
Method Article
Modelando um site ativo enzimático usando freeware de visualização molecular
Neste Artigo
Resumo
Uma habilidade fundamental na modelagem biomolecular é exibir e anotar locais ativos em proteínas. Esta técnica é demonstrada usando quatro programas gratuitos populares para visualização macromolecular: iCn3D, Jmol, PyMOL e UCSF QuimeraX.
Resumo
As habilidades de visualização biomolecular são primordiais para a compreensão de conceitos-chave nas ciências biológicas, como relações estrutura-função e interações moleculares. Vários programas permitem que um aluno manipule estruturas 3D, e a modelagem biomolecular promove a aprendizagem ativa, constrói habilidades computacionais e faz a ponte entre as imagens bidimensionais dos livros didáticos e as três dimensões da vida. Uma habilidade crítica nessa área é modelar um local ativo de proteína, exibindo partes da macromolécula que podem interagir com uma pequena molécula, ou ligante, de uma forma que mostre interações vinculantes. Neste protocolo, descrevemos este processo usando quatro programas de modelagem macromolecular disponíveis livremente: iCn3D, Jmol/JSmol, PyMOL e UCSF QuimeraX. Este guia destina-se a estudantes que buscam aprender o básico de um programa específico, bem como instrutores incorporando modelagem biomolecular em seu currículo. O protocolo permite que o usuário modele um site ativo usando um programa de visualização específico ou prove vários dos programas gratuitos disponíveis. O modelo escolhido para este protocolo é a glucokinase humana, uma isoforme da enzima hexokinase, que catalisa o primeiro passo da glicólise. A enzima está ligada a um de seus substratos, bem como a um analógico substrato não reativo, que permite ao usuário analisar interações no complexo catalítico.
Introdução
Compreender representações do mundo molecular é fundamental para se tornar um especialista em ciências biomoleculares1, pois a interpretação de tais imagens é fundamental para entender a função biológica2. A introdução de um aluno às macromoléculas geralmente vem na forma de imagens bidimensionais de membranas celulares, organelas, macromoléculas, etc. mas a realidade biológica é que estas são estruturas tridimensionais, e uma compreensão de suas propriedades requer maneiras de visualizar e extrair significado a partir de modelos 3D.
Assim, o desenvolvimento da alfabetização visual biomolecular nos cursos de ciências da vida molecular da divisão superior tem ganhado atenção, com uma série de artigos relatando a importância e as dificuldades de ensinar e avaliar habilidades de visualização1,3,4,5,6,7,8,9 . A resposta a esses artigos tem sido um aumento no número de intervenções em sala de aula, tipicamente dentro de um semestre em uma única instituição, em que programas e modelos de visualização molecular são usados para atingir conceitos difíceis2,10,11,12,13,14,15 . Além disso, os pesquisadores têm procurado caracterizar como os alunos utilizam programas de visualização biomolecular e/ou modelos para abordar um tópico específico16,17,18,19. Nosso próprio grupo, BioMolViz, descreveu um Quadro que subdivide temas abrangentes na alfabetização visual em objetivos de aprendizagem e objetivos para orientar tais intervenções20,21, e lideramos oficinas que treinam professores para usar o Framework no projeto retrógrado de avaliações para medir habilidades de alfabetização visual22.
No centro de todo esse trabalho está uma habilidade crítica: a capacidade de manipular estruturas de macromoléculas usando programas para visualização biomolecular. Essas ferramentas foram desenvolvidas de forma independente usando uma variedade de plataformas; portanto, eles podem ser bastante únicos em sua operação e uso. Isso requer instruções específicas do programa, e a identificação de um programa com o que um usuário se sente é importante para facilitar a implementação contínua.
Além do básico de manipular estruturas em 3D (girar, selecionar e alterar o modelo), um grande objetivo é modelar o local ativo de uma proteína. Esse processo permite que um aluno desenvolva sua compreensão em três temas abrangentes descritos pelo Quadro BioMolViz: interações moleculares, ligantes/modificações e relações estrutura-função20,21.
Quatro opções populares de programas para visualização biomolecular incluem: Jmol/JSmol23, iCn3D24, PyMOL25e UCSF Quimera26,27. Encorajamos os novos da Quimera a usar a UCSF QuimeraX, a próxima geração do programa de visualização molecular Quimera, que é a versão atualmente suportada do programa.
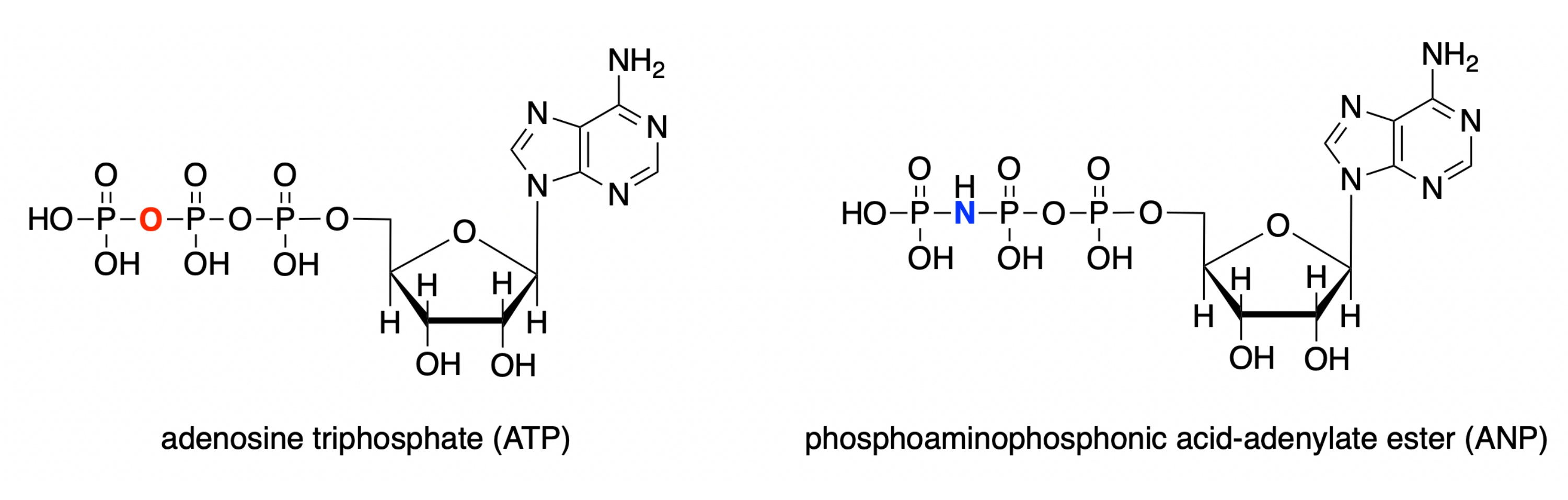
Neste protocolo, demonstramos como usar cada um desses quatro programas para modelar o local ativo da glucokinase humana com um complexo analógico substrato vinculado (PDB ID: 3FGU) e para exibir medidas para ilustrar interações vinculantes específicas28. O modelo representa um complexo catalítico da enzima. Para capturar o local ativo no estado pré-catálise, um análogo não hidrolisável de ATP estava vinculado ao local ativo da glucokinase. Este éster ácido fosfoaminofosfônico-adeníto (ANP) contém uma ligação fosforosa-nitrogênio em vez da ligação fósforo-oxigênio usual nesta posição. O local ativo também contém glicose (denotado BCG no modelo) e magnésio (mg denotado). Além disso, há um íon de potássio (K) na estrutura, resultante do cloreto de potássio usado no solvente de cristalização. Este íon não é crítico para a função biológica e está localizado fora do local ativo.

Figura 1: Estruturas ATP/ANP. Estrutura de adenosina triphosfato (ATP) em comparação com o éster ácido fosfoaminofosfônico-adeníto (ANP). Clique aqui para ver uma versão maior desta figura.
{kind=link}
O protocolo demonstra a seleção dos ligantes vinculados do complexo analógico substrato e a identificação de resíduos ativos dentro de 5 Å do complexo vinculado, que captura aminoácidos e moléculas de água capazes de fazer interações moleculares relevantes, incluindo interações hidrofóbicas e van der Waals.
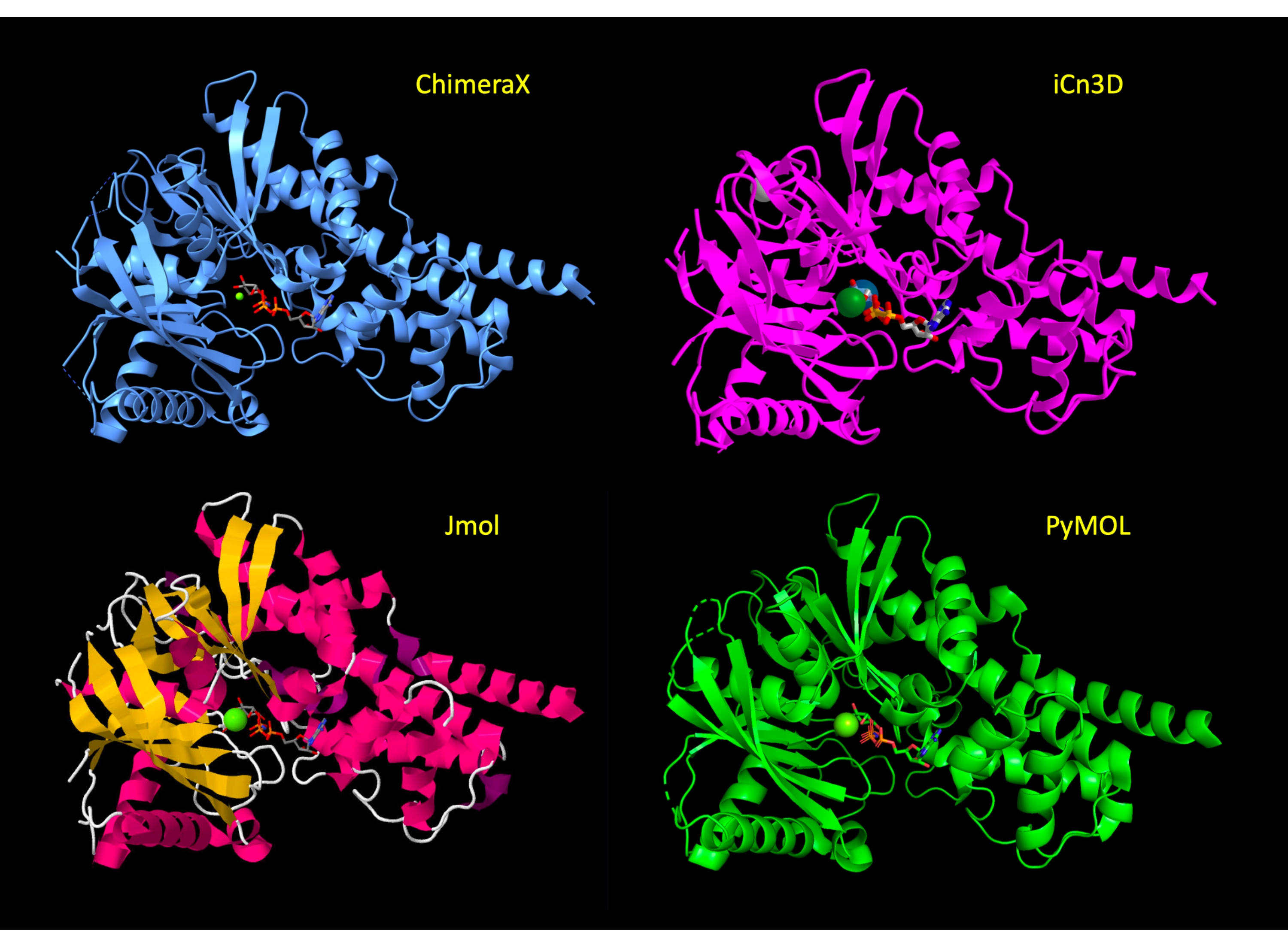
O display é inicialmente manipulado para mostrar a maior parte da proteína em uma representação de desenho animado, com o local ativo resíduos de aminoácidos em representação de vara para mostrar os átomos relevantes da proteína e destacar as interações moleculares. Após a etapa 3 do protocolo para cada programa, essas representações foram aplicadas e a visão da proteína é semelhante entre os programas(Figura 2). No final do protocolo, o desenho animado de proteínas é escondido para simplificar a visão e focar no site ativo.

Figura 2: Comparação de estrutura entre programas. Comparação da estrutura do 3FGU em cada programa seguindo a etapa ajustar a representação (etapa 2 ou 3 de cada protocolo). Clique aqui para ver uma versão maior desta figura.
{kind=link}
A coloração CPK é aplicada aos aminoácidos do local ativo e ligantes vinculados29,30. Este esquema de coloração distingue átomos de diferentes elementos químicos em modelos moleculares mostrados em linha, pau, bola e vara, e representações de preenchimento espacial. Hidrogênio é branco, nitrogênio é azul, oxigênio é vermelho, enxofre é amarelo, e fósforo é laranja no esquema de coloração CPK. Tradicionalmente, o preto é usado para carbono, embora no uso moderno, a coloração de carbono pode variar.
Átomos de hidrogênio não são visíveis em estruturas cristalinas, embora cada um desses programas seja capaz de prever sua localização. Adicionar os átomos de hidrogênio a uma grande estrutura macromolecular pode obscurecer a visão, assim eles não são exibidos neste protocolo. Assim, as ligações de hidrogênio serão mostradas medindo a partir do centro de dois heteroatomas (por exemplo, oxigênio ao oxigênio, oxigênio a nitrogênio) nessas estruturas.
Visões gerais do programa
Interfaces gráficas de usuário (GUIs): PyMOL (Versão 2.4.1), QuimeraX (Versão 1.2.5) e Jmol (Versão 1.8.0_301) são ferramentas de modelagem molecular baseadas em GUI. Essas três interfaces possuem linhas de comando para inserir código digitado; muitas das mesmas capacidades estão disponíveis através de menus e botões na GUI. Uma característica comum na linha de comando desses programas é que o usuário pode carregar e re-executar comandos anteriores usando as teclas de seta para cima e para baixo no teclado.
GUIs baseado na Web: iCn3D (I-see-in-3D) é um visualizador baseado em WebGL para visualização interativa de estruturas macromoleculares tridimensionais e produtos químicos na Web, sem a necessidade de instalar um aplicativo separado. Ele não usa uma linha de comando, embora a versão web completa tenha um registro de comando editável. JSmol é uma versão JavaScript ou HTML5 do Jmol para uso em um site ou em uma janela de navegador da Web, e é muito semelhante em operação ao Jmol. O JSmol pode ser usado para criar tutoriais online, incluindo animações.
Proteopedia31,32, FirstGlance em Jmol33, e a interface web JSmol (JUDE) no Milwaukee School of Engineering Center for BioMolecular Modeling são exemplos desses ambientes de design on-line baseados em Jmol34. O Wiki Proteopedia é uma ferramenta de ensino que permite ao usuário modelar uma estrutura de macromolécula e criar páginas com esses modelos dentro do site35. A ferramenta de autoria de cena Proteopedia, construída com o JSmol, integra uma GUI com recursos adicionais não disponíveis na GUI Jmol.
Jmol e iCn3D são baseados na linguagem de programação Java; JSmol usa Java ou HTML5, e PyMOL e ChimeraX são baseados na linguagem de programação Python. Cada um desses programas carrega arquivos de banco de dados de proteína, que podem ser baixados do RCSB Protein Data Bank sob um PDB ID alfanumérico de 4 dígitos36,37. Os tipos de arquivos mais comuns são os arquivos PDB (Protein Data Bank, banco de dados de proteína) contendo a extensão .pdb e o Arquivo de Informações Cristalográficas (CIF ou mmCIF) contendo a extensão .cif. O CIF substituiu o PDB como o tipo de arquivo padrão do Protein Data Bank, mas ambos os formatos de arquivo funcionam nesses programas. Pode haver pequenas diferenças na forma como a sequência/estrutura é exibida ao usar cif em oposição aos arquivos PDB; no entanto, os arquivos funcionam de forma semelhante e as diferenças não serão abordadas em detalhes aqui. O Banco de Dados de Modelagem Molecular (MMDB), produto do Centro Nacional de Informações de Biotecnologia (NCBI), é um subconjunto de estruturas PDB às quais as informações categóricas têm sido associadas (por exemplo, características biológicas, domínios de proteína conservada)38. O iCn3D, um produto do NCBI, é capaz de carregar arquivos PDB contendo os dados do MMDB.
Para visualizar um modelo, o usuário pode baixar o arquivo desejado na página dedicada do Protein Data Bank para a estrutura (por exemplo, https://www.rcsb.org/structure/3FGU), e, em seguida, usar o menu de arquivo suspensa do programa para abrir a estrutura. Todos os programas também são capazes de carregar um arquivo de estrutura diretamente através da interface, e esse método é detalhado dentro dos protocolos.
Os GUIs QuimeraX, Jmol e PyMOL contêm cada uma ou mais janelas do console que podem ser redimensionadas arrastando o canto. ICn3D e JSmol estão inteiramente contidos em um navegador da Web. Ao usar o iCn3D, o usuário pode precisar rolar dentro das janelas pop-up para revelar todos os itens do menu, dependendo do tamanho e resolução da tela.
Os protocolos aqui detalhados fornecem um método simples para exibir o site ativo da enzima usando cada programa. Deve-se notar que existem várias maneiras de executar as etapas em cada programa. Por exemplo, em QuimeraX, a mesma tarefa pode ser executada usando menus suspensos, a barra de ferramentas na parte superior ou a linha de comando. Os usuários interessados em aprender um programa específico em detalhes são encorajados a explorar os tutoriais online, manuais e Wikis disponíveis para esses programas39,40,41,42,43,44,45,46.
Os manuais e tutoriais existentes para esses programas apresentam os itens deste protocolo como tarefas discretas. Para exibir um site ativo, o usuário deve sintetizar as operações necessárias a partir dos vários manuais e tutoriais. Este manuscrito aumenta os tutoriais existentes disponíveis, apresentando um protocolo linear para modelar um site ativo rotulado com interações moleculares, fornecendo ao usuário uma lógica para modelagem ativa do site que pode ser aplicada a outros modelos e programas.

Figura 3: QuimeraX GUI. Interface gui quimeraX com os menus suspensos, barra de ferramentas, visualizador de estrutura e linha de comando rotulados. Clique aqui para ver uma versão maior desta figura.
{kind=link}

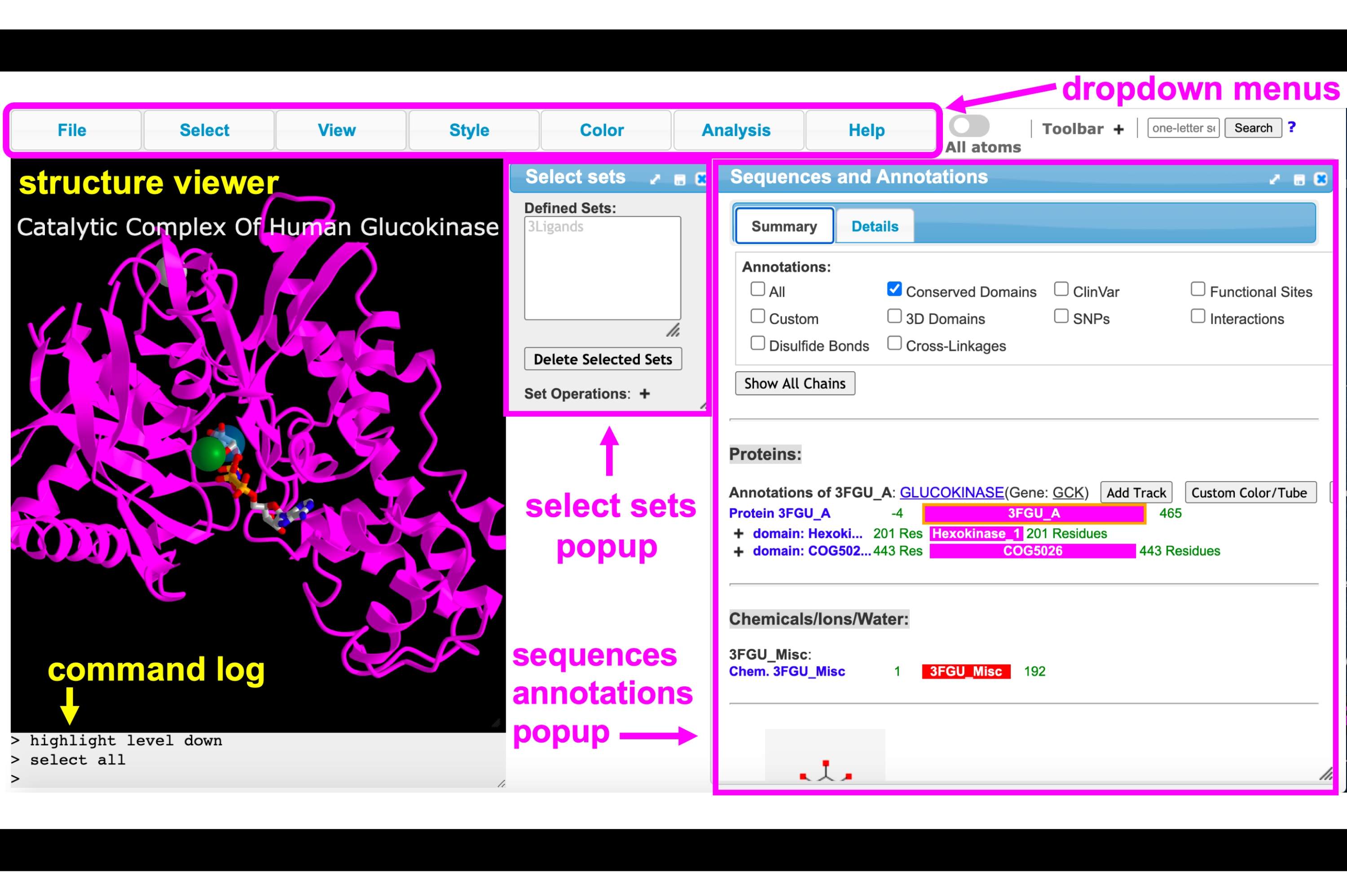
Figura 4: interface de GUI iCn3D com os menus suspensos, barra de ferramentas, visualizador de estrutura, log de comando, configurações selecionadas pop-up e menus pop-up e sequências e anotações rotulados. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Jmol GUI. Interface Jmol GUI com os menus suspensos, barra de ferramentas, visualizador de estrutura, menu pop-up e linha de console/comando rotulada. Clique aqui para ver uma versão maior desta figura.
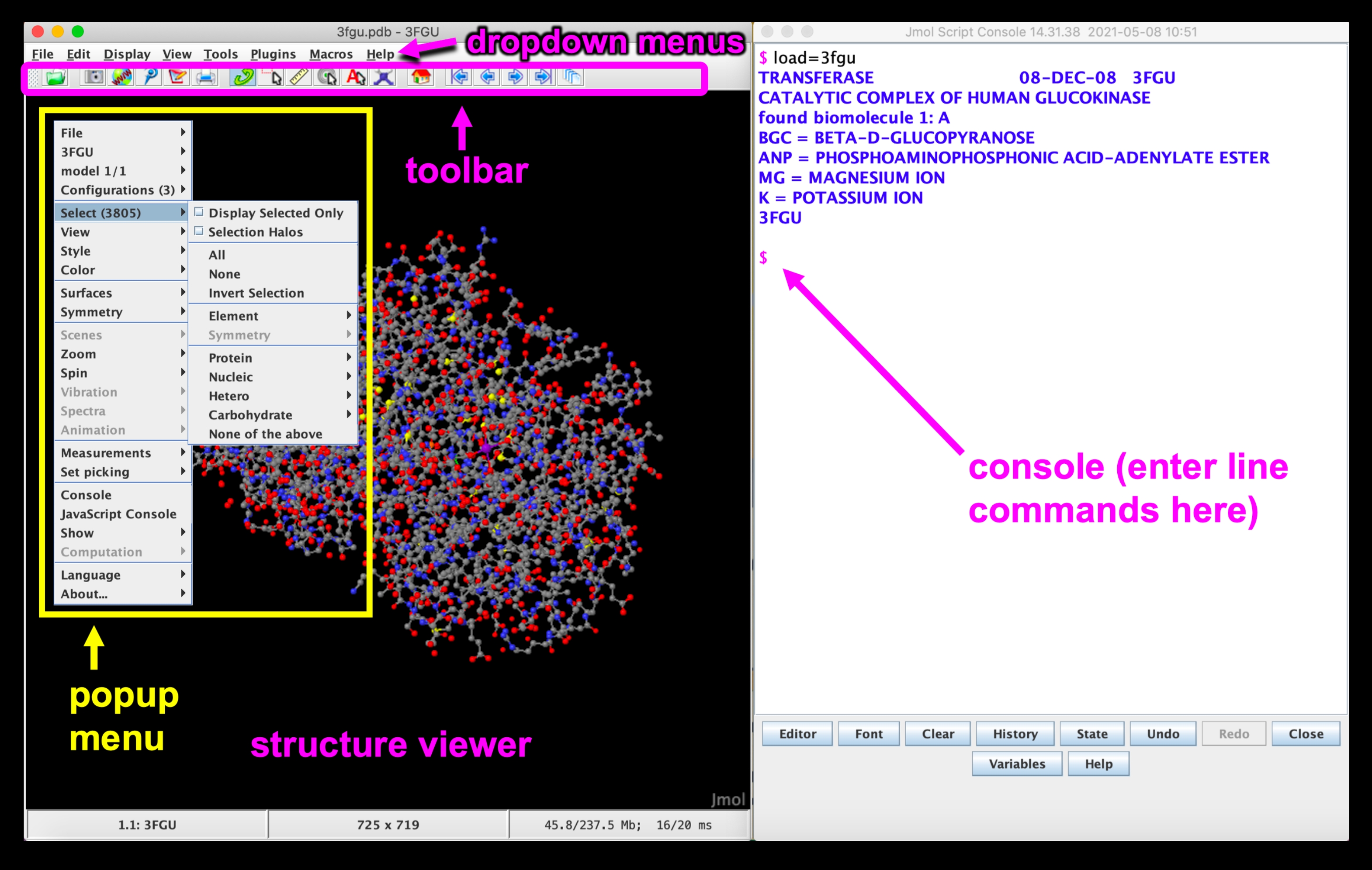{kind=link}

Figura 6: PymOL GUI. Interface PyMOL GUI com os menus suspensos, visualizador de estrutura, nomes/painel de objetos, menu de controles do mouse e linha de comando rotulado. Clique aqui para ver uma versão maior desta figura.
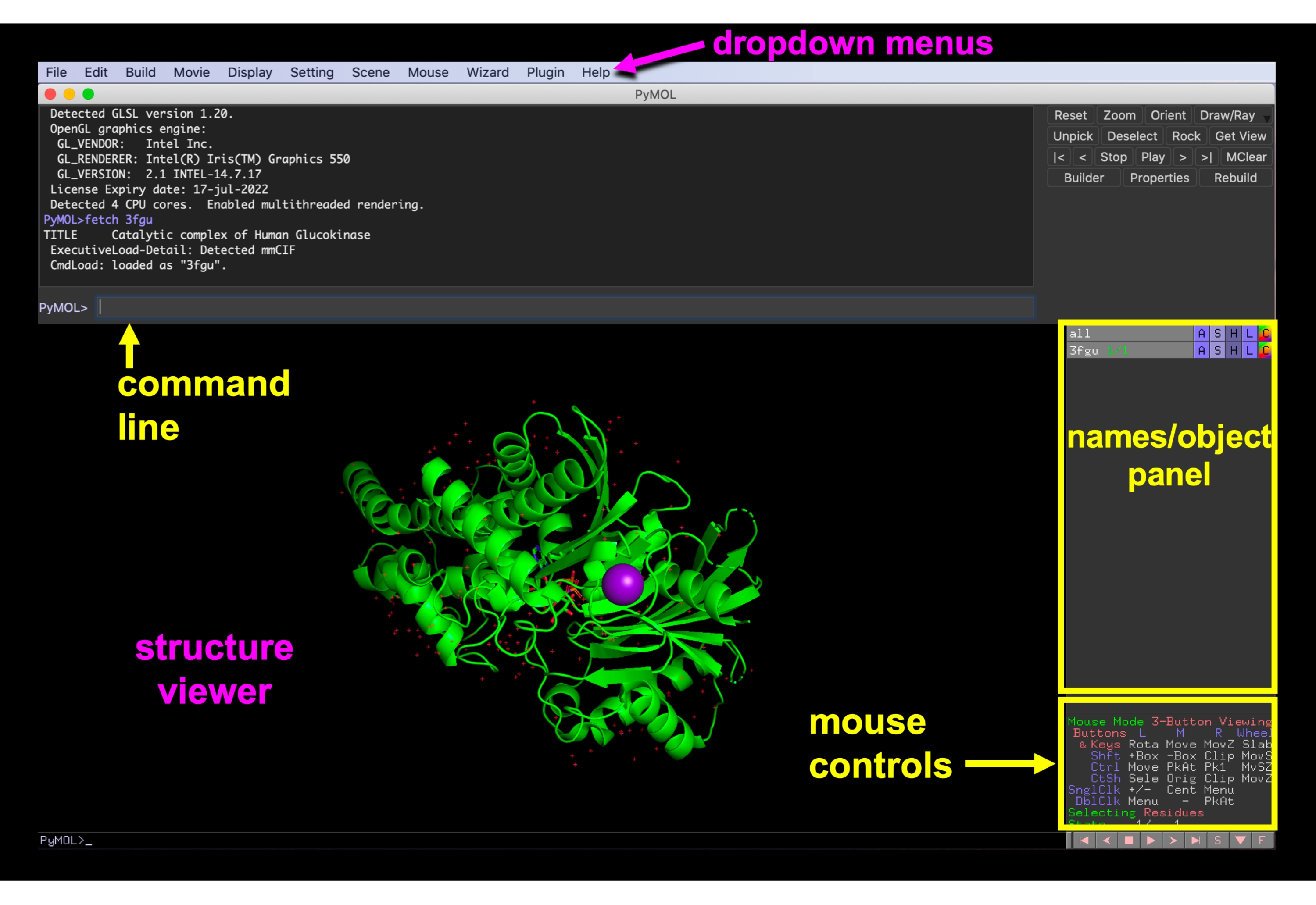{kind=link}
Protocolo
NOTA: O protocolo para cada programa é delineado em dez etapas abrangentes, (1) Carregar a estrutura no programa, (2) Identificar os ligantes no local ativo, (3) Ajustar a representação, (4) Selecionar resíduos dentro de 5 Å para definir um local ativo, (5) Mostrar as interações da enzima com os ligantes ativos do local, (6) Exibir as correntes laterais como varas e mostrar/ajustar as moléculas de água do local ativo, (7) Simplificando a estrutura, (8) Rotulagem de ligantes e cadeias laterais ligadas a hidrogênio, (9) Salvar a renderização a qualquer momento para voltar a trabalhar nela ou compartilhar com outros, (10) Salvar uma imagem para incorporar ou imprimir. As etapas 1, 4 e 7-10 são idênticas para cada protocolo; no entanto, devido ao funcionamento único de cada programa, alguns protocolos são executados de forma mais eficiente quando as etapas 2/3 e 5/6 são intercambiadas.
1. Protocolo QuimeraX da UCSF
NOTA: Controles de trackpad e mouse. Para girar, clique e arraste ou use arrastar dois dedos (mouse: clique esquerdo e arraste). Para ampliar, beliscar e espalhar (Mac) ou controlar + movimento de dois dedos (PC) (mouse: roda de rolagem). Para traduzir (ou seja, mover toda a estrutura) pressione a opção + clique e arraste (Mac) ou mude + clique e arraste (PC) (mouse: clique com o botão direito do mouse e arraste). Para recentar, use os menus suspensos na parte superior da interface para clicar em Ações > Exibir.
- Carregando a estrutura em QuimeraX: Na linha de comando localizada na parte inferior da GUI que é precedida por "Comando:", digite:
aberto 3fgu
NOTA: Após inserir qualquer comando de linha digitado, pressione o retorno no teclado para executá-lo. - Identificação dos ligantes no site ativo: Certifique-se de que há duas representações, uma fita de desenho animado e paus. Usando o mouse, gire/amplie a proteína para melhor visualizar os ligantes exibidos perto do centro da proteína, que são mostrados como palitos. Paire sobre um ligante para mostrar seu nome.
- Ajuste da representação: Use os comandos nos subpassos abaixo para recolorir a proteína e os ligantes, aplicar a coloração CPK em átomos não-carbono e, em seguida, desmarcar a seleção. Partes selecionadas da molécula ficam destacadas em verde.
- Use os menus suspensos na parte superior da interface para alterar a coloração: Clique em Ações > Color > Cornflower Blue. Em seguida, clique em Selecionar > Estrutura > Ligand. Para selecionar a cor, clique em Ações > Cor > Cinza. Para aplicar a coloração cpk, clique em Selecionar > Alle clique em Ações > Color > Por Heteroatom. Por fim, limpe a seleção clicando em Select > Clear.
NOTA: A seleção também pode ser limpa pressionando o controle e clicando no fundo preto do visualizador da estrutura ou na linha de comando digitando: ~selecionar. Por padrão, para a maioria das estruturas contendo cadeias 1-4, quimeraX mostrará automaticamente moléculas de água e resíduos de aminoácidos dentro de 3,6 Å de ligantes e íons. - Use o menu suspenso para ocultar os átomos exibidos atualmente clicando em Ações > Átomos/Títulos > Ocultar.
- Use o menu suspenso para mostrar os ligantes e mgion no site ativo clicando em Selecionar > Estrutura > Ligand. Em seguida, clique em Actions > Atoms/Bonds > Show. Em seguida, clique em Selecionar resíduos > > MGe, em seguida, em Ações > Átomos/Títulos > Show. Para limpar a seleção, clique em Selecionar > Limpar.
NOTA: Depois de fazer a seleção com o menu suspenso, a etapa 1.3.3 pode ser executada clicando nos botões Ocultar e Mostrar na barra de ferramentas Átomos.
- Use os menus suspensos na parte superior da interface para alterar a coloração: Clique em Ações > Color > Cornflower Blue. Em seguida, clique em Selecionar > Estrutura > Ligand. Para selecionar a cor, clique em Ações > Cor > Cinza. Para aplicar a coloração cpk, clique em Selecionar > Alle clique em Ações > Color > Por Heteroatom. Por fim, limpe a seleção clicando em Select > Clear.
- Selecionando resíduos dentro de 5 Å para definir um local ativo: No visualizador da estrutura, para selecionar os ligantes pressionam controle + shift e executar o mouse clique em qualquer átomo ou vínculo em cada um dos três ligantes, ou seja, BCG, ANPe Mg.
- Em seguida, pressione a tecla de seta para cima no teclado até que todos os átomos dos três ligantes sejam destacados com um brilho verde. Defina esta seleção para uso futuro clicando no menu suspenso Selecione > Define Seletor. No menu pop-up, digite:
ligantes para nomear a seleção atual e, em seguida, clique em OK.
NOTA: Clicar na seta para cima muitas vezes na etapa 1.4.1 selecionará toda a proteína. Nesse caso, clique no botão de seta para baixo até que apenas os átomos dos três ligantes sejam selecionados. - Use o menu suspenso para selecionar os resíduos dentro de 5 Å dos ligantes: Clique em Selecionar > Zona. Na janela pop-up que aparece, alterne o menu 'Selecionar' para Resíduose certifique-se de que a caixa superior seja verificada (verifique a distância menor que (<) e esteja definida como 5.0 Å). Em seguida, clique em OK. Serão destacados apenas resíduos com menos de 5 Å de distância.
NOTA: As etapas 1.4-1.4.2 podem ser simplificadas usando extensivamente a linha de comando, digitando:
ligantes congelados nome :BGC:MG:ANP
selecionar ligantes de zona 5 estender resíduos verdadeiros verdadeiros
- Em seguida, pressione a tecla de seta para cima no teclado até que todos os átomos dos três ligantes sejam destacados com um brilho verde. Defina esta seleção para uso futuro clicando no menu suspenso Selecione > Define Seletor. No menu pop-up, digite:
- Exibindo as cadeias laterais como varas e mostrando/ajustando as moléculas de água do local ativo: Use o menu suspenso para exibir a seleção e centralizar e ampliar a seleção clicando em Ações > Átomos/Títulos > Mostrar para mostrá-las. Para centralizar a seleção, clique em Ações > Ver. Em seguida, para limpar a seleção, clique em Selecionar > Limpar ou clique em qualquer lugar no espaço vazio .
- Mostrando as interações da enzima com os ligantes ativos do site: Use os menus suspensos e clique em Selecionar > seletores definidos pelo usuário > Ligands. Em seguida, clique em Ferramentas > Análise de Estrutura > de títulos H. Na janela pop-up, certifique-se de que o Limite por Seleção seja verificado, o menu suspenso esteja definido como Com pelo menos uma extremidade selecionada e selecione átomos e, em seguida, clique em OK. Para limpar a seleção, clique em Selecionar > Limpar.
NOTA: Verifique a caixa De etiqueta de distância para ver os comprimentos do título em Å; no entanto, isso torna a vista muito ocupada. Finalmente, você pode alterar a cor dos títulos H clicando na caixa Color e selecionando uma nova cor na janela pop-up. - Simplificando a estrutura: usando a barra de ferramentas de desenho animado para ocultar o desenho animado ou clicar no menu suspenso: Ações > Cartoon > Hide.
- Ligantes de rotulagem e cadeias laterais ligadas a hidrogênio: Use o mouse para selecionar resíduos que estejam ligados aos ligantes (conectados pelas linhas tracejadas), como na etapa 1.4. Em seguida, nos menus suspensos, clique em Ações > Rótulo > Resíduos > Name Combo. Em seguida, clique em Selecionar > seletores definidos pelo usuário > Ligands. Em seguida, clique em Actions > Label > Residues > Off. Por fim, limpe a seleção, clicando em Selecionar > Limpar.
- Salvar a renderização a qualquer momento para voltar a trabalhar nele ou compartilhar com outros: no menu suspenso clique em Arquivo > Salvar. Selecione um local, digite um nome de arquivo e clique em Salvar.
NOTA: Certifique-se de que o formato está definido para: Sessão QuimeraX *.cxs. - Salvando uma imagem para incorporar ou imprimir: Primeiro use o mouse para orientar a molécula conforme desejado. Altere a cor de fundo para branca digitando na linha de comando:
definir bgColor branco
Por fim, clique no ícone de snapshot na barra de ferramentas. A imagem será salva na área de trabalho.
NOTA: A cor de fundo também está disponível no menu suspenso; em um Mac, clique em UCSF QuimeraX > Preferences; em um PC, clique em Favorites > Configurações > Fundo.
2. protocolo iCn3D
NOTA: Controles de trackpad e mouse: Para girar, clicar e arrastar (mouse: clique esquerdo e arraste). Para ampliar, aperte e espalhe (mouse: gire a roda de rolagem). Para traduzir (ou seja, mover toda a estrutura) clique e arraste com dois dedos (mouse: clique direito e arraste). Para recentar, passe o mouse sobre o View nos menus suspensos superiores e, em seguida, clique em Seleção de Centro.
- Carregando a estrutura em iCn3D: Navegue para o visualizador de estrutura 3D baseado na Web iCn3D e digite 3FGU na caixa de identidade do ID De entrada MMDB ou PDB para carregar o arquivo.
- Identificando os ligantes no site ativo: Passe o mouse sobre análise no menu suspenso e clique em Seq. e Anotações. As sequências, neste caso Proteínas e Íons Químicos/Íons/Água, são mostradas em uma tabela empilhada. Role para baixo para ver os ligantes do site ativo ANP, BGC e Mg listados. No espectador da estrutura, paire sobre os ligantes no site ativo (mostrado como varas no centro do desenho animado de proteínas) para ver seus nomes.
- Ajustando a representação: Não são necessários ajustes iniciais para este protocolo.
- Selecionando resíduos dentro de 5 Å para definir um site ativo: Para selecionar os ligantes, use o menu 'Selecionar's', clique em Selecionar em 3D. Certifique-se de que o resíduo seja verificado.
- Para selecionar os ligantes, mantenha pressionado o botão ALT em um PC ou no botão Opção em um Mac e clique no primeiro ligante (por exemplo, BCG) usando o mouse ou trackpad. Em seguida, pressione o controle e clique em ligantes ANP e MG para adicioná-los à seleção.
NOTA: Os ligantes serão destacados em amarelo à medida que forem selecionados. - Salve esta seleção usando o menu suspenso: Clique em Selecionar > Salvar seleção e use o teclado para inserir um nome na janela pop-up (por exemplo, 3Ligands),e clique em Salvar. A janela pop-up Select Sets será agora exibida.
NOTA: Se a seleção estiver incorreta, clique em Selecionar > Seleção Clara. - Selecione os resíduos dentro de 5 Å dos ligantes: No menu suspenso, clique em Selecionar > por distância. No menu pop-up que aparece, altere o segundo item (Esfera com um raio), para 5 Å digitando no bloco. Clique na palavra boxed Displaye, em seguida, feche a janela clicando no sinal cruzado no canto superior direito.
NOTA: No menu pop-up que aparece na etapa 2.4.3, deixe o primeiro conjunto com a entrada "selecionada" e o segundo conjunto como "não selecionado". Observe que os átomos/estruturas dentro de 5 Å ficam destacados com um brilho amarelo quando o display é clicado. - Salve o site ativo de 5 Å usando o menu suspenso: Passe do mouse sobre Selecionar e clique em Salvar seleção,insira um nome na janela pop-up usando o teclado (por exemplo, 5Ang) e clique em Salvar.
- Em seguida, crie uma nova seleção que combine os dois conjuntos (5Ang e 3Ligands): No menu pop-up Select Sets, ctrl-click (PC) ou command-click (Mac) 5Ang e 3Ligands. Clique em Selecionar > Salvar seleção,use o teclado para digitar um nome (por exemplo, 5AFull)e clique em Salvar.
- Para selecionar os ligantes, mantenha pressionado o botão ALT em um PC ou no botão Opção em um Mac e clique no primeiro ligante (por exemplo, BCG) usando o mouse ou trackpad. Em seguida, pressione o controle e clique em ligantes ANP e MG para adicioná-los à seleção.
- Mostrando as interações da enzima com os ligantes ativos do site, como ligações de hidrogênio: Passe o mouse sobre análise no menu suspenso e clique em Interações. Um menu pop-up abrangente de todas as interações não covalentes aparecerá.
- Desmarque tudo, exceto as caixas de seleção "Hydrogen" e "Salt Bridge/Ionic". Clique em 3Ligands para selecionar o primeiro conjunto e 5Ang para o segundo conjunto. Clique no texto encaixotado que lê interações de exibição 3D. Feche a janela clicando no sinal cruzado no canto superior direito.
NOTA: O Contato/Interações presumivelmente representam interação de dipolo induzido por dipolo induzido por dipolo, o que muitas vezes torna o display ocupado. Se desejar, altere a distância para qualquer tipo de interação. - Para mostrar apenas as ligações de hidrogênio, clique em 5Afull na janela de configurações selecionadas. Em seguida, passe o mouse sobre análise no menu suspenso e, em seguida, clique em Chem. Binding > Show.
- Desmarque tudo, exceto as caixas de seleção "Hydrogen" e "Salt Bridge/Ionic". Clique em 3Ligands para selecionar o primeiro conjunto e 5Ang para o segundo conjunto. Clique no texto encaixotado que lê interações de exibição 3D. Feche a janela clicando no sinal cruzado no canto superior direito.
- Exibindo as cadeias laterais como varas e mostrando/ajustando as moléculas de água do local ativo: Use o menu pop-up de conjuntos selecionados e clique em 5AFull. Em seguida, nos menus suspensos, clique em Style > Side Chains > Stick. Para aplicar a coloração cpk clique em Color > Atom. Por fim, clique em Style > Water > Spheres (se preferir moléculas de água maiores).
- Simplificando a estrutura: No menu pop-up de conjuntos selecionados, clique em 5AFull. Em seguida, nos menus suspensos, clique em Exibir > Exibir seleção (para ver apenas o site de vinculação 5AFull). Em seguida, clique em Style > Proteins > Stick (para mostrar a cadeia proteica como vara em vez de fita).
- Para colorir os átomos de carbono dos ligantes uma cor contrastante, clique em Chemicals na janela pop-up Select Sets. Em seguida, no menu suspenso, clique na seleção Exibir > Ver. Clique em Selecionar > Selecionar em 3D (certifique-se de que "átomo" seja verificado). Usando os controles descritos na etapa 2.4.1, use o mouse e o teclado para selecionar todos os átomos de carbono no BGC e ANP. Em seguida, no menu suspenso clique em Color > Unicolor > Cyan > Cyan.
- Para reexibir todo o site ativo, use a janela pop-up Select Sets para clicar em 5AFull. Em seguida, no menu suspenso, clique em Exibir > Ver seleção.
- Ligantes de rotulagem e cadeias laterais ligadas a hidrogênio: Use a janela pop-up de conjuntos selecionados para selecionar Interface_alle, em seguida, no menu suspenso, clique em Análise > Label > Por Resíduo & Número.
NOTA: Você terá que resmarcar Por Resíduo & Número cada vez que desejar adicionar um rótulo, mesmo que o item do menu já seja verificado a partir de um rótulo anterior. - Salvar a renderização a qualquer momento para voltar a trabalhar nele ou compartilhar com outros: No menu suspenso, clique em Arquivo > Compartilhar Link. Copie a URL curta (por exemplo: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?r83NqCz41bu7cmcs8) e cole-a em um navegador.
- Salvando uma imagem para incorporar ou imprimir: No menu suspenso, clique em Selecionar > destaque de Alternar. Em seguida, clique em Style > Fundo > Branco. Finalmente, clique em Arquivo > Salvar arquivos > imagem PNG iCn3D e escolha o tamanho desejado.
3. Protocolo Jmol
NOTA: Controles de trackpad e mouse: Para girar, clique e arraste (mouse: clique esquerdo e arraste). Para ampliar: role verticalmente usando dois dedos (mouse: shift + left click + drag vertically). Para traduzir (ou seja, mover toda a estrutura) controle + alt + clique e arraste (PC), controle + opção + clique e arraste (Mac). Para recentrar: mude + clique duas vezes no espaço vazio da janela do visualizador da estrutura.
- Carregando a estrutura em Jmol: Use o menu suspenso na parte superior da GUI para configurar o espaço de trabalho com a estrutura clicando em Arquivo > Console. Em seguida, clique em Arquivo > obter PDB. Na janela pop-up, tipo: 3fgu
Em seguida, clique em OK.
NOTA: Alternativamente, use o console Jmol para carregar a estrutura, digitando: carga = 3fgu
NOTA: Após inserir qualquer comando de linha digitado, pressione o retorno no teclado para executá-lo. - Ajustando a representação: Abra o menu pop-up clicando com o botão direito do mouse (ou controle + clique) em qualquer lugar da janela do visualizador da estrutura.
- Para alterar a proteína para representação de desenho animado, no menu pop-up, clique em Selecionar halos de seleção >. Em seguida, clique em Selecionar > Proteína > Tudo. Por fim, clique em Style > Scheme > Cartoon.
NOTA: Os halos de seleção colocam um contorno amarelo (brilho) em torno de todos os átomos selecionados. - Use o menu suspenso superior para ocultar as águas clicando em Exibir > Selecione > Água. Em seguida, clique em Exibir > Átomo > Nenhume, finalmente, clique em Exibir > Selecione > Nenhum.
- Para alterar a proteína para representação de desenho animado, no menu pop-up, clique em Selecionar halos de seleção >. Em seguida, clique em Selecionar > Proteína > Tudo. Por fim, clique em Style > Scheme > Cartoon.
- Identificação dos ligantes no site ativo: Use o mouse para ampliar o site ativo e, em seguida, use os comandos nos subpassos para exibir os ligantes como sticks.
NOTA: Nomes de ligantes aparecem no console Jmol quando você carrega o arquivo. Você também pode visualizar nomes de ligantes vinculados usando o menu pop-up, clicando em Selecionar > hetero > pelo HETATM.- Passe o mouse sobre os ligantes com o mouse para ver seus nomes. O local ativo fica perto do centro da estrutura; os ligantes MG, BGC e ANP estão localizados no local ativo.
- Selecione os ligantes BCG e ANP: Usando o console Jmol, digite:
selecione BGC, ANP - Para exibir os ligantes BCG e ANP como sticks, use o menu pop-up e clique em Style > Scheme > Sticks.
- Selecionando resíduos dentro de 5 Å para definir um site ativo: No console Jmol, digite o seguinte comando para selecionar átomos dentro de 5 Å dos três ligantes:
selecionar dentro (5, (bgc,anp,mg))- Para selecionar resíduos completos de aminoácidos, digite o seguinte no console e pressione Enter
selecionar dentro (grupo, selecionado)
NOTA: O console Jmol é a melhor maneira de selecionar os resíduos dentro de 5 Å.
- Para selecionar resíduos completos de aminoácidos, digite o seguinte no console e pressione Enter
- Exibindo as correntes laterais como varas e mostrando/ajustando as moléculas de água do local ativo: Clique com o botão direito do mouse para trazer o menu pop-up e passe o mouse sobre o Esquema de > estilo > Sticks.
NOTA: O passo 3.5 mostra as cadeias laterais do site ativas na representação da vara. Ainda haverá alguns halos vazios na estrutura que representam as moléculas de água no local ativo.- No console Jmol, re-execute o seguinte comando:
selecionar dentro (5, (bgc,anp,mg))
NOTA: Para re-executar um comando, clique dentro do console e use as teclas de seta no teclado até que esse comando apareça e clique em enter para re-executá-lo. - Para exibir os átomos da molécula de água, remova os ligantes e a proteína da seleção digitando os seguintes dois comandos:
selecionar remover proteína de grupo
selecionar remover grupo hetero e não água - Para exibir as moléculas de água, clique no menu suspenso Display. Passe o mouse sobre o Átomo e clique em 20% van der Waals. Os íons verdes de Magnésio ainda serão mostrados como paus. Exibir o íon magnésio na representação da esfera mais comum digitando os seguintes comandos no console Jmol:
selecionar Mg
preenchimento espacial 50% - Recolorir os ligantes para distingui-los da proteína: No console Jmol, digite o seguinte para executar um comando que recolora os ligantes em um esquema de cores mais claro:
selecionar (bgc,anp) e carbono; cor [211.211.211]
selecionar (bgc,anp) e oxigênio; cor [255.185.185]
selecionar (bgc,anp) e nitrogênio; cor [150.210.255]
selecionar (bgc,anp) e fósforo; cor [255.165,75]
selecionar Mg; cor palegreen
- No console Jmol, re-execute o seguinte comando:
- Mostrando as interações da enzima com os ligantes ativos do site: Usando o console Jmol, execute cada linha do seguinte comando:
definir ligbind (ANP, BGC, MG)
selecionar dentro (5, (bgc,anp,mg))
selecionar remover grupo hetero e não água- Para mostrar linhas para ilustrar ligações de hidrogênio, digite este comando no console Jmol:
conectar 3.3 (ligbind e (oxigênio ou nitrogênio)) (selecionado e (oxigênio ou nitrogênio)) strut amarelo
Em seguida, modifique a espessura das linhas digitando o seguinte comando no console:
selecionar todos; strut 0.1; selecionar nenhum
- Para mostrar linhas para ilustrar ligações de hidrogênio, digite este comando no console Jmol:
- Simplificando a estrutura: Para esconder o desenho animado da proteína e limpar a seleção, no console Jmol, digite:
selecionar todos; desenho animado fora; selecionar nenhum - Rotulagem de ligantes e cadeias laterais ligadas a hidrogênio: Na janela pop-up, clique em Set Picking > Select Atom. Clique em um átomo em um dos resíduos ligados ao hidrogênio. Os números de aminoácidos e resíduos aparecem no console. Em seguida, use o console para digitar um rótulo, por exemplo:
rótulo Glu-256 - Salvar a renderização a qualquer momento para voltar a trabalhar nele ou compartilhar com outros: No menu superior, clique no ícone da câmera. Digite um nome de arquivo e selecione um local para salvar.
NOTA: Um arquivo JPEG exportado (.jpg) contém as informações tanto para uma imagem quanto aparece na janela de exibição no momento da exportação, bem como para o estado atual do modelo. Para recarregar o modelo, abra jmol e arraste o arquivo JPEG salvo para a janela de exibição Jmol. - Salvando uma imagem para incorporar ou imprimir: No console Jmol, recolorir o fundo para branco, digitando:
fundo branco
Como na etapa 3.9, clique no ícone da câmera e salve o arquivo.
4. Protocolo PyMOL
NOTA: Controles de trackpad e mouse: Para girar, clique e arraste (mouse: clique esquerdo e arraste). Para ampliar, beliscar e espalhar (mouse: clique com o botão direito do mouse e arraste). Para traduzir (ou seja, mover toda a estrutura), controlar + clicar e arrastar (mouse: comando + clique esquerdo e arraste). Para centralizar de acordo com o painel de objetos à direita e clique em Um > Oriente ou Centro.
- Carregando a estrutura em PyMOL: Na linha de comando perto da parte superior da GUI (precedida por "PyMOL>"), digite:
buscar 3FGU
NOTA: Após inserir qualquer comando de linha digitado, pressione o retorno no teclado para executá-lo. - Ajuste da representação: No painel nomes/objetos no lado direito da janela PyMOL, à direita de "3FGU" clique em H > Waters.
- Identificação dos ligantes no site ativo: Primeiro ligue o visualizador de sequência clicando no menu suspenso superior: Exibir > Sequência.
- Role a barra cinza para a direita até encontrar os nomes de ligantes (BCG, ANP, MG, K).
NOTA: Há duas representações, uma fita de desenho animado e paus; os ligantes são mostrados como paus. Certifique-se de que o modo de seleção nos controles do mouse no painel inferior direito está definido como Resíduo e modo de visualização de 3 botões clicando nesses nomes para alternar as opções. - Usando o mouse, gire e amplie para tornar os ligantes visíveis.
- Role a barra cinza para a direita até encontrar os nomes de ligantes (BCG, ANP, MG, K).
- Selecionando resíduos dentro de 5Å para definir um site ativo: Para selecionar os ligantes no site ativo, clique em cada um deles (BCG, ANP, MG) no visualizador da estrutura. Uma nova seleção aparece no painel nomes/objetos; à direita deste novo objeto chamado "sele", clique no botão A e clique em Renomear no menu pop-up.
NOTA: Para limpar uma seleção indesejada, clique no espaço vazio no visualizador da estrutura para desmarcar.- Usando o teclado, exclua as letras "sele" que aparecem no lado superior esquerdo da janela do visualizador da estrutura e, no lugar delas, digite:
Ligands
NOTA: As etapas 4.4-4.4.1 podem ser feitas usando a linha de comando; tipo:
ligantes seles, resn BGC+ANP+MG - Use esta seleção para definir a área ao redor dos ligantes, primeiro duplicando-a, clique em ligantes > A > Duplicate. Em seguida, clique em sel01 > A > Rename
Usando o teclado, exclua as letras "se101" e digite:
ativo - Modifique esta seleção para mostrar resíduos dentro de 5 Å: No painel nomes/objetos, clique em ativo > A > Modificar > Expandir > por 5 A, Resíduos. Em seguida, para mostrar esses resíduos como varas, clique em > ativo S > Licorice > Sticks. Por fim, clique no espaço vazio no Visualizador de Estrutura para limpar a seleção.
NOTA: O passo 4.4.3 pode ser feito usando a linha de comando, digite:
sele ativo, byres todos dentro de 5 de ligantes
mostrar varas, ativo
- Usando o teclado, exclua as letras "sele" que aparecem no lado superior esquerdo da janela do visualizador da estrutura e, no lugar delas, digite:
- Exibindo as correntes laterais como varas e mostrando/ajustando as moléculas de água do local ativo: Nos nomes/painel do objeto clique em ligantes > A > Duplicado. Para renomear a seleção, clique em Sel02 > A > Rename Selection. Exclua as letras no menu de renomeação que aparece no canto superior direito do visualizador da estrutura e digite:
active_water- Para ajustar a nova seleção para conter moléculas de água ativas do local, clique em active_water > A > Modifique > em torno de átomos > dentro de 4 Angstroms. Para modificar isso ainda mais e limitar as moléculas de água, clique em active_water > A > Modificar > Restringir > ao Solvente. Por fim, clique em active_water > A > Preset > ball and Stick.
NOTA: A GUI permite a seleção dentro de 4 Å; os comandos de linha permitem a seleção de uma distância mais apropriada de 3,3 Å para moléculas de água de ligação de hidrogênio. O raio van der Waals das esferas não pode ser definido na GUI, mas a seleção "bola e pau" está perto de 0,5 Å.
NOTA: As etapas 4.5-4.5.1 podem ser executadas usando a linha de comando, digitando cada linha do seguinte código:
selecionar active_water ((ligantes)em torno de 3,3) e (resn HOH)
mostrar esferas, active_water
alterar active_water, vdw=0,5
reconstruir
- Para ajustar a nova seleção para conter moléculas de água ativas do local, clique em active_water > A > Modifique > em torno de átomos > dentro de 4 Angstroms. Para modificar isso ainda mais e limitar as moléculas de água, clique em active_water > A > Modificar > Restringir > ao Solvente. Por fim, clique em active_water > A > Preset > ball and Stick.
- Mostrando as interações da enzima com os ligantes ativos do site. Zoom no site ativo clicando em > ativo A > Zoom. Para encontrar os contatos polares entre os ligantes e o site ativo, clique em ligantes > A > Encontrar contatos polares > > a quaisquer átomos. Mostre distâncias como rótulos clicando em ligands_polar_contacts > S > Labels.
- Simplificando a estrutura: Esconda o desenho animado da proteína, que esconde a parte da proteína que não está no site ativo, clicando no 3FGU > H > Cartoon no painel nomes/objetos. Em seguida, esconda as etiquetas do comprimento da ligação de hidrogênio clicando em ligands_polar_contacts > H > Labels no painel nomes/objetos.
- Para colorir os ligantes para diferenciá-los da proteína, clique em ligantes > C > Por Elemento > CHNOS e selecione a opção onde "C" é ciano (um azul claro).
NOTA: A etapa 4.7.1 pode ser executada usando a linha de comando. Tipo:
Ciano cor, ligantes
cor atômica, ligantes & !elem C
- Para colorir os ligantes para diferenciá-los da proteína, clique em ligantes > C > Por Elemento > CHNOS e selecione a opção onde "C" é ciano (um azul claro).
- Ligantes de rotulagem e cadeias laterais ligadas a hidrogênio: No painel nomes/objetos, nos botões à direita de qualquer nome do objeto, clique em > ativoS L > Resíduos.
- Salvar a renderização a qualquer momento para voltar a trabalhar nele ou compartilhar com outros: No menu suspenso clique em Arquivo > Salvar sessão como. Em seguida, selecione um local na janela pop-up, digite um nome de arquivo e clique em Salvar.
- Salvando uma imagem para incorporar ou imprimir: Primeiro, altere o fundo para branco no menu suspenso clicando em Exibir > Fundo > Branco. Exporte a imagem como um novo arquivo, clicando em File > Export Image As > PNG.
Resultados
Um protocolo executado com sucesso para cada um dos programas resultará em um modelo molecular ampliado no local ativo, com resíduos ativos do local e ligantes mostrados como varas, o desenho animado de proteína escondido e ligantes exibidos com um esquema de cores contrastante. Os resíduos de aminoácidos interagindo devem ser rotulados com seus identificadores, e a ligação de hidrogênio e interações iônicas mostradas com linhas. A presença dessas características pode ser determinada pela inspeção visual do modelo.
Para facilitar essa inspeção e permitir que o usuário determine se executou corretamente as etapas do protocolo, fornecemos figuras animadas que apresentam uma imagem da estrutura após cada etapa. Para QuimeraX, iCn3D, Jmol e PyMOL, isso é ilustrado nas Figuras 7-10, respectivamente.
Figura 7: Saída doprotocolo QuimeraX. Figura animada ilustrando os passos 1.1-1.8 do protocolo QuimeraX. Clique aqui para baixar este número.
Figura 8: saída do protocolo iCn3D. Figura animada ilustrando as etapas 2.1-2.8 do protocolo iCn3D. Clique aqui para baixar este número.
Figura 9: Saída do protocolo Jmol. Figura animada ilustrando os passos 3.1-3.8 do protocolo Jmol. Clique aqui para baixar este número.
Figura 10: Saída do protocolo PyMOL. Figura animada ilustrando as etapas 4.1-4.8 do protocolo PyMOL. Clique aqui para baixar este número.
O erro mais comum que pode influenciar o resultado desses protocolos é a seleção errônea, resultando em parte da estrutura sendo exibida em uma renderização indesejada. Isso normalmente é resultado de um clique errado, seja na própria estrutura, ou em um dos botões do menu de exibição. Um exemplo de resultado subótimo seria um modelo contendo resíduos fora do local ativo exibidos como varas. O usuário pode começar a analisar se esse erro ocorreu inspecionando visualmente os resíduos exibidos como varas e garantindo que eles estejam nas proximidades dos ligantes ativos do local. Um método avançado para avaliar se os resíduos exibidos estão ou não dentro de 5Å dos ligantes ativos do local é usar as ferramentas de medição incorporadas em cada programa para medir a distância entre um ligante próximo e o resíduo ativo do local. As ferramentas de medição estão além do escopo deste manuscrito; no entanto, encorajamos os usuários interessados a explorar os muitos tutoriais online detalhando esse tipo de análise.
Apresentamos um exemplo específico de uma execução abaixo do ideal deste protocolo, resultante de um clique errado no painel nomes/objetos no PyMOL. Este erro exibe toda a proteína como varas, em vez de mostrar apenas o site ativo usando essa representação, conforme ilustrado na Figura 11.

Figura 11: Resultado negativo. Exemplo de resultado negativo. Selecionando mal o desenho animado completo em PyMOL e exibindo varas. Clique aqui para ver uma versão maior desta figura.
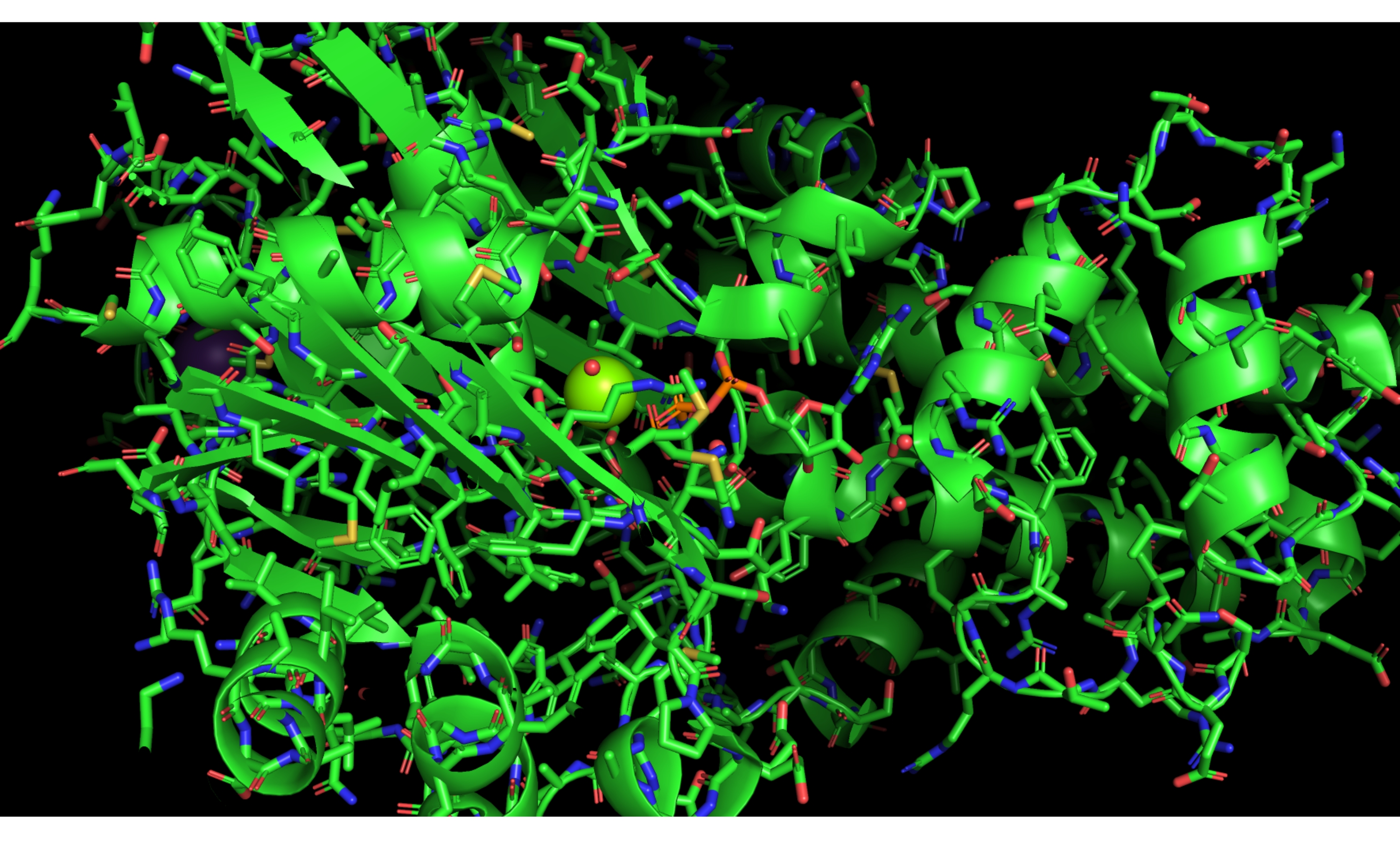{kind=link}
Para solucionar problemas, o usuário precisará ocultar os bastões de todo o modelo (rotulado 3FGU no painel nomes/objetos) e, em seguida, mostrar a representação do bastão apenas para a seleção chamada "ativo", usando os botões/comandos de ocultação e exibição no PyMOL. Recuperar o modelo desse tipo de erro é relativamente simples uma vez que o usuário é capaz de criar seleções apropriadas para diferentes partes do modelo e exibi-los e escondê-los efetivamente. É tentador reiniciar o protocolo e trabalhar as etapas outra vez; no entanto, encorajamos o usuário a não ter medo de sair do script e experimentar o modelo. Em nossa experiência, trabalhar através de erros de exibição facilita o progresso na compreensão do programa de modelagem.
Uma exibição lado a lado da saída final de um protocolo executado com sucesso para cada programa é mostrada na Figura 12. As visualizações são orientadas da mesma forma para permitir que o usuário compare a aparência dos modelos criados em diferentes programas.

Figura 12: Comparação final da estrutura entre os programas. Comparação da estrutura de cada renderização de site ativo no final do protocolo. R: QuimeraX, B: iCn3D, C: Jmol, D: PyMOL. O rótulo do local ativo PyMOL inclui todos os resíduos ativos do local e os ligantes. As outras saídas têm apenas cadeias laterais ligadas a hidrogênio rotuladas. Clique aqui para ver uma versão maior desta figura.
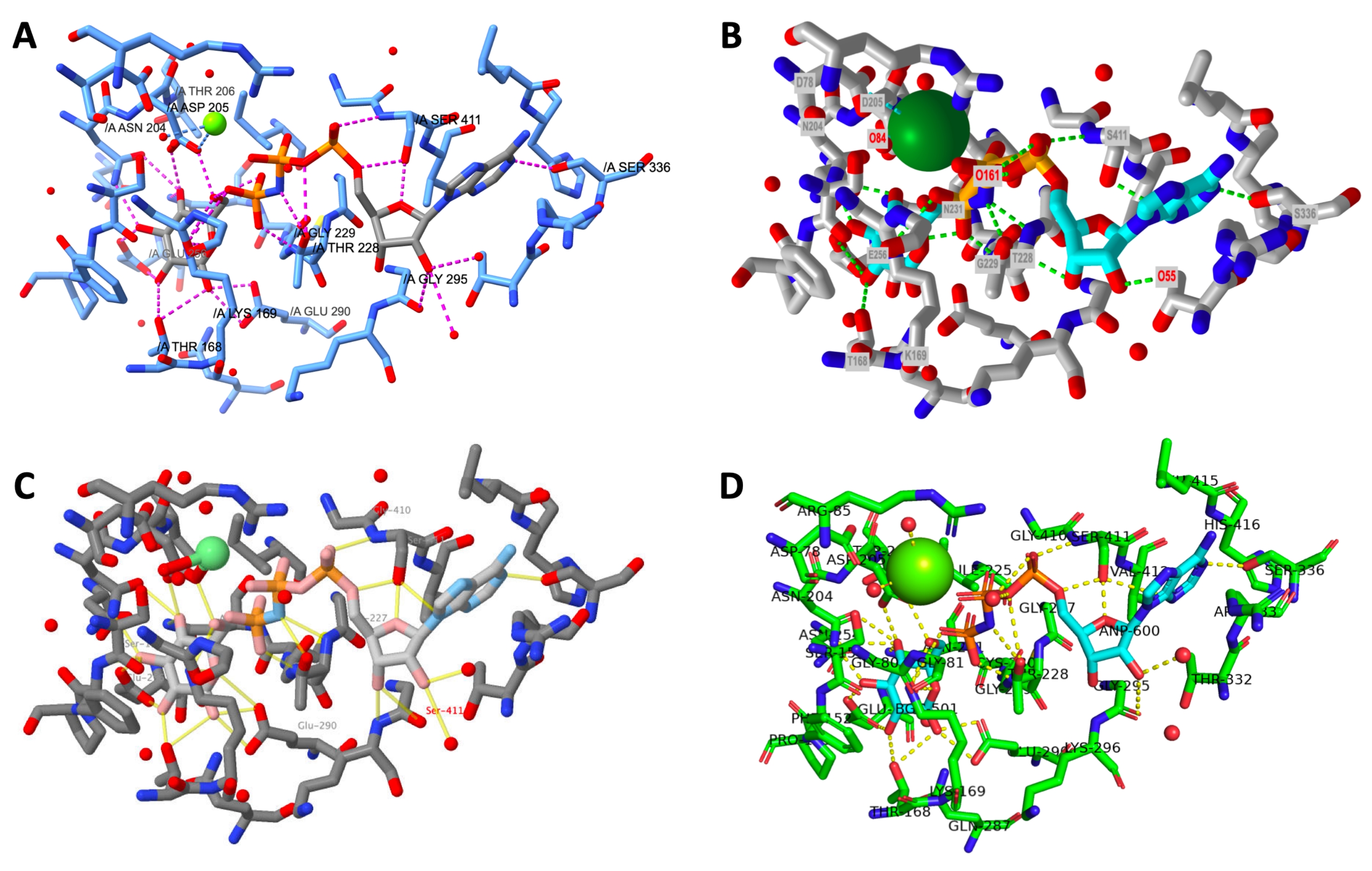{kind=link}
Discussão
Este protocolo descreve um processo de dez etapas para a modelagem de um site ativo enzimáculo, aplicado a quatro programas populares para modelagem biomolecular. As etapas críticas do protocolo são: identificar os ligantes no local ativo, selecionar resíduos dentro de 5 Å para definir um local ativo e mostrar as interações da enzima com os ligantes ativos do local. Distinguir os ligantes relevantes para a função biológica é primordial, pois isso permite ao usuário definir os resíduos de aminoácidos dentro de 5 Å que podem desempenhar um papel na ligação dos ligantes. Finalmente, o uso do programa para exibir interações moleculares permite ao usuário desenvolver as habilidades necessárias para entender as interações moleculares que promovem a vinculação.
Uma limitação dos protocolos de modelagem molecular baseados em computador é a dependência de comandos específicos e sintaxe. Embora os protocolos bioquímicos possam ser tolerantes a pequenas mudanças no procedimento, investigações baseadas em computador podem produzir produtos finais extremamente diferentes se o procedimento não for intimamente respeitado. Isso é particularmente importante ao usar interfaces de linha de comando onde a sintaxe específica do programa é necessária para alcançar uma determinada saída, e uma mudança aparentemente insignificante na pontuação ou capitalização pode fazer com que um comando falhe. Existem vários Wikis e manuais para cada programa, onde um usuário pode encontrar e solucionar problemas entradas de linha de comando; o usuário deve prestar atenção aos detalhes da sintaxe de comando. Embora a maioria dos programas de visualização molecular inclua comandos de desfazer, devido à complexidade das interfaces, o comando de desfazer nem sempre inverte fielmente a última etapa executada. Portanto, salvar o estado de trabalho atual muitas vezes é incentivado, especialmente para novos usuários.
Outras limitações podem surgir dos dados utilizados para criar o próprio modelo. Embora os padrões inerentes ao Banco de Dados de Proteínas garantam um certo nível de consistência, os usuários de programas de visualização molecular muitas vezes encontrarão efeitos inesperados em uma renderização de proteínas. Em primeiro lugar, a maioria das estruturas são determinadas por meio da cristalografia de raios-X, que fornece um único modelo da proteína; no entanto, as estruturas NMR são frequentemente compostas de múltiplos modelos que podem ser visualizados um de cada vez. Em segundo lugar, estruturas determinadas a partir de experimentos de cristalografia ou microscopia eletrônica criogênica podem conter átomos cuja posição não pode ser elucidada e aparecem como lacunas em certas representações da proteína. Estruturas proteicas podem ter conformações alternativas de cadeias laterais, que, quando exibidas na renderização de vara, aparecem como dois grupos salientes da mesma espinha dorsal de aminoácidos. Mesmo seções curtas de espinha dorsal podem ter tais conformações alternativas, e às vezes ligantes são sobrepostos no local ativo em mais de uma conformação de ligação.
Para uma estrutura cristalina, as coordenadas 3D depositadas incluem todos os componentes da unidade assimétrica, que fornece informações suficientes para reproduzir a unidade repetitiva de um cristal proteico. Às vezes, essa estrutura conterá cadeias proteicas adicionais em comparação com a forma biologicamente ativa da proteína (por exemplo, mutante da hemoglobina fetal, PDB ID: 4MQK). Por outro lado, alguns programas podem não carregar automaticamente todas as cadeias da unidade biologicamente ativa. Por exemplo, o sars-cov2 protease principal (PDB ID: 6Y2E) carrega metade do dimer biologicamente ativo (composto por duas cadeias proteicas) quando buscado usando os comandos descritos neste protocolo em QuimeraX, PyMOL e Jmol. Embora uma pequena modificação do comando carregue o dimer biologicamente ativo, essa consideração pode não ser simples para o usuário do programa de modelagem novato. Uma questão diferente que pode surgir está na identificação do site ativo ou do próprio substrato. Experimentos cristalográficos são realizados usando uma variedade de moléculas, que podem ser modeladas na estrutura final. Por exemplo, moléculas de sulfato podem ligar locais de ligação de fosfato no local ativo, ou podem ligar outras regiões que não são relevantes ao mecanismo. Essas moléculas podem obscurecer a identificação correta do próprio local ativo e podem até sugerir ao aluno que elas fazem parte do mecanismo.
Presumivelmente, o usuário desejará aplicar este procedimento em outros sites ativos/vinculativos. Para aplicar este protocolo no futuro trabalho envolvendo a análise de novos sites ativos proteicos, o usuário precisará identificar quais dos ligantes vinculados são relevantes para funcionar. Alguns ligantes não estão associados à função proteica e, em vez disso, são resultado das condições de solvente ou cristalização utilizadas para conduzir o experimento (por exemplo, o íon potássio presente no modelo 3FGU). Os ligantes-chave devem ser identificados consultando o manuscrito original. Com a prática e, quando aplicável, uma compreensão da sintaxe de comando da linha, o usuário poderá aplicar o protocolo para o programa de modelagem desejado a qualquer site ativo enzimáculo, e modelar outras macromoléculas de sua escolha.
Identificar e analisar substratos e ligantes vinculados é central para a elucidação de mecanismos moleculares e esforços de design de medicamentos baseados em estrutura, que levaram diretamente a melhorias nos tratamentos para doenças, incluindo síndrome da imunodeficiência adquirida (AIDS) e COVID-1947,48,49,50,51,52 . Enquanto programas individuais de visualização molecular oferecem diferentes interfaces e experiências do usuário, a maioria oferece recursos comparáveis. É importante para o desenvolvimento da alfabetização biomolecular que os alunos de bioquímica de nível superior se familiarizem com a visualização da estrutura e as ferramentas para gerar tais imagens4,20,53. Isso permite que os alunos ultrapassem a interpretação de imagens bidimensionais em livros didáticos e artigos de revistas e desenvolvam mais facilmente suas próprias hipóteses a partir de dados estruturais54, que prepararão cientistas em desenvolvimento para abordar futuros problemas de saúde pública e melhorar a compreensão dos processos bioquímicos.
Em resumo, este protocolo detalha a modelagem ativa do site usando quatro programas de modelagem macromolecular gratuitos líderes. Nossa comunidade, BioMolViz, adota uma abordagem não específica de software para modelagem biomolecular. Evitamos especificamente uma crítica ou comparação de recursos do programa, embora um usuário que amostra cada programa provavelmente descobrirá que prefere certos aspectos da modelagem macromolecular em um programa versus outro. Convidamos os leitores a utilizar o BioMolViz Framework, que detalha os objetivos e objetivos de aprendizagem baseados em visualização biomolecular direcionados a este protocolo, e explorar recursos para ensino e aprendizagem de visualização biomolecular através do site da comunidade BioMolViz em http://biomolviz.org.
Divulgações
Os autores declaram que não possuem interesses financeiros relevantes ou materiais relacionados à pesquisa descrita neste artigo.
Agradecimentos
O financiamento para este trabalho foi fornecido pela Fundação Nacional de Ciência:
Melhoria do Subsídio de Educação STEM de Graduação (Prêmio #1712268)
Redes de Coordenação de Pesquisa em Graduação em Educação em Biologia de Graduação (Prêmio # 1920270)
Somos gratos a Karsten Theis, PhD, Westfield University, por discussões úteis sobre Jmol.
Materiais
| Name | Company | Catalog Number | Comments |
| ChimeraX (Version 1.2.5) https://www.rbvi.ucsf.edu/chimerax/ | |||
| Computer | Any | ||
| iCn3D (web-based only: https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html) | |||
| Java (for Jmol) https://java.com/en/download/ | |||
| Jmol (Version 1.8.0_301) http://jmol.sourceforge.net/ | |||
| Mouse (optional) | Any | ||
| PyMOL (Version 2.4.1 - educational): https://pymol.org/2 educational use only version: https://pymol.org/edu/?q=educational |
Referências
- Loertscher, J., Green, D., Lewis, J. E., Lin, S., Minderhout, V. Identification of threshold concepts for biochemistry. CBE Life Sciences Education. 13 (3), 516-528 (2014).
- Jaswal, S. S., O’Hara, P. B., Williamson, P. L., Springer, A. L. Teaching structure: Student use of software tools for understanding macromolecular structure in an undergraduate biochemistry course: Teaching structure in undergraduate biochemistry. Biochemistry and Molecular Biology Education. 41 (5), 351-359 (2013).
- Tibell, L. A. E., Rundgren, C. -. J. Educational challenges of molecular life science: Characteristics and implications for education and research. CBE Life Sciences Education. 9 (1), 25-33 (2010).
- Schönborn, K. J., Anderson, T. R. The importance of visual literacy in the education of biochemists. Biochemistry and Molecular Biology Education. 34 (2), 94-102 (2006).
- Anderson, T. R. Bridging the educational research-teaching practice gap: The importance of bridging the gap between science education research and its application in biochemistry teaching and learning: Barriers and strategies. Biochemistry and Molecular Biology Education. 35 (6), 465-470 (2007).
- Schönborn, K. J., Anderson, T. R. Bridging the educational research-teaching practice gap: Foundations for assessing and developing biochemistry students’ visual literacy. Biochemistry and Molecular Biology Education. 38 (5), 347-354 (2010).
- Bateman, R. C., Craig, P. A. Education corner: A proficiency rubric for biomacromolecular 3D literacy. PDB Newsletter. 45, 5-7 (2010).
- Mnguni, L., Schönborn, K., Anderson, T. Assessment of visualization skills in biochemistry students. South African Journal of Science. 112, 1-8 (2016).
- Craig, P. A., Michel, L. V., Bateman, R. C. A survey of educational uses of molecular visualization freeware. Biochemistry and Molecular Biology Education. 41 (3), 193-205 (2013).
- Loertscher, J., Villafañe, S. M., Lewis, J. E., Minderhout, V. Probing and improving student’s understanding of protein α-Helix structure using targeted assessment and classroom interventions in collaboration with a faculty community of practice. Biochemistry and Molecular Biology Education. 42 (3), 213-223 (2014).
- Abualia, M., et al. Connecting protein structure to intermolecular interactions: A computer modeling laboratory. Journal of Chemical Education. 93 (8), 1353-1363 (2016).
- Carvalho, I., Borges, A. D. L., Bernardes, L. S. C. Medicinal chemistry and molecular modeling: An integration to teach drug structure–activity relationship and the molecular basis of drug action. Journal of Chemical Education. 82 (4), 588 (2005).
- Forbes-Lorman, R. M., et al. Physical models have gender-specific effects on student understanding of protein structure-function relationships. Biochemistry and Molecular Biology Education. 44 (4), 326-335 (2016).
- Terrell, C. R., Listenberger, L. L. Using molecular visualization to explore protein structure and function and enhance student facility with computational tools. Biochemistry and Molecular Biology Education. 45 (4), 318-328 (2017).
- Zhang, S., et al. Structure-based drug design of an inhibitor of the SARS-CoV-2 (COVID-19) main protease using free software: A tutorial for students and scientists. European Journal of Medicinal Chemistry. 113390, (2021).
- Roberts, J. R., Hagedorn, E., Dillenburg, P., Patrick, M., Herman, T. Physical models enhance molecular three-dimensional literacy in an introductory biochemistry course. Biochemistry and Molecular Biology Education. 33 (2), 105-110 (2005).
- Jenkinson, J., McGill, G. Visualizing protein interactions and dynamics: Evolving a visual language for molecular animation. CBE Life Sciences Education. 11 (1), 103-110 (2012).
- Bussey, T. J., Orgill, M. What do biochemistry students pay attention to in external representations of protein translation? The case of the Shine–Dalgarno sequence. Chemistry Education Research and Practice. 16 (4), 714-730 (2015).
- Harle, M., Towns, M. H. Students’ understanding of primary and secondary protein structure: Drawing secondary protein structure reveals student understanding better than simple recognition of structures. Biochemistry and Molecular Biology Education. 41 (6), 369-376 (2013).
- Dries, D. R., et al. An expanded framework for biomolecular visualization in the classroom: Learning goals and competencies. Biochemistry and Molecular Biology Education. 45 (1), 69-75 (2017).
- Procko, K., et al. Meeting report: BioMolViz workshops for developing assessments of biomolecular visual literacy. Biochemistry and Molecular Biology Education. 49 (2), 278-286 (2021).
- Wang, J., et al. iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics. 36 (1), 131-135 (2020).
- PyMOL . . The PyMOL Molecular Graphics System. Version 2.0. , (2021).
- Goddard, T. D., et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science. 27 (1), 14-25 (2018).
- Pettersen, E. F., et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science. 30 (1), 70-82 (2021).
- Petit, P., et al. The active conformation of human glucokinase is not altered by allosteric activators. Acta Crystallographica. Section D. 67 (11), 929-935 (2011).
- Corey, R. B., Pauling, L. Molecular models of amino acids, peptides and proteins. Review of Scientific Instruments. 24, 621-627 (1953).
- Koltun, W. L. Precision space-filling atomic models. Biopolymers. 3 (6), 665-679 (1965).
- Hodis, E., et al. Proteopedia - a scientific 'wiki' bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biology. 9 (8), 1-10 (2008).
- Prilusky, J., et al. Proteopedia: A status report on the collaborative, 3D web-encyclopedia of proteins and other biomolecules. Journal of Structural Biology. 175 (2), 244-252 (2011).
- . FirstGlance in Jmol Available from: https://www.bioinformatics.org/firstglance/fgij/ (2021)
- Jmol User Design Environment (JUDE). MSOE Centerfor BioMolecular Modeling Available from: https://cbm.msoe.edu/modelingResources/jmolUserDesignEnvironment/#forward (2021)
- Castro, C. R., et al. A practical guide to teaching with Proteopedia. Biochemistry and Molecular Biology Education. 49 (5), 707-719 (2021).
- Berman, H. M., et al. The protein data bank. Nucleic Acids Research. 28, 235-242 (2000).
- . The Protein Data Bank Available from: https://www.rcsb.org/ (2021)
- Wang, Y., et al. MMDB: 3D structure data in Entrez. Nucleic Acids Research. 28 (1), 243-245 (2000).
- . iCn3D Help Page Available from: https://www.ncbi.nlm.nih.gov/Structure/icn3d/docs/icn3d_help.html (2021)
- . MSOE Center for BioMolecular Modeling Jmol Training Guide Available from: https://cbm.msoe.edu/modelingResources/jmolTrainingGuide/started.html (2021)
- . Jmol/JSmol Interactive Scripting Documentation Available from: https://chemapps.stolaf.edu/jmol/docs/ (2021)
- . PyMOL Wiki Available from: https://pymolwiki.org/index.php/Main_Page (2021)
- . PyMOL Advanced Scripting Workshop by Schrödinger Available from: https://pymol.org/tutorials/scripting/index.html (2021)
- . UCSF ChimeraX User Guide Available from: https://www.cgl.ucsf.edu/chimerax/docs/user/index.html (2021)
- . UCSF ChimeraX Tutorials Available from: https://www.rbvi.ucsf.edu/chimerax/tutorials.html (2021)
- Kuntz, I. D. Structure-based strategies for drug design and discovery. Science. 257 (5073), 1078-1082 (1992).
- Hubbard, R. E. . Structure-based drug discovery: an overview. , (2006).
- Patrick, G. L. . An introduction to medicinal chemistry, 6th ed. , (2017).
- Van Montfort, R. L., Workman, P. Structure-based drug design: aiming for a perfect fit. Essays in Biochemistry. 61 (5), 431-437 (2017).
- Holdgate, G. A., Meek, T. D., Grimley, R. L. Mechanistic enzymology in drug discovery: a fresh perspective. Nature Reviews. Drug Discovery. 17 (2), 115-132 (2018).
- Wang, M. Y., et al. SARS-CoV-2: structure, biology, and structure-based therapeutics development. Frontiers in Cellular and Infection Microbiology. 10, (2020).
- White, B., Kim, S., Sherman, K., Weber, N. Evaluation of molecular visualization software for teaching protein structure differing outcomes from lecture and lab: Differing outcomes from lecture and lab. Biochemistry and Molecular Biology Education. 30 (2), 130-136 (2002).
- Canning, D. R., Cox, J. R. Teaching the structural nature of biological molecules: Molecular visualization in the classroom and in the hands of students. Chemistry Education Research and Practice. 2 (2), 109-122 (2001).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados