatore e l'accettore. Inoltre, il donatore e l'accettore devono essere in prossimità (di solito meno di 10 nm) affinché si verifichi la FRET. Come tale, FRET può essere sfruttato per misurare il legame diretto tra un recettore proteico marcato fluorescentmente e un ligando fluorescente.
Diverse proteine diverse sono regolate o attivate legando nucleotidi adenina intracellulari o extracellulari (ATP, ADP, AMP, cAMP). Molte proteine trasportatrici richiedono l'idrolisi dell'ATP per il loro ciclo di reazione, compresi i trasportatori di cassette leganti l'ATP e le ATPasi di tipo P come la pompa Na+/K+ 4,5. I canali K+ (K ATP) sensibiliall'ATP, il regolatore della conduttanza transmembrana della fibrosi cistica (CFTR) e i canali regolati dai nucleotidi ciclici sono tutti canali ionici che sono controllati dal legame dei nucleotidi adenina intracellulari, rendendoli squisitamente sensibili ai cambiamenti nel metabolismo cellulare e nella trasduzione del segnale 6,7,8. I recettori purinergici P2X e P2Y rispondono ai cambiamenti nell'ATP extracellulare, che può essere rilasciato come neurotrasmettitore o come risultato di danni tissutali9. Abbiamo sviluppato un saggio basato su FRET per la misura del legame dei nucleotidi adenina alle proteine di membrana in tempo reale. Abbiamo precedentemente applicato questo metodo per studiare il legame nucleotidico ai canali KATP 10,11.
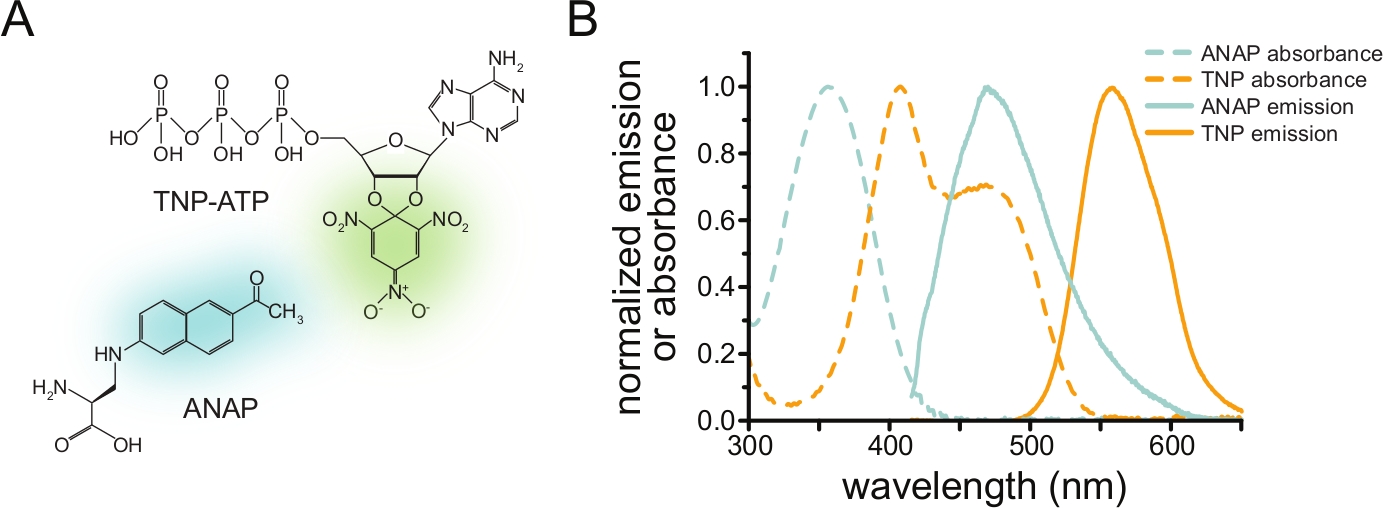
Per misurare il legame nucleotidico tramite FRET, una proteina di interesse deve prima essere marcata con un fluoroforo. Il tag fluorescente deve essere inserito in modo specifico nella proteina di interesse in modo che sia abbastanza vicino al sito di legame del ligando perché si verifichi la FRET, con particolare attenzione per garantire che il tag non influenzi la struttura e la funzione generale della proteina. Per fare ciò, impieghiamo una tecnica sviluppata da Chatterjee et al., utilizzando la soppressione del codone di arresto ambrato per inserire un amminoacido fluorescente non canonico (l-3-(6-acetilnaftalene-2-ilammino)-2-amminopropionico; ANAP) nel sito desiderato12. Misuriamo il legame nucleotidico come FRET tra proteine marcate con ANAP e derivati nucleotidici fluorescenti del trinitrofenile (TNP) (Figura 1A). Lo spettro di emissione per ANAP si sovrappone allo spettro di assorbanza dei nucleotidi TNP, una condizione necessaria affinché si verifichi la FRET (Figura 1B). Qui delineiamo due diversi tipi di esperimento di legame. Nel primo, il legame nucleotidico al lato intracellulare dei canali KATP marcati con ANAP viene misurato in cellule che sono state scoperte dalla sonicazione lasciando frammenti aderenti di membrana plasmatica su un vetrino di copertura10,11,13,14.
Nel secondo metodo, il legame nucleotidico ai canali KATP marcati con ANAP viene misurato in un patch di membrana sotto morsetto di tensione, consentendo la misurazione simultanea delle correnti ioniche e della fluorescenza. Combinando questi due approcci sperimentali, i cambiamenti nel legame possono essere direttamente correlati con i cambiamenti nella funzione del canale11. Vengono discussi i risultati tipici, le potenziali insidie e l'analisi dei dati.
Protocollo
1. Preparazione dei vetrini di copertura
NOTA: Questi passaggi devono avvenire in una cappa sterile per coltura tissutale. Le quantità sono indicate per la preparazione di 10 piatti.
- Posizionare dieci vetrini di vetro borosilicato da 30 mm autoclavati singolarmente in dieci piatti sterili non trattati da 35 mm e risciacquare una volta con 2 ml di acqua distillata sterile.
- Diluire 1 mL di soluzione di poli-L-lisina allo 0,1% p/v in acqua distillata sterile fino ad un volume totale di 10 mL (concentrazione finale dello 0,01% p/v). Mescolare bene, quindi pipettare 1 mL su ciascun vetrino di copertura e incubare a temperatura ambiente per 20 minuti.
- Aspirare la poli-L-lisina e lavare due volte ogni vetrino di copertura con almeno 2 ml di acqua distillata sterile. Lasciare asciugare completamente cioè almeno 3 ore.
2. Semina di cellule HEK-293T
NOTA: Questi passaggi devono avvenire in una cappa di coltura tissutale. Le cellule HEK-293T sono state scelte per il loro background a bassa corrente e la facilità di crescita in coltura. Questo protocollo può essere adattato ad altri tipi di cellule.
- Risciacquare un matraccio T75 confluente all'80-90% di cellule HEK-293T una volta con 12 mL di soluzione salina tamponata fosfato (PBS) prima di incubare con 2 mL di tripsina per 2-5 minuti, o fino a quando le cellule sono completamente staccate e quasi completamente dissociate.
- Risospendere le cellule aggiungendo 10 mL di Modified Eagle Medium (DMEM) di Dulbecco integrati con siero bovino fetale al 10%, 100 U/mL di penicillina e 100 μg/mL di streptomicina. Pipettare delicatamente contro il fondo del matraccio per rompere i rimanenti grumi di cellule.
- Aggiungere 2 ml di DMEM integrato al numero desiderato di piatti da 35 mm contenenti vetrini di copertura rivestiti. Aggiungere 100 μL di celle risospese a ciascun piatto. Incubare per una notte a 37 °C.
3. Trasfezione
NOTA: Questi passaggi devono avvenire in una cappa di coltura tissutale. Le quantità sono indicate per la trasfezione di 10 piatti. Per l'incorporazione ANAP specifica del sito, il codone DNA nella posizione destinata all'etichettatura deve essere sostituito con il codone di arresto ambra (TAG). Questo costrutto è co-trasfettato con due plasmidi: pANAP e peRF1-E55D12,15. pANAP codifica diverse copie di una coppia tRNA/tRNA sintetasi specifica per ANAP. In presenza di ANAP, la trasfezione di questo plasmide produce tRNA caricato con ANAP che riconosce il codone di stop ambrato. peRF1-E55D codifica per un fattore di rilascio ribosomiale negativo dominante che aumenta la resa della proteina a lunghezza intera marcata con ANAP.
- Preparare una provetta da 1,5 ml con 10 μg di pANAP, 10 μg di peRF1-E55D e DNA per il costrutto destinato all'etichettatura con ANAP. Portare ad un volume finale di 500 μL con DMEM non integrato.
- In una provetta separata, preparare 3 μL di reagente di trasfezione a base lipidica (vedere Tabella dei materiali) per ogni 1 μg di DNA e portare ad un volume finale di 500 μL con DMEM non integrato.
- Unire le miscele di DNA e reagenti di trasfezione in un'unica provetta e incubare per 20 minuti a temperatura ambiente.
- Aggiungere 400 μL dello stock di 1 mM ANAP (sale di trifluoroacetato in 30 mM NaOH) a 20 mL di DMEM integrato per una concentrazione finale di 20 μM ANAP. Sostituire i vecchi supporti delle celle placcate con 2 ml di terreno contenente ANAP per piatto.
- Pipettare il 10% della miscela di trasfezione del DNA su ogni piatto. Incubare a 33 °C per 2-4 giorni prima degli esperimenti. L'incubazione a 33 °C rallenta la divisione cellulare e aumenta la resa proteica per cellula16.
4. Esperimenti su membrane senza tetto
- Usa un paio di pinze per rompere un foglietto di copertura con cellule trasfettate in frammenti più piccoli.
- Seguire una delle procedure riportate di seguito per smontare le celle del tetto.
- Se si utilizzano vetrini di copertura preverniciati, sciacquare un frammento con PBS, quindi posizionarlo sul fondo di un piatto da 35 mm contenente 2 ml di PBS. Sonicare brevemente utilizzando un sonicatore a sonda (50 W, 20%-40% di ampiezza, sonda da 3 mm) posizionato 3-5 mm sopra il campione per aprire il tetto delle cellule e lasciare frammenti di membrana plasmatica aderenti (Figura 2A,C).
NOTA: la potenza, la durata e l'altezza della sonda del sonicatore al di sopra del campione possono essere variate per ottenere un'elevata resa di membrane non coperte senza denudare completamente il vetrino di copertura. - Se non si utilizzano vetrini di copertura pre-rivestiti, sciacquare un frammento di vetrino di copertura con PBS, quindi immergerlo in un tubo contenente lo 0,1% p/v di poli-L-lisina per ~30 s prima di sonicare brevemente (come nel passaggio 4.2.1) per aprire le celle e lasciare dietro di sé frammenti di membrana plasmatica senza tetto / parzialmente senza tetto (Figura 2A, C, D). È stato dimostrato che brevi esposizioni alla poli-L-lisina migliorano l'aderenza al foglietto13.
- Se si utilizzano vetrini di copertura preverniciati, sciacquare un frammento con PBS, quindi posizionarlo sul fondo di un piatto da 35 mm contenente 2 ml di PBS. Sonicare brevemente utilizzando un sonicatore a sonda (50 W, 20%-40% di ampiezza, sonda da 3 mm) posizionato 3-5 mm sopra il campione per aprire il tetto delle cellule e lasciare frammenti di membrana plasmatica aderenti (Figura 2A,C).
- Posizionare il frammento sonicato in un piatto di vetro da 35 mm contenente 2 ml di soluzione da bagno e montarlo su un microscopio invertito dotato di un obiettivo ad immersione in acqua ad alto NA, 60x. La porta della telecamera del microscopio è collegata ad uno spettrografo in serie con una camera CCD ad alta sensibilità. Perfondere la camera da bagno (0,5 – 1 mL/min) con tampone utilizzando una pompa peristaltica. La composizione del tampone varierà a seconda della proteina in studio.
NOTA: Se l'utente non ha accesso a un obiettivo con una lunga distanza di lavoro, potrebbe essere impossibile concentrarsi sui frammenti di membrana non coperti a causa dell'altezza extra della slitta di copertura. Un'alternativa è quella di seminare le cellule direttamente su piatti con fondi di vetro di poli-L-lisina (vedi Tabella dei materiali per un esempio). Ciò ridurrà anche le potenziali aberrazioni nell'immagine associate alla messa a fuoco attraverso due pezzi di vetro. Queste aberrazioni non influenzano la forma degli spettri acquisiti. - Identificare i frammenti di membrana senza tetto che esprimono il canale marcato con ANAP cercando la fluorescenza del canale (Figura 2C,D).
NOTA: Si raccomanda di utilizzare un'etichetta fluorescente aggiuntiva (in cui lo spettro di emissione è distinguibile dallo spettro di emissione ANAP) per aiutare a identificare le membrane senza tetto contenenti la proteina di interesse. Gli esperimenti in Figura 2C,D sono stati eseguiti su canali marcati con ANAP con tag proteici fluorescenti C-terminali. - Inserire parzialmente la maschera dello spettrometro (aumentare ~ 10%) tra la porta della fotocamera sul microscopio e lo spettrografo. L'ombra della maschera apparirà sull'immagine della fotocamera. Allineare la membrana senza tetto con la maschera dello spettrometro, regolando lo stadio del microscopio. Acquisire un campo luminoso e un'immagine di fluorescenza della membrana senza tetto. Questi saranno utilizzati per selezionare una regione di interesse per l'analisi.
- Avvicinare la punta del sistema di perfusione a microvolume alla membrana non coperta.
NOTA: Per ridurre la fluorescenza di fondo, il deflusso del sistema di perfusione è stato sostituito con una punta personalizzata in vetro borosilicato. - Per visualizzare spettri di fluorescenza, eccitare la membrana con un LED da 385 nm attraverso un filtro di eccitazione passa banda 390/18 nm e un bordo dicroico di 416 nm. Raccogliere la luce emessa attraverso un filtro di emissione passa-lungo 400 nm (Figura 2B).
- Attivare la maschera dello spettrometro e assicurarsi che la luce emessa venga attraversata. Inserire i reticoli dello spettrometro (300 scanalature/mm). Con i reticoli in posizione, la luce diffratta dallo spettrometro verrà proiettata sul chip della camera CCD per produrre immagini spettrali (Figura 3A). Queste immagini conservano le informazioni spaziali nella dimensione y . La dimensione x viene sostituita con la lunghezza d'onda.
- Facoltativamente, se la proteina di interesse è marcata con una proteina fluorescente, acquisire un'immagine spettrale della proteina fluorescente utilizzando il set di filtri appropriato.
- Prendere una o più esposizioni di 0,1-10 s all'inizio dell'esperimento mentre si perfonde la soluzione tampone priva di nucleotidi. Questi verranno utilizzati per correggere e normalizzare i dati durante il resto dell'esperimento (vedere la sezione 5 di seguito).
NOTA: La scelta del tempo di esposizione dipenderà dal livello di espressione raggiunto, dalla luminosità del fluoroforo e dall'ottica. Il tempo di esposizione dovrebbe essere scelto per massimizzare il segnale e ridurre al minimo il tasso di sbiancamento osservato. L'intervallo di tempo di esposizione indicato al punto 4.10 è adatto per misure di legame all'equilibrio, ma può essere utile per misurare variazioni cinetiche più lente10. La capacità di utilizzare tempi di esposizione brevi per tracciare cinetiche più veloci sarà limitata dai livelli di espressione proteica e dal fotosbiancamento, piuttosto che dall'hardware. - Applicare un intervallo di concentrazioni di TNP-ATP (di solito preparato in soluzione di bagno) per stabilire una curva concentrazione-risposta. Perfondere ogni soluzione per almeno 1 minuto per assicurarsi che venga raggiunto uno stato stazionario e lavare ogni concentrazione con la soluzione da bagno per almeno 1 minuto.
NOTA: È importante assicurarsi che il sistema di perfusione possa raggiungere rapidamente l'equilibrio (Figura 2E) e raggiungere la corretta concentrazione locale di TNP-ATP (Figura 2F). - Assumere un'esposizione (della stessa durata utilizzata al punto 4.10) ad ogni concentrazione e alla fine di ogni dilavamento.
5. Analisi spettrale
NOTA: queste istruzioni sono scritte per l'uso con il codice di analisi "pcf.m", disponibile su GitHub. https://github.com/mpuljung/spectra-analysis10. Codice aggiuntivo e alternativo può essere trovato in https://github.com/smusher/KATP_paper_201911. Abbiamo descritto qui le operazioni eseguite dal software in modo che l'utente possa creare il proprio codice o scegliere di analizzare i dati manualmente.
- Avviare il programma di analisi digitando il nome del programma ("pcf") nella riga di comando.
- Quando si apre una finestra di dialogo Apri file/cartella con il messaggio: "Seleziona file per ROI", seleziona i nomi dei file associati alle immagini in campo chiaro e fluorescenza della membrana non coperta. Nella riga di comando verrà visualizzato un prompt per digitare il nome del file di output.
- Digita il nome del file e premi invio.
- Quando il software visualizza le immagini in campo chiaro e fluorescenza, selezionare una regione di interesse (ROI) nell'immagine spettrale corrispondente alla posizione del frammento di membrana senza tetto o del cerotto asportato (vedere la sezione 6) seguendo le istruzioni del software. Selezionare una regione di sfondo nella stessa immagine spettrale (che rappresenta lo stesso intervallo di lunghezze d'onda del ROI) corrispondente a una sezione di vetrino di copertura o piatto senza membrana attaccata (Figura 3A). Il software chiederà di fare clic sulla parte superiore del ROI e premere Invio, fare clic sulla parte inferiore del ROI e premere Invio e quindi ripetere questo processo per l'area di background.
- Quando si apre una finestra di dialogo Apri file/cartella con il messaggio: "Seleziona file per FP Spectrum", selezionare il nome del file associato allo spettro delle proteine fluorescenti (FP) (passaggio facoltativo 4.9). Se non è stato acquisito alcuno spettro FP, selezionare un file di spettro diverso. Lo spettro FP funge da controllo di qualità per distinguere tra proteine marcate e fluorescenza di fondo.
- Quando si apre una finestra di dialogo Apri file/cartella con il prompt: "Seleziona file per l'analisi", selezionare tutti i file corrispondenti agli spettri ANAP (dai passaggi 4.10 a 4.12), inclusi i file necessari per la correzione della candeggina.
- Quando si apre una finestra di dialogo Apri file/cartella con il messaggio: "Seleziona file per la raccolta di sbiancamento", selezionare il sottoinsieme di file dal punto 5.6 corrispondente agli spettri iniziali acquisiti in soluzione priva di nucleotidi all'inizio dell'esperimento o agli spettri acquisiti durante i lavaggi in soluzione priva di nucleotidi da utilizzare per la correzione (dai punti 4.10 a 4.12).
- Calcola la media delle linee di ogni immagine per produrre spettri, cioè media dell'intensità per tutti i pixel nella dimensione y di un ROI o di una regione di sfondo a ciascuna lunghezza d'onda. (Figura 3B). Sottrarre lo spettro di fondo medio risultante dallo spettro medio acquisito dal ROI per rimuovere la fluorescenza di fondo e la fluorescenza dal TNP-ATP non legato (Figura 3C). Questi passaggi vengono eseguiti automaticamente dal software.
- Determinare l'intensità ANAP per ciascuna esposizione calcolando la media dell'intensità di una finestra di 5 nm centrata attorno al picco ANAP degli spettri sottratti (tipicamente ~470 nm ma può variare a seconda del microambiente locale del residuo ANAP).
NOTA: La Figura 3D mostra 6 spettri ottenuti da esposizioni consecutive di 10 s di un frammento di membrana non coperta che esprime canali marcati ANAP. L'inserto mostra l'intensità media del picco di ogni spettro. Il software troverà automaticamente la lunghezza d'onda di picco nel primo spettro acquisito e utilizzerà questo valore in tutto. L'intensità verrà calcolata automaticamente dal software. - Normalizzare le intensità ANAP per ciascun esperimento dividendo l'intensità ANAP di una data esposizione (F) per l'intensità ANAP della prima esposizione nella serie temporale, che è stata presa nel passaggio 4.10 (Fmax). Ancora una volta, il software esegue automaticamente questi calcoli.
- Eseguire i passaggi seguenti per ottenere i dati.
- Per correggere il fotosbiancamento ANAP, adattare prima un singolo decadimento esponenziale, (F/Fmax) = A*exp(-t/τ)+(1-A), dove t è il tempo di esposizione cumulativo, τ è la costante di tempo e A è l'ampiezza) alle fasi intermedie di lavaggio tra applicazioni TNP-ATP o a esposizioni iniziali multiple effettuate prima del lavaggio su TNP-ATP (Figura 3D, riquadro).
NOTA: il software visualizzerà questo adattamento e chiederà di accettarlo o rifiutarlo. Se l'adattamento viene rifiutato, verrà fornita un'altra opportunità per selezionare i file per la correzione dello sbiancamento. - Dividere gli spettri ANAP normalizzati (nel passo 5.10) per il valore previsto dell'adattamento esponenziale dal punto 5.11.1 in ciascun punto temporale (Figura 3E).
NOTA: Per l'esempio mostrato, il picco di fluorescenza normalizzato osservato a 50 s è 0,65 e la fluorescenza prevista dall'adattamento esponenziale è 0,64. Per correggere lo sbiancamento, dividere il valore osservato (0,65, riquadro Figura 3E, cerchio vuoto) per il valore previsto (0,64, riquadro Figura 3E, linea tratteggiata) per produrre il valore corretto (~1, riquadro Figura 3E, cerchio colorato). Se la correzione dello sbiancamento è adeguata, l'intensità di ANAP da tutte le esposizioni acquisite in assenza di nucleotidi deve essere approssimativamente uguale (Figura 3E). Questi calcoli vengono eseguiti automaticamente dal software. - Ottenere l'output come un'immagine che traccia i dati e un foglio di calcolo a schede contenente gli spettri grezzi, gli spettri sottratti, gli spettri corretti per il fotosbiancamento e i dati di picco per ciascun file in modo che possano essere condotte ulteriori analisi.
- Per correggere il fotosbiancamento ANAP, adattare prima un singolo decadimento esponenziale, (F/Fmax) = A*exp(-t/τ)+(1-A), dove t è il tempo di esposizione cumulativo, τ è la costante di tempo e A è l'ampiezza) alle fasi intermedie di lavaggio tra applicazioni TNP-ATP o a esposizioni iniziali multiple effettuate prima del lavaggio su TNP-ATP (Figura 3D, riquadro).
6. Esperimenti di fluorometria patch-clamp
- Tirare pipette patch da capillari di vetro borosilicato a parete spessa a una resistenza da 1,5 MΩ a 2,5 MΩ quando riempite con soluzione di pipetta. La composizione della soluzione di pipetta varia a seconda della proteina in studio.
- Trasferire un vetrino di copertura con cellule trasfettate su un piatto di vetro da 35 mm contenente 2 mL di soluzione da bagno e montarlo su un microscopio invertito dotato di un obiettivo ad immersione in acqua ad alto NA, 60x. Perfondere la camera da bagno (0,5 – 1 mL/min) con una soluzione da bagno utilizzando una pompa peristaltica. Per quanto riguarda la soluzione di pipetta, la soluzione del bagno varierà a seconda della proteina in studio.
- Identificare una cellula che esprime canali marcati con ANAP cercando la fluorescenza sulla membrana cellulare.
- Riempire una pipetta patch con una soluzione per pipette. Applicare una leggera pressione positiva sulla pipetta e posizionarla nella camera da bagno. Premere la pipetta contro la membrana della cella e applicare una leggera aspirazione per ottenere una tenuta GΩ (Figura 4A).
- Eliminare il cerotto allontanando rapidamente il portapipette dalla cella (Figura 4A).
NOTA: L'asmissione del cerotto in questo modo dovrebbe formare una patch inside-out, con i domini citosolici della proteina esposti al sistema di perfusione. Se la posizione del sito di legame nucleotidico in studio non è citosolica, sarà necessario utilizzare cerotti esterni o registrazioni di cellule intere per eseguire esperimenti PCF. - Avvicinare la punta della pipetta del cerotto alla punta del sistema di perfusione e verificare che il cerotto si trovi all'interno della fessura della maschera dello spettrometro (Figura 4A).
- Applicare TNP-ATP e spettri di immagine come nei passaggi 4.10-4.12, registrando contemporaneamente la risposta della corrente ionica all'applicazione nucleotidica.
NOTA: Il vetro per pipette può introdurre aberrazioni e riflessi spaziali nelle immagini acquisite. Tuttavia, queste aberrazioni non influenzeranno la forma degli spettri acquisiti e la luce di eccitazione riflessa è facilmente separata dalla fluorescenza utilizzando lo spettrografo o un filtro ad emissione passa-lungo. - Analizza gli spettri. Gli spettri ripresi da cerotti asportati possono mostrare un'eccessiva sottrazione della fluorescenza TNP-ATP non legata a causa dell'esclusione di TNP-ATP dal vetro della pipetta patch (Figura 4C-E). Questa eccessiva sottrazione non influisce sullo spettro di emissione ANAP e quindi può essere ignorata.
NOTA: Poiché il segnale di fluorescenza nelle chiazze asportate sarà inferiore rispetto alle membrane senza tetto, è importante utilizzare un tempo di esposizione che fornisca un rapporto segnale-rumore sufficientemente elevato senza sbiancare l'ANAP troppo rapidamente.
Risultati
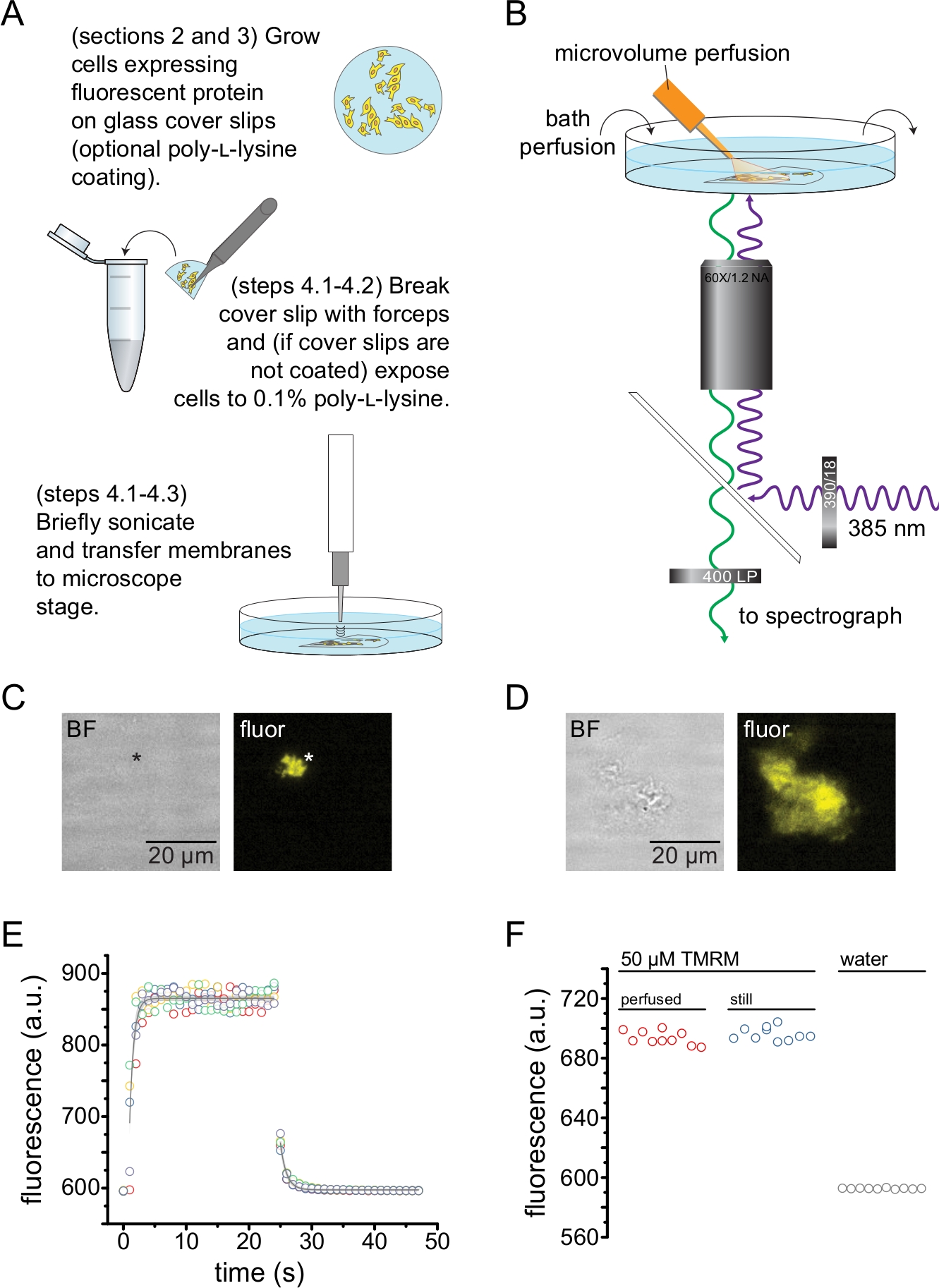
La Figura 2 illustra la configurazione sperimentale di base per misurare il legame nucleotidico alle proteine fluorescenti in frammenti di membrana senza tetto ottenuti mediante sonicazione (Figura 2A,B). Sono stati utilizzati due diversi approcci per ottenere membrane non coperte, coltivando direttamente le cellule su vetrini di copertura rivestiti di poli-L-lisina o coltivando cellule su vetro non trattato ed esponendoli brevemente alla poli-L-lisina (0,1% in acqua) prima di scoperchiare. La Figura 2C mostra un tipico frammento di membrana senza tetto proveniente da una cellula HEK-293T che esprime canali K ATP marcati con proteina fluorescente arancione (OFP). Le membrane senza tetto erano praticamente invisibili nelle immagini a campo chiaro e sono state identificate dalla fluorescenza delle proteine di membrana marcate o dalla controcolorazione con un colorante di membrana come l'ottadecilrodamina B13. Oltre alle membrane senza tetto, la sonicazione delle cellule HEK-293T ha prodotto anche frammenti di cellule parzialmente senza tetto (Figura 2D)10,17. Questi frammenti erano visibili in campo chiaro. Questo potrebbe essere il risultato di membrane plasmatiche arruffate che sono solo scarsamente aderenti al vetro di copertura. In alternativa, questi frammenti possono contenere vescicole e membrane da organelli intracellulari. Pertanto, è preferibile acquisire immagini solo da membrane "vere" senza tetto, poiché la proteina bersaglio marcata associata alle membrane intracellulari può riflettere le fasi intermedie dell'elaborazione e dell'assemblaggio post-traduzionale. Si raccomanda di coltivare le cellule su vetro rivestito di poli-L-lisina in quanto ciò ha comportato una maggiore resa di membrane "vere" senza tetto dopo la sonicazione.
Un sistema di perfusione in microvolume è stato applicato ai nucleotidi fluorescenti per ridurre al minimo le quantità necessarie in un tipico esperimento (Figura 2B). La punta in vetro rivestita di poliimmide fornita è stata sostituita con una punta in vetro borosilicato tirata a mano nella nostra configurazione di perfusione, che ha ridotto lo sfondo di fluorescenza. Per ridurre al minimo l'accumulo di nucleotidi attorno alle membrane non coperte da riprendere, l'intera camera da bagno è stata lentamente perfusa con tampone. Pertanto, abbiamo voluto misurare la velocità di variazione della soluzione dal nostro sistema di perfusione a microvolume e verificare che fossimo in grado di raggiungere la concentrazione di ligando prevista nella nostra regione di interesse, cioè che il ligando del nostro sistema di perfusione non fosse diluito direttamente nel mezzo di balneazione prima di raggiungere la membrana senza tetto. Per controllare queste possibilità, sono stati misurati il wash-in e il wash-out di una soluzione 50 μM di tetrametilrhodamina-5-maleimmide (TMRM) dal nostro sistema di perfusione di microvolume diretto sulla superficie di un piatto con fondo di vetro di copertura perfuso con acqua (Figura 2E). La cinetica di scambio della soluzione era riproducibile e ben descritta da un singolo decadimento esponenziale con costanti di tempo inferiori a 1 s sia per il wash-in che per il wash-out. Tali tempi di scambio della soluzione limitano la nostra capacità di misurare la cinetica del legame e dell'unbinding del ligando nella nostra configurazione attuale. Per verificare che siamo stati in grado di raggiungere la concentrazione desiderata di ligando sulla superficie del vetrino di copertura, abbiamo confrontato l'intensità di fluorescenza di 50 μM TMRM erogata al vetrino di copertura dal nostro sistema di perfusione a microvolume con 50 μM TMRM in un bagno fermo (Figura 2F). Non è stata osservata alcuna differenza di intensità, verificando che sia possibile ottenere concentrazioni appropriate di ligando sulla superficie del vetrino di copertura con il nostro sistema di perfusione a microvolume, anche quando il bagno è perfuso.
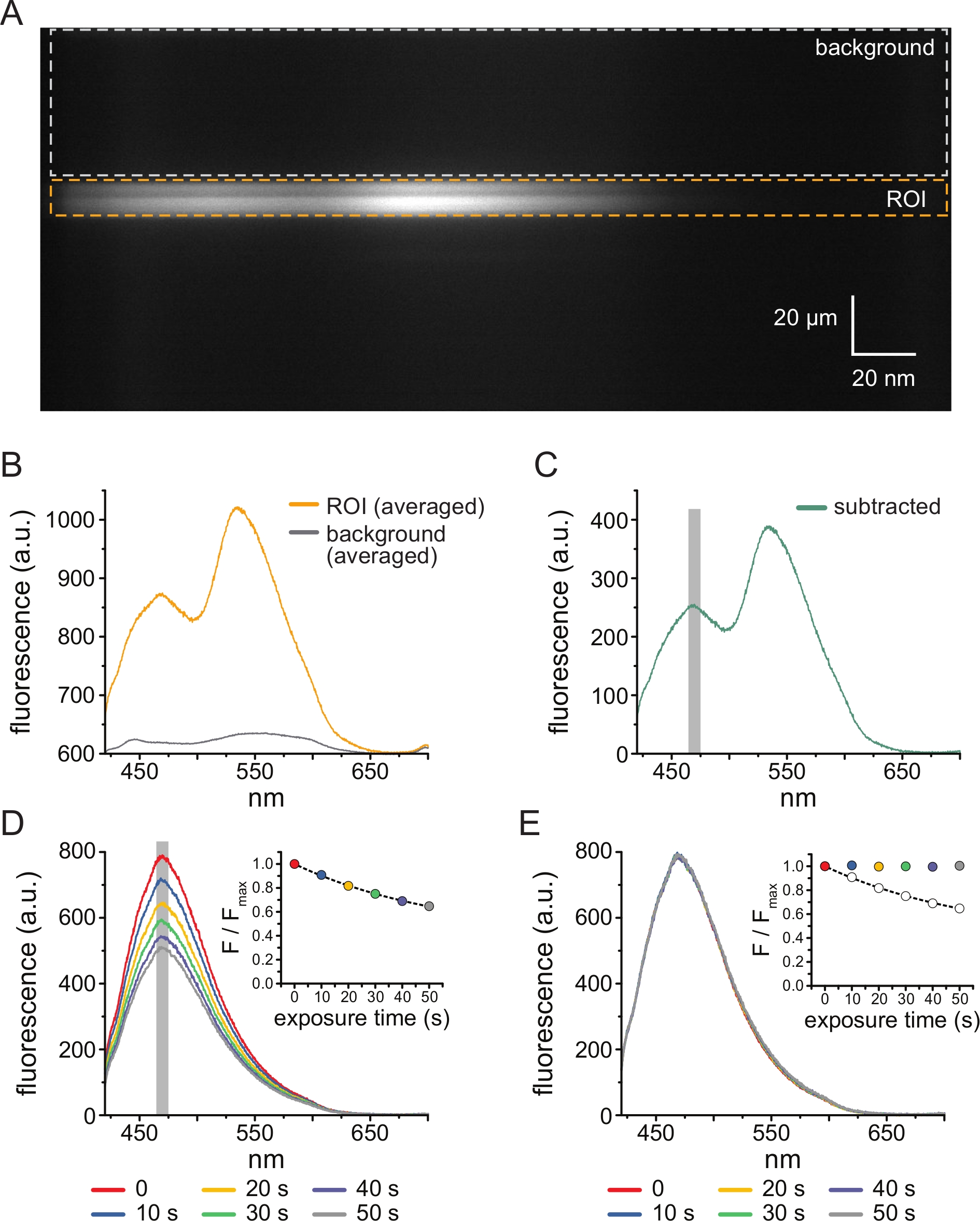
La figura 3A mostra un'immagine spettrale ottenuta dai canali K ATP marcati con ANAP in una membrana senza tetto da una cella HEK-239T esposta a 5 μM TNP-ATP. Per ottenere tali immagini, la luce emessa dalla membrana senza tetto è stata diretta attraverso uno spettrometro in serie con una camera CCD. La fluorescenza emessa è stata diffratta dai reticoli e proiettata sul chip della fotocamera, producendo spettri. Le immagini risultanti mantengono le informazioni spaziali nella dimensione y, ma la dimensione x è stata sostituita con la lunghezza d'onda. La regione di interesse (ROI), corrispondente alla membrana non coperta, è delineata in arancione. Nell'immagine sono evidenti due regioni di alta intensità, corrispondenti al picco di emissione di ANAP e TNP-ATP. Ciò è stato meglio apprezzato nello spettro mediato lunghezza d'onda per lunghezza d'onda (sull'intero ROI) mostrato nella Figura 3B. Il picco ~470 nm corrisponde all'ANAP incorporato in KATP; il picco ~535 nm corrisponde a TNP-ATP. Per correggere la fluorescenza di fondo e l'eccitazione diretta di TNP-ATP in soluzione, è stata selezionata una regione di fondo (Figura 3A, grigio) da ciascuna immagine. Lo spettro di fondo medio è mostrato nella Figura 3B. Lo spettro finale è stato ottenuto sottraendo lo spettro di fondo medio dallo spettro ROI medio (Figura 3C).
ANAP è incline a artefatti fotosbiancanti. La figura 3D mostra la riduzione del picco di fluorescenza ANAP dopo esposizioni multiple. Il picco di fluorescenza da diverse esposizioni in assenza di TNP-ATP (o da lavaggi tra le concentrazioni di TNP-ATP) è stato adattato a un decadimento esponenziale singolo e questo è stato utilizzato per correggere gli artefatti fotosbiancanti (Figura 3E). Si raccomanda di eseguire esperimenti concentrazione-risposta da concentrazioni di nucleotidi da basse a alte e alte-basse. Se la correzione dello sbiancamento non introduce artefatti aggiuntivi, i risultati dovrebbero essere comparabili11.
La figura 5A mostra immagini spettrali rappresentative da una membrana senza tetto ottenuta da una cellula che esprime canali K ATP marcati con ANAP in assenza e presenza di TNP-ATP. Gli spettri corretti sono mostrati nella figura 5B. Osservando gli spettri di emissione, c'era una netta separazione tra l'emissione di fluorescenza del donatore e dell'accettore. Poiché è stato osservato un legame non specifico di TNP-ATP alle membrane plasmatiche naïve da cellule HEK-293T non trasfettate, si raccomanda di quantificare FRET come riduzione della fluorescenza del donatore (ANAP)10,11. Questo picco era specifico per il recettore marcato.
Per i ligandi che inducono un cambiamento conformazionale nel loro recettore, gli studi di legame isolati non forniscono informazioni dirette e meccanicamente significative sul processo di legame del ligando18. La relazione concentrazione-risposta per il legame del ligando dipende non solo dall'affinità intrinseca di legame, ma anche dal cambiamento conformazionale indotto dal legame del ligando e dalla propensione intrinseca del recettore a cambiare conformazione in assenza di ligando. Per comprendere meglio i processi alla base delle interazioni ligando-recettore, le misurazioni di legame possono essere abbinate a esperimenti che forniscono una lettura della funzione proteica. A tal fine, i canali ionici sono un sistema modello ideale, in quanto le loro correnti possono essere misurate con risoluzione temporale inferiore a quella dei ms fino al livello di singola molecola utilizzando il morsetto di tensione. Storicamente, le misurazioni accoppiate di corrente e fluorescenza hanno fornito informazioni significative sull'apertura e la chiusura (gating) dei canali ionici voltaggio e ligando-dipendenti 19,20,21. Sono stati condotti esperimenti per misurare simultaneamente le correnti ioniche e il legame del nucleotide ciclico fluorescente a vari canali regolati da nucleotidi ciclici22,23,24. Questi studi hanno impiegato un ligando che ha aumentato la sua resa quantica al momento del legame. La fluorescenza dal ligando non legato nel volume della soluzione vicino al cerotto può essere sottratta mediante imaging dei cerotti utilizzando la microscopia confocale22,23. Nei nostri studi, il legame è stato misurato utilizzando la riduzione della fluorescenza ANAP. Poiché questo segnale è specifico per il canale e FRET tra ANAP e TNP-ATP è fortemente dipendente dalla distanza (metà massimale a ~ 43 Å), è stata evitata la contaminazione del nostro segnale da nucleotidi non specificamente legati e non legati.
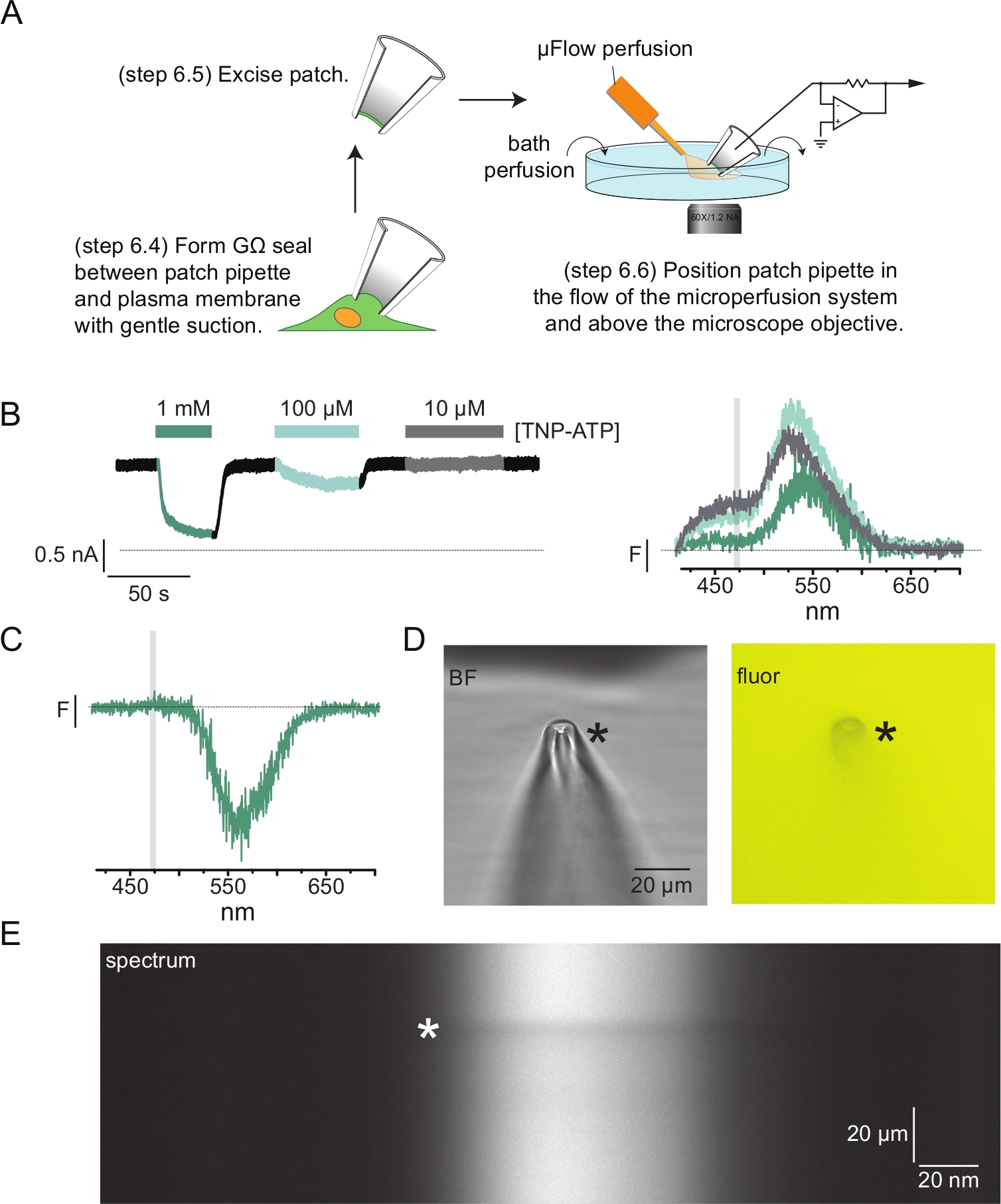
La Figura 4A mostra un tipico esperimento di fluorometria patch-clamp (PCF). Una guarnizione ad alta resistenza (GΩ) è stata formata tra una pipetta di vetro borosilicato riempita di soluzione salina (collegata a un amplificatore a pinza magnetica) e una cella che esprime KATP marcata ANAP. Dopo la formazione del sigillo, la pipetta è stata allontanata dalla cellula, consentendo l'accesso ai siti di legame dei nucleotidi intracellulari. La pipetta è stata quindi posizionata sopra l'obiettivo del microscopio, centrato sulla fessura della maschera dello spettrometro e il deflusso del sistema di perfusione a microvolume (modificato con una punta in vetro borosilicato) è stato portato vicino alla pipetta (Figura 4D). La tensione è stata controllata e le correnti sono state misurate dai canali nella patch. Le correnti e gli spettri rappresentativi dei canali KATP marcati con ANAP sono mostrati nella Figura 4B, codificati a colori per abbinare gli spettri alle correnti. Gli spettri di emissione sono stati corretti per lo sfondo e lo sbiancamento come per le membrane senza tetto.

Figura 1: ANAP e TNP-ATP formano una coppia FRET adatta. a) Strutture di ANAP e TNP-ATP. Le parti fluorescenti sono evidenziate. (B) Spettri di emissione di assorbanza e fluorescenza di ANAP e TNP-ATP. Per la FRET è richiesta la sovrapposizione tra emissione ANAP e assorbanza TNP-ATP. Adattato da Puljung et al. (pubblicato sotto la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)10. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 2: Misurazione del legame nucleotidico in membrane plasmatiche senza tetto. (A) Schema per la preparazione di membrane plasmatiche senza tetto da cellule aderenti che esprimono una proteina fluorescente di membrana. Le istruzioni sono fornite per le cellule coltivate su vetrini di copertura rivestiti di poli-L-lisina o non trattati. (B) Messa a punto sperimentale per misurare il legame nucleotidico in membrane non coperte. (C) Campo luminoso e immagini fluorescenti di una membrana plasmatica completamente scoperchiata derivata da una cellula che esprime una proteina fluorescente arancione (OFP) marcata con canali KATP. L'asterisco segna la posizione della membrana, che è quasi invisibile nell'immagine a campo chiaro. OFP è stata eccitata con un ampio LED da 565 nm attraverso un filtro passa banda 531/40 nm e una luce dicroica a 562 nm ed emessa attraverso un filtro passa banda 593/40 nm. (D) Campo luminoso e immagini fluorescenti di un frammento di membrana parzialmente scoperchiato derivato da una cellula che esprime una proteina fluorescente arancione (OFP) marcata con canali KATP. (E) Corso di tempo di scambio della soluzione acquisito utilizzando la configurazione descritta in B. Vengono mostrate cinque repliche tecniche. Il sistema di perfusione in microvolume è stato caricato con 50 μM di tetrametilrhodamina-5-maleimmide (TMRM). Il bagno è stato perfuso con acqua ad una velocità di ~ 0,5 ml / min. I dati dei cicli di wash-on (fluorescenza crescente) e wash-out (fluorescenza decrescente) sono stati adattati con un singolo decadimento esponenziale della forma F = A * exp (-x / τ) + y0. La costante di tempo (τ) per il wash-in era ~0,6 s. La costante di tempo per il wash-out era ~ 1,0 s. TMRM è stato eccitato con un ampio LED da 565 nm attraverso un filtro passa-banda 540/25 nm e una luce dicroica del bordo di 565 nm e la luce emessa è stata raccolta attraverso un filtro passa banda 605/55 nm. (F) Confronto dell'intensità di fluorescenza di una soluzione di 50 μM di TMRM applicata utilizzando il sistema di perfusione in microvolume come in B e di un bagno fermo contenente 50 μM di TMRM. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 3: Sottrazione dello sfondo e correzione dello sbiancamento. (A) Immagine spettrale (informazioni spaziali nella dimensione y, lunghezza d'onda nella dimensione x) di una membrana plasmatica senza tetto da una cellula che esprime canali KATP marcati con ANAP. 5 μM TNP-ATP è stato applicato utilizzando la configurazione descritta nella Figura 2B. La scatola arancione indica la regione di interesse (ROI), corrispondente alla membrana senza tetto. La casella grigia indica la regione di sfondo utilizzata per correggere lo spettro. (B) Spettri di emissione derivati dalle medie lunghezza d'onda per lunghezza d'onda del ROI e delle regioni di fondo in A. (C) Spettro derivato sottraendo lo spettro di fondo medio dallo spettro ROI medio in B. La finestra di 5 nm attorno al picco ANAP utilizzata per determinare l'intensità media è mostrata come un'area ombreggiata grigia. (D) Spettri acquisiti da sei esposizioni consecutive 10-s di una membrana plasmatica senza tetto da una cellula che esprime canali KATP marcati con ANAP. Si noti il decremento della fluorescenza derivante dal fotosbiancamento. L'inserto mostra l'adattamento del picco di fluorescenza normalizzato con un singolo decadimento esponenziale della forma F/Fmax = A*exp(-t/τ) + (1-A). I simboli nel riquadro sono codificati a colori per corrispondere agli spettri. (E) Gli stessi spettri di cui al punto D corretti per il fotosbiancamento. L'inserto mostra la fluorescenza di picco normalizzata da D come cerchi aperti, con la fluorescenza di picco corretta mostrata usando cerchi pieni. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 4: Misure simultanee del legame nucleotidico e delle correnti di canale mediante fluorometria patch-clamp (PCF). (A) Schema che mostra la configurazione sperimentale per misurare il legame nucleotidico e le correnti ioniche. (B) Esempi di correnti (a sinistra) e spettri (a destra) acquisiti da un cerotto di membrana asportato da una cellula che esprime canali KATP marcati con ANAP. Le correnti sono state registrate a un potenziale di mantenimento di -60 mV, digitalizzate a 20 kHz e filtrate a 5 kHz. L'area ombreggiata grigia corrisponde all'intervallo di lunghezze d'onda da cui è stata quantificata l'intensità ANAP. Adattato da Usher et al. (pubblicato sotto la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (C) Spettro acquisito da un cerotto di membrana asportato da una cellula che esprime canali K ATP marcati con ANAP esposti a 1 mM TNP-ATP. Si noti il picco negativo corrispondente all'intervallo di lunghezze d'onda su cui si osserva la fluorescenza TNP-ATP. L'area ombreggiata grigia indica l'intervallo di lunghezze d'onda utilizzato per quantificare la fluorescenza ANAP come in B. Adattato da Usher et al. (pubblicato sotto la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. (D) Immagini in campo chiaro e fluorescenti di una pipetta patch esposta a 1 mM TNP-ATP. L'asterisco indica la punta della pipetta. (E) Immagine spettrale della stessa pipetta patch in 1 mM TNP-ATP. L'asterisco indica la posizione della pipetta. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

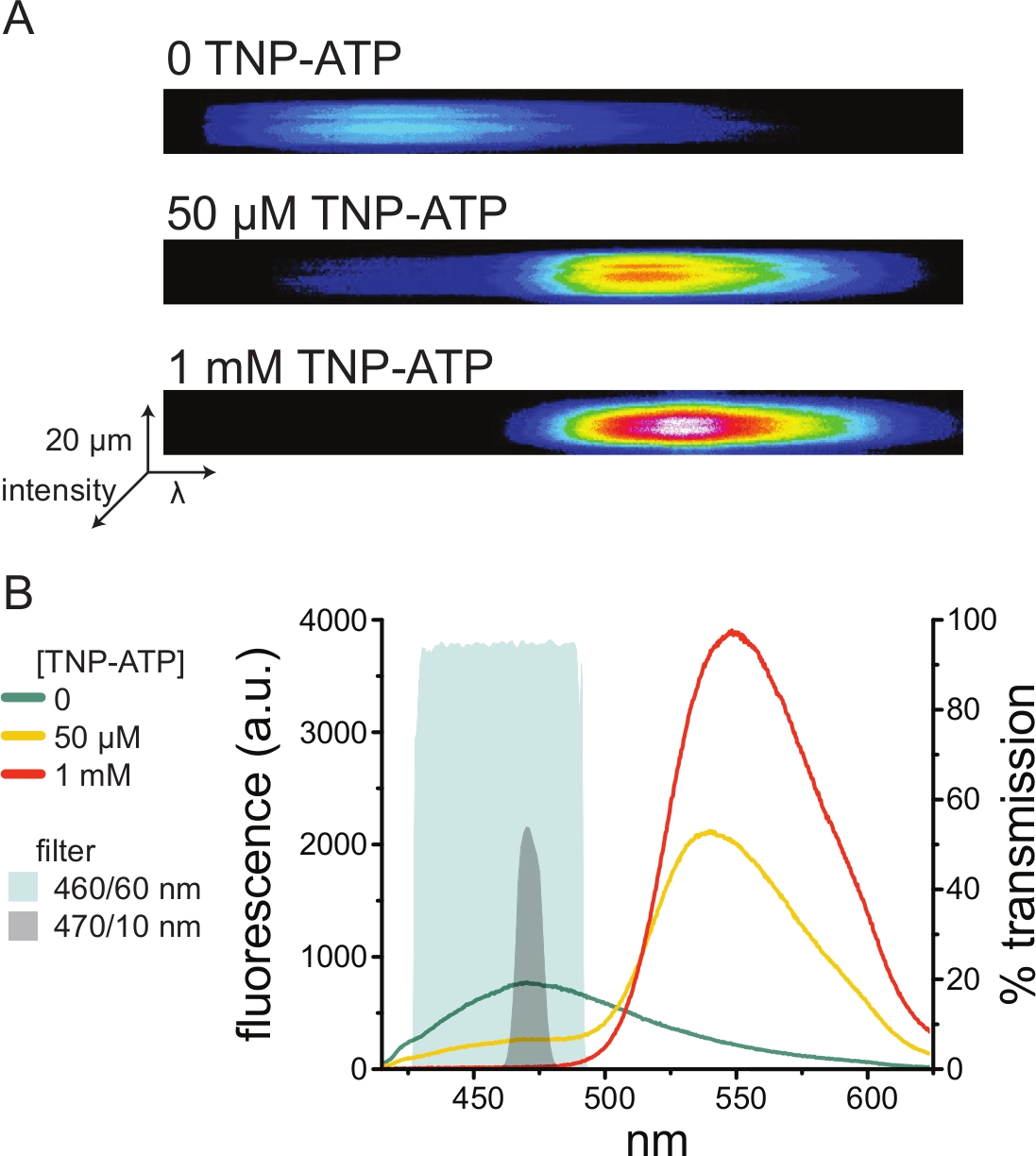
Figura 5: Legame TNP-ATP ai canali KATP marcati ANAP. (A) Immagini spettrali di una membrana plasmatica senza tetto da una cellula che esprime canali K ATP marcati con ANAP in assenza di TNP-ATP o in presenza di 50 μM o 1 mM TNP-ATP. Le intensità sono mostrate come una mappa di calore. (B) Spettri mediati lunghezza d'onda per lunghezza d'onda dalle immagini in A che mostrano l'estinzione della fluorescenza ANAP da parte di TNP-ATP. Le aree ombreggiate rappresentano due diversi filtri passa-banda che possono essere utilizzati per misurare la tempra ANAP se non è disponibile uno spettrometro. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

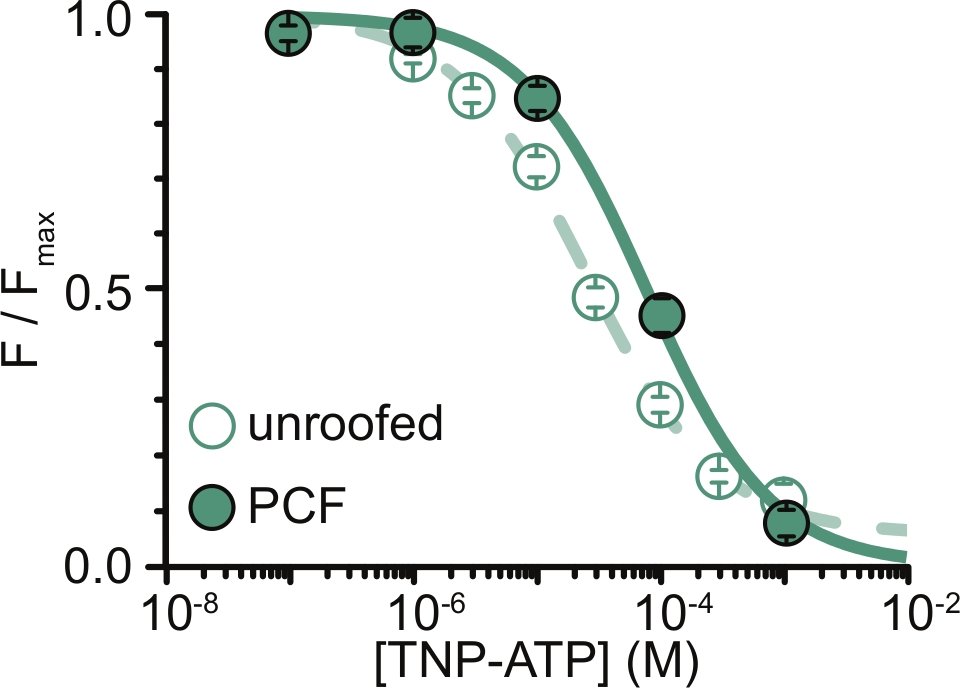
Figura 6: Tempra dei canali K ATP marcati con ANAP medianteTNP-ATP in membrane senza tetto e PCF. Sovrapposizione di dati da Usher et al. (pubblicato sotto la Creative Commons Attribution License, https://creativecommons.org/licenses/by/4.0/)11. I dati erano adatti all'equazione di Hill: F / F max = E max + (1 – E max) / (1+10(EC50 –[TNP-ATP])*h). F è la fluorescenza misurata, F max è la fluorescenza massima in assenza di nucleotide, Emax è la massima tempra a saturare le concentrazioni nucleotidiche e h è la pendenza di Hill. EC50, (la concentrazione nucleotidica alla quale la tempra è la metà massima) e [TNP-ATP] sono valori logaritmici. Membrane senza tetto: EC50 = -4,59 (25,7 μM), h = 0,82, Emax = 0,93. PCF: EC50 = -4,11 (77,6 μM), h = 0,87, Emax = 1,00. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Discussione
Abbiamo sviluppato un metodo per misurare il legame dei nucleotidi adenina in tempo reale alle proteine di membrana intatte. Il nostro metodo si basa su diverse altre tecniche consolidate, tra cui l'etichettatura delle proteine con ANAP utilizzando la soppressione del codone di arresto ambrato 12, lo scoperchio delle cellule 14 e la fluorometria a morsetto di tensione / PCF 19,20,21,22,23,24,25 . La sintesi di questi approcci consente la misurazione del legame nucleotidico con un'elevata risoluzione spaziale e temporale. Infatti, nel nostro lavoro precedente, siamo stati in grado di distinguere tra diversi siti di legame sullo stesso complesso proteico usando questo approccio10,11. È importante sottolineare che questa tecnica può essere applicata direttamente a piccole quantità di proteine in un ambiente cellulare in condizioni che preservano la funzione proteica. L'utilizzo del nostro metodo di legame in combinazione con la lettura elettrofisiologica diretta delle correnti dei canali ionici ci consente di ottenere informazioni approfondite sulle basi molecolari del canale gating11.
Poiché gli spettrometri sono un'apparecchiatura di laboratorio non standard, l'intensità ANAP può anche essere monitorata in relativo isolamento utilizzando filtri passa-banda. La Figura 5B illustra le proprietà spettrali di due di questi filtri. Il filtro passa banda 470/10 nm scherma efficacemente il segnale di fluorescenza da TNP-ATP e si sovrappone bene alla fluorescenza ANAP di picco. Tuttavia, la trasmittanza di picco di questo filtro è solo del 50% circa, il che può rendere difficile ottenere buoni segnali da membrane deboli (o in patch di membrana asportate sotto morsetto di tensione). Un'altra opzione è un filtro passa banda 460/60 nm. C'è una sovrapposizione leggermente maggiore tra il filtro 460/60 nm e il piede del picco di emissione TNP-ATP rispetto al filtro 470/10 nm. Tuttavia, il passaggio di banda 460/60 nm ha una trasmittanza del 90-95% su un ampio intervallo del picco ANAP, che dovrebbe aumentare il segnale di emissione di fluorescenza.
ANAP è un fluoroforo 12,26,27 sensibile all'ambiente. L'emissione di picco e la resa quantica variano a seconda del sito di incorporazione sulla proteina di interesse e possono cambiare quando la proteina cambia conformazione. Tali cambiamenti sarebbero immediatamente evidenti dagli spettri di emissione, ma non sarebbero altrettanto evidenti quando l'intensità ANAP viene misurata usando filtri. In ogni caso, sono necessari controlli appropriati per dimostrare che il segnale di fluorescenza non varia a causa dei cambiamenti nell'ambiente locale intorno all'ANAP successivi al legame nucleotidico. Esperimenti di controllo con nucleotidi non marcati possono aiutare a verificare che eventuali cambiamenti nell'intensità di ANAP siano il risultato di FRET tra nucleotidi ANAP e TNP. I nucleotidi TNP possono legarsi in modo non specifico alle membrane derivate da cellule non trasfettate (alla membrana plasmatica o alle proteine native della membrana)10. Quantifichiamo il legame come un decremento nella fluorescenza del donatore, poiché questo segnale è specifico per il canale etichettato. Tuttavia, raccomandiamo di eseguire ulteriori esperimenti di controllo per ciascuna coppia agonista/recettore, ad esempio mutando il sito di legame nucleotidico se noto, per verificare che il cambiamento nella fluorescenza del donatore sia realmente il risultato del legame diretto al recettore marcato11. Infine, si raccomanda di lavorare con costrutti che contengono un tag proteico fluorescente oltre all'etichetta ANAP. Questo aiuta a differenziare la fluorescenza del recettore marcato dallo sfondo / autofluorescenza. La fluorescenza di fondo può essere distinta dall'ANAP dal picco e dalla forma degli spettri di emissione10, ma tali determinazioni possono essere molto difficili quando vengono utilizzati solo set di filtri. Inoltre, le cellule e le membrane senza tetto che esprimono recettori fluorescenti possono essere identificate utilizzando il tag proteico fluorescente senza dover eccitare l'ANAP e rischiare un eccessivo fotosbiancamento.
In molti dei nostri record PCF, abbiamo osservato un forte picco negativo nei nostri spettri ad alte concentrazioni di TNP-ATP (Figura 4C). Questo picco negativo è un artefatto del nostro protocollo di sottrazione dello sfondo. La Figura 4D mostra immagini fluorescenti e in campo chiaro di una pipetta patch esposta a 1 mM TNP-ATP. È evidente un'ombra sulla punta della pipetta, risultante dall'esclusione di TNP-ATP dal volume delle pareti della pipetta, che è più evidente all'interno del piano di messa a fuoco. L'immagine spettrale nella Figura 4E mostra una banda scura, corrispondente a questa ombra. Quando una regione sopra o sotto questa banda scura viene utilizzata per la sottrazione dello sfondo, produce un picco negativo. È importante sottolineare che questo picco si è verificato su un intervallo di lunghezze d'onda corrispondente all'emissione di TNP-ATP e non ha influenzato le nostre misurazioni della tempra ANAP.
La principale limitazione dei nostri esperimenti è stata quella di ottenere un'adeguata espressione della membrana plasmatica di costrutti marcati con ANAP per misurare la fluorescenza. In genere era più facile acquisire spettri di alta qualità da membrane non coperte che in PCF, grazie alle loro dimensioni maggiori e alla nostra capacità di scansionare rapidamente un intero piatto di membrane non coperte, a differenza del PCF dove i cerotti possono essere ottenuti solo uno alla volta. Nei nostri esperimenti, i dati delle membrane senza tetto e degli esperimenti PCF erano simili ma non equivalenti (Figura 6) 11. Tuttavia, non vi è alcuna ragione a priori per cui questa dovrebbe essere un'osservazione universale poiché le proteine in una pipetta patch possono essere in uno stato funzionale diverso da quelle nelle membrane senza tetto.
Qui, sono stati fatti tentativi per massimizzare l'espressione dei nostri costrutti marcati ANAP, in particolare abbassando la temperatura della coltura cellulare a 33 ° C10,11,16. Nella nostra esperienza, il tentativo di identificare i siti nella proteina in cui ANAP sarebbe una sostituzione conservativa non ha portato costantemente a costrutti che si esprimevano bene. Abbiamo avuto più successo scansionando sistematicamente intere regioni proteiche per i siti di incorporazione ANAP e selezionando i candidati per l'espressione superficiale10. Il sistema di marcatura ANAP funziona anche negli ovociti di Xenopus laevis, che consente di asportare patch di membrana molto più grandi, aumentando così il segnale al rumore26,27,28.
Mentre ci si aspetta che livelli di espressione più elevati si traducano in segnali più luminosi, il numero minimo di canali richiesto per misurare la fluorescenza dipende da diversi fattori, tra cui la luminosità del fluoroforo, il grado di fotosbiancamento, l'intensità della luce di eccitazione e il piano di messa a fuoco. In teoria, le stime potrebbero essere fatte correlando l'intensità della fluorescenza e la corrente del canale come è stato mostrato in precedenza28,29. Tuttavia, l'affidabilità di tali stime richiede una certa conoscenza della conduttanza a canale singolo e della probabilità aperta del canale. Oltre ai fattori sopra elencati, il segnale di fluorescenza sarà influenzato anche da canali associati a vescicole o sezioni della membrana plasmatica attaccate al vetro della pipetta che non sono sotto morsetto di tensione.
Questo metodo è facilmente adattabile allo studio di altri canali ionici sensibili ai nucleotidi. CFTR è strutturalmente simile alla subunità accessoria del recettore della sulfonilurea di KATP30,31. Come il gating KATP CFTR è controllato dal legame nucleotidico, rendendolo un ovvio obiettivo futuro del nostro metodo7. I recettori purinergici P2X sono canali ionici controllati dall'ATP9 extracellulare. TNP-ATP agisce come antagonista per i recettori P2X32,33. Pertanto, non sarà utile per studiare l'attivazione P2X, sebbene possa essere utilizzato in saggi di competizione con agonisti P2X. In alternativa, altri derivati fluorescenti dell'ATP con sufficiente sovrapposizione spettrale con emissione ANAP possono essere utilizzati per studiare l'attivazione. Alexa-647-ATP è un agonista fluorescente P2X34. L'R0 calcolato tra Alexa-647 e ANAP è ~ 85 Å, il che significa che il legame diretto a P2X dovrebbe comportare una sostanziale tempra di ANAP incorporato nel canale. Tuttavia, un R0 così lungo comporterà anche la tempra da Alexa-647-ATP legato alle subunità vicine e aumenta la probabilità che il legame nucleotidico non specifico si traduca in FRET. Poiché il sito di legame del ligando nei recettori P2X è extracellulare, le misurazioni di legame verrebbero eseguite su cellule intatte, in morsetti di tensione dell'intera cellula o in patch di membrana esterne. Il nostro metodo può anche essere esteso allo studio del legame e dell'attivazione di trasportatori e pompe elettrogenici e non elettrogeni che dipendono dall'ATP per il loro ciclo di reazione e dei recettori P2Y accoppiati a proteine G. Infine, anche se abbiamo sviluppato questo metodo per misurare il legame nucleotidico dell'adenina (TNP-ATP, TNP-ADP, TNP-AMP), lo stesso approccio può essere utilizzato per studiare il legame praticamente a qualsiasi recettore per il quale è stato identificato un ligando fluorescente adatto.
Divulgazioni
Gli autori non dichiarano conflitti di interesse.
Riconoscimenti
Si ringrazia Raul Terron Exposito per l'eccellente assistenza tecnica. Questo lavoro è stato finanziato dal Biotechnology and Biological Sciences Research Council (BB/R002517/1; MCP e FMA) e il Wellcome Trust (203731/Z/16/A; SGU)
Materiali
| Name | Company | Catalog Number | Comments |
| T75 tissue-culture treated flask | StarLab | CC7682-4875 | |
| 0.1% w/v poly-L-lysine | Sigma-Aldrich | P8920 | |
| 30 mm borosilicate cover glass slips | VWR | 631-0174 | |
| 35 mm non-treated sterile dishes | CytoOne | CC7672-3340 | |
| 35 mm cover glass bottom dish | WPI | FD35-PDL-100 | |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | 31966021 | |
| Foetal bovine serum (FBS) | Gibco | 10500-064 | |
| Penicillin/Streptomycin | Gibco | 15140-122 | |
| TrypLE select (tryosin) | Gibco | 12563-011 | Trypsin/EDTA reagent |
| Phosphate buffered saline (PBS) | Gibco | 14040-091 | |
| UltraPure distilled water | Invitrogen | 10977-035 | |
| HEK293T cells | ATTC | CRL-3216 | Used between passages 5-30 |
| ANAP-TFA | AsisChem | ASIS-0014 | Reconstituted in 30 mM NaOH to a final concentration of 1 mM |
| pANAP expression plasmid | Addgene | Plasmid #48696 | Encodes tRNA/tRNA synthetase pair for expression of ANAP-tagged protein |
| peRF1-E55D | Chin Lab (MRC Laboratory of Molecular Biology, Cambridge, UK) | Jason Chin: DOI: 10.1021/ja5069728 | Encodes dominant-negative eukaryotic ribosomal release factor |
| TransIT-LT1 | Mirus Bio | MIR 2300 | Lipopolyplex transfection reagent |
| Thick-walled borosilicate glass capillaries | Harvard Apparatus | GC150F-15 | |
| Tetramethylrhodamine-5-maleimide | Sigma-Aldrich | 94506 | |
| TNP-ATP | Jena Bioscience | NU-221L | Delivered at 10 mM in water |
| Nikon Eclipse TE2000-U inverted microscope microscope | Nikon | ||
| 60x water immersion objective (1.4 NA) | Nikon | MRD07602 | |
| 4-Wavelength High-Power LED Head | ThorLabs | LED4D245 | 385/490/565/625 nm LEDs |
| Four-Channel LED Driver | ThorLabs | DC4100 | |
| 390/18 nm band-pass excitation filter | ThorLabs | MF390-18 | For ANAP excitation |
| 400 nm long-pass emission filter | ThorLabs | FEL0400 | For imaging ANAP spectra |
| 416 nm edge dichroic | ThorLabs | MD416 | For imaging ANAP spectra |
| 460/60 nm band-pass emission filter | ThorLabs | MF460-60 | Suggested wide band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 470/10 nm band-pass emission filter | ThorLabs | FB470-10 | Suggested narrow band-pass filter for imaging ANAP fluorescence (Figure 4B) |
| 531/40 band-pass excitation filter | Brightline | FF01-531/40-25 | For orange fluorescent protein (OFP) excitation |
| 540/25 nm band-pass excitation filter | Chroma | D540/25X | For tetramethylrhodamine-5-maleimide (TMRM) excitation |
| 562 nm edge dichroic | Semrock | FF562-Di03 | For imaging OFP fluorescence |
| 565 nm edge dichroic | Chroma | 565DC | For imaging TMRM fluorescence |
| 593/40 nm band-pass excitation filter | Brightline | FF01-387/11-25 | For imaging OFP fluorescence |
| 605/55 nm band-pass emission filter | Chroma | D605/55M | For imaging TMRM fluorescence |
| IsoPlane-160 Imaging Spectrometer | Princeton Instruments | IsoPlane-160 | |
| PIXIS 400BR_eXcelon Camera | Princeton Instruments | PIXIS: 400BR_eXcelon | |
| Axopatch 200B amplifier | Molecular Devices | Axopatch 200B-2 | |
| Digidata 1440A digitizer | Molecular Devices | Digidata 1440A | |
| Probe sonicator | Sonics & Materials | VC-50 | For unroofing |
| REGLO digital peristaltic pump | Ismatec | ISM 832 | For bath perfusion |
| Microvolume perfusion system | ALA Scientific Instruments | ALA μFlow-8 | For TNP-ATP perfusion |
| pClamp 10.6.2 | Molecular Devices | Recording and analysing currents | |
| Lightfield 5.20.1507 | Princeton Instruments | Acquisition software for images and spectra | |
| Matlab | Mathworks | For data analysis | |
| Python 3.8.1 | Python Software Foundation | For data analysis |
Riferimenti
- Garcia, M. L., Kaczorowski, G. J. Ion channels find a pathway for therapeutic success. Proceedings of the National Academy of Sciences of the United States of America. 113 (20), 5472-5474 (2016).
- Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B., Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. 16 (12), 829-842 (2017).
- Lakowicz, J. R. Principles of fluorescence spectroscopy. 3rd edn. , Springer. (2006).
- Higgins, C. F., Linton, K. J. The ATP switch model for ABC transporters. Nature Structural & Molecular Biology. 11 (10), 918-926 (2004).
- Toyoshima, C., Cornelius, F. New crystal structures of PII-type ATPases: excitement continues. Current Opinion in Structural Biology. 23 (4), 507-514 (2013).
- Craven, K. B., Zagotta, W. N. CNG and HCN channels: two peas, one pod. Annual Review of Physiology. 68, 375-401 (2006).
- Csanady, L., Vergani, P., Gadsby, D. C. Strict coupling between CFTR's catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proceedings of the National Academy of Sciences of the United States of America. 107 (3), 1241-1246 (2010).
- Vedovato, N., Ashcroft, F. M., Puljung, M. C. The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophysical Journal. 109 (12), 2452-2460 (2015).
- Burnstock, G. Introduction to the Special Issue on Purinergic Receptors. Advances in Experimental Medicine and Biology. 1051, 1-6 (2017).
- Puljung, M., Vedovato, N., Usher, S., Ashcroft, F. Activation mechanism of ATP-sensitive K(+) channels explored with real-time nucleotide binding. Elife. 8, 41103(2019).
- Usher, S. G., Ashcroft, F. M., Puljung, M. C. Nucleotide inhibition of the pancreatic ATP-sensitive K+ channel explored with patch-clamp fluorometry. Elife. 9, 52775(2020).
- Chatterjee, A., Guo, J., Lee, H. S., Schultz, P. G. A genetically encoded fluorescent probe in mammalian cells. Journal of the American Chemical Society. 135 (34), 12540-12543 (2013).
- Gordon, S. E., Senning, E. N., Aman, T. K., Zagotta, W. N. Transition metal ion FRET to measure short-range distances at the intracellular surface of the plasma membrane. Journal of General Physiology. 147 (2), 189-200 (2016).
- Heuser, J. The production of 'cell cortices' for light and electron microscopy. Traffic. 1 (7), 545-552 (2000).
- Schmied, W. H., Elsasser, S. J., Uttamapinant, C., Chin, J. W. Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. Journal of the American Chemical Society. 136 (44), 15577-15583 (2014).
- Lin, C. Y., et al. Enhancing Protein Expression in HEK-293 Cells by Lowering Culture Temperature. PloS One. 10 (4), 0123562(2015).
- Usukura, J., et al. Use of the unroofing technique for atomic force microscopic imaging of the intra-cellular cytoskeleton under aqueous conditions. Journal of Electron Microscopy. 61 (5), 321-326 (2012).
- Colquhoun, D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. British Journal of Pharmacology. 125 (5), 924-947 (1998).
- Mannuzzu, L. M., Moronne, M. M., Isacoff, E. Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271 (5246), 213-216 (1996).
- Zheng, J., Zagotta, W. N. Gating rearrangements in cyclic nucleotide-gated channels revealed by patch-clamp fluorometry. Neuron. 28 (2), 369-374 (2000).
- Zheng, J., Zagotta, W. N. Patch-clamp fluorometry recording of conformational rearrangements of ion channels. Science's STKE. 2003 (176), 7(2003).
- Biskup, C., et al. Relating ligand binding to activation gating in CNGA2 channels. Nature. 446 (7134), 440-443 (2007).
- Kusch, J., et al. Interdependence of receptor activation and ligand binding in HCN2 pacemaker channels. Neuron. 67 (1), 75-85 (2010).
- Wu, S., et al. State-dependent cAMP binding to functioning HCN channels studied by patch-clamp fluorometry. Biophysical Journal. 100 (5), 1226-1232 (2011).
- Cha, A., Bezanilla, F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 19 (5), 1127-1140 (1997).
- Kalstrup, T., Blunck, R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proceedings of the National Academy of Sciences of the United States of America. 110 (20), 8272-8277 (2013).
- Kalstrup, T., Blunck, R. S4-S5 linker movement during activation and inactivation in voltage-gated K(+) channels. Proceedings of the National Academy of Sciences of the United States of America. 115 (29), 6751-6759 (2018).
- Dai, G., Aman, T. K., DiMaio, F., Zagotta, W. N. The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nature Structural & Molecular Biology. 26 (8), 686-694 (2019).
- Liu, C., et al. Patch-clamp fluorometry-based channel counting to determine HCN channel conductance. Journal of General Physiology. 148 (1), 65-76 (2016).
- Hwang, T. C., et al. Structural mechanisms of CFTR function and dysfunction. Journal of General Physiology. 150 (4), 539-570 (2018).
- Puljung, M. C. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. Journal of General Physiology. 150 (5), 653-669 (2018).
- Kasuya, G., et al. Structural insights into the competitive inhibition of the ATP-gated P2X receptor channel. Nature Communications. 8 (1), 876(2017).
- Virginio, C., Robertson, G., Surprenant, A., North, R. A. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Molecular Pharmacology. 53 (6), 969-973 (1998).
- Bhargava, Y., Nicke, A., Rettinger, J. Validation of Alexa-647-ATP as a powerful tool to study P2X receptor ligand binding and desensitization. Biochemical and Biophysical Research Communications. 438 (2), 295-300 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati