
Method Article
TMT Пример ы подготовки к протеомики объекта представления и последующего анализа данных
В этой статье
Резюме
Мы представляем оптимизированный протокол маркировки тандемной массы (TMT), который включает подробную информацию по каждому из следующих шагов: экстракция белка, количественная оценка, осадки, пищеварение, маркировка, представление на объект протеомики и анализ данных.
Аннотация
Протеомные технологии являются мощными методологиями, которые могут помочь нашему пониманию механизмов действия в биологических системах, предоставляя глобальное представление о воздействии болезни, лечения или других состояний на протеом в целом. В настоящем докладе содержится подробный протокол для извлечения, количественной оценки, осадков, пищеварения, маркировки и последующего анализа данных образцов белков. Наш оптимизированный протокол маркировки TMT требует более низкой концентрации меток метки и обеспечивает стабильно надежные данные. Мы использовали этот протокол для оценки профилей экспрессии белка в различных тканях мыши (т.е. сердце, скелетные мышцы и мозг), а также клетки, культивированные в пробирке. Кроме того, мы демонстрируем, как оценить тысячи белков из полученного набора данных.
Введение
Термин "протеомика" был впервые определен как крупномасштабная характеристика всего белкового дополнения клетки, ткани или организма1. Протеомный анализ позволяет иссмотреть механизмы и клеточные процессы, участвующие в развитии болезни, терапевтические пути, и здоровые системы с использованием методов для выполнения относительной количественной оценки уровня экспрессии белка2. Первоначальные описания таких исследований были опубликованы в 1975 году и продемонстрировали использование двухмерного полиакриламида геля электрофорусис (2D-PAGE) для этой цели1,3. 2D метод отделяет белки на основе заряда (изоэлектрическая фокусировка, IEF) и молекулярной массы (натрий dodecyl сульфат полиакриламимид гель электрофорексис, или SDS-PAGE)4. В течение многих лет, сочетание 2D-PAGE и последующего тандема масс-спектрометрии выполняется на каждом компоненте геля был наиболее распространенным нецелевым методом анализа экспрессии белка выполняется и определены многочисленные ранее неизвестные профили экспрессии белка5,6. Общие недостатки 2D-PAGE подход являетсято,что это отнимает много времени, не работает хорошо для гидрофобных белков, и Есть ограничения в общем количестве белков оценивается из-за низкой чувствительности7,8.
Стабильная маркировка изотопов аминокислотами в клеточной культуре (SILAC) стала следующим популярным подходом к выявлению и количественной оценке изобилия белка в образцах9. Она состоит из метаболической маркировки клеток, которые инкубируются в среде, не хватает стандартной незаменимой аминокислоты и дополнены изотоп-маркированной версии этой специфической аминокислоты10. Преимуществом этой техники является ее эффективность и точная маркировка9. Основным ограничением подхода SILAC является в первую очередь снижение темпов роста клеток, вызванных включением изотопных этикеток, что может быть особенно сложным в относительно чувствительных клеточных линиях, моделирующих заболевания человека11.
В 2003 году на поле12была введена новая и надежная техника протеомики с использованием тандемных массовых меток (TMT) изобобариата. TMT маркировки является мощным методом из-за его повышенной чувствительности для обнаружения относительных уровней экспрессии белка и постпереводных модификаций13. На эту дату публикации были разработаны наборы TMT, которые могут одновременно маркировать 6, 10, 11 или 16 образцов. В результате можно измерить изобилие пептидов в нескольких условиях с биологическими репликациями одновременно14,,15,,16. Недавно мы использовали TMT для характеристики сердечного протеомный профиль мыши модели синдрома Барта (BTHS)17. При этом, мы смогли продемонстрировать широкое улучшение в сердечных профилей bTHS мышей, обработанных с помощью генной терапии и определить новые белки, пострадавших от BTHS, которые показали новые терапевтические пути, участвующие в кардиомиопатии.
Здесь мы описываем подробный метод для выполнения мультиплекса TMT количественного протеомики анализов с использованием образцов тканей или клеточных гранул. Это может быть полезно для выполнения подготовки образца и маркировки до представления в ядро, потому что помеченные триптические пептиды являются более стабильными, чем сырые замороженные образцы, не все ядра имеют опыт обработки всех типов образцов, и подготовка образцов в лаборатории может сэкономить время для ядер, которые часто имеют длинные отставания. Для подробного описания масс-спектроскопии часть этого процесса, пожалуйста, см Kirshenbaum и др. и Перумал и др.18,19.
Протокол подготовки выборки состоит из следующих основных шагов: добыча, количественная оценка, осадки, пищеварение и маркировка. Основными преимуществами этого оптимизированного протокола являются то, что он снижает затраты на маркировку, улучшает экстракцию белка и последовательно генерирует высококачественные данные. Кроме того, мы описываем, как анализировать данные TMT для проверки тысяч белков в короткий промежуток времени. Мы надеемся, что этот протокол поощряет другие исследовательские группы рассмотреть вопрос о включении этой мощной методологии в свои исследования.
протокол
Институциональный комитет по уходу за животными и использованию животных из Университета Флориды одобрил все исследования на животных.
1. Подготовка реагентов
- Приготовьте CHAPS Lysis Buffer (150 мм KCl, 50 мМ HEPES pH - 7,4, 0,1% CHAPS и 1 таблетка для ингибитора протеазы на 50 мл буфера). Буфер без ингибиторов протеазы может храниться при 4 градусах По Цельсию в течение 6 месяцев или буфер с ингибитором протеазы, хранящийся при -20 градусов по Цельсию до 1 года.
- Приготовьте 100 мМ триэтиламмония бикарбоната (TEAB): Добавьте 500 л из 1 М ТЭБа до 4,5 мл ультрачистой воды.
- Приготовьте 200 мМ три (2-карбоксетил) фосфиновый гидрохлорид (TCEP): Добавьте 70 л TCEP 0,5 М, денатурный реагент, до 70 л ультрачистой воды. Затем добавьте 35 зл из 1 M TEAB.
- Подготовка 5% гидроксиламин: Добавить 50 л 50% гидроксиламин до 450 л 100 мМ TEAB.
2. Экстракция белка
- Изолировать четырехглавую мышцу от эвтаназии мыши в соответствии с протоколом, утвержденным IACUC. Заморозить и поддерживать при -80 градусов или продолжить с протоколом для немедленного использования.
- Вырезать, чтобы изолировать примерно 10 мг свежей или замороженной четырехглавой ткани мыши. Отдельные волокна с помощью пинцета при работе со скелетной мышцей. Кроме того, при работе с клеточными культурами, повторно приостановите 3 х 106 ячеек в 300 л из буфера lysis CHAPS и перейдите к шагу 2.4.
- Гомогенизировать ткани с помощью бисера разрушитель с помощью 2 мл труб заполнены примерно 200 л 1 мм zirconia / кремнезема шарики и 500 Л буфера лизиса CHAPS. Масштабируйте вверх или вниз по мере необходимости (например, 5 мг ткани в 250 л из буфера лиза CHAPS).
- Выполните звукование (10x на 10 с каждый с 50% амплитуды и 30 s интервалы на льду) для высвобождения белка, связанного с ДНК. Те же результаты деградации ДНК могут быть достигнуты либо с помощью лысиса шприца, передавая лизат 10x через 21 G иглу, прикрепленную к шприцу 1 мл, либо с помощью инкубации бензоназа (44 U/mL) при 37 градусах по Цельсию в течение 30 мин.
- Центрифуга лизат на 16000 х г в течение 10 мин при 4 градусах По Цельсию и передать супернатант в новую центрифугу трубки.
3. Измерение белка
- Определить концентрацию белка супернатанта с помощью установленных протоколов (см. Таблицу Материалов).
ПРИМЕЧАНИЕ: Лучше всего использовать образцы при 2 мкг/Л, но могут также использоваться менее концентрированные образцы. При использовании менее концентрированной выборки необходимо будет надлежащим образом отрегулировать объемы редукторных/алкилирующих реагентов в шаге 5.1. - Подготовьте разбавление стандартной кривой BSA с помощью буфера lysis CHAPS.
- Следуйте инструкциям производителя, и после 15 мин, читать абсорбции на 750 нм.
4. Снижение/алкилирование реагента
- Перенесите 200 мкг белка на состояние в новую центрифужную трубку и приспособите к окончательному объему 100 л с помощью буфера лиза CHAPS. Можно масштабировать до 200 кл, когда концентрация белка слишком низка, но не забудьте соответствующим образом отрегулировать объем сокращения / алкилирования реагента.
- Добавьте 5 qL из 200 мМ TCEP и инкубировать образцы при 55 градусах по Цельсию на 1 ч.
- Непосредственно перед использованием, подготовить 375 мМ йодоацетамид путем растворения одной трубки йодоацетамида (т.е., 9 мг) в 132 Л 100 мм TEAB. Защитите это решение от света.
- Добавьте 5 йодоацетамида 375 мМ в образец и инкубировать в течение 30 минут при комнатной температуре (RT), защищенной от света.
5. Метанол/хлороформное количество осадков20
- Добавьте 400 qL метанола к каждому 100 qL протеина и кратко образцы вихря.
- Центрифуга при 9000 х г на 10 с на RT. Это необходимо для включения жидкостей, депонированных по бокам образца трубки.
- Добавьте в смесь 100 л хлороформа и кратко евки. Используйте 200 л хлороформа, если в образце высокая концентрация фосфолипидов.
- Центрифуга при 9000 х г на 10 с на RT. Это необходимо для включения жидкостей, депонированных по бокам образца трубки.
- Добавить 300 л воды и вихря энергично. Важно получить однородное решение.
- Центрифуга при 9000 х г в течение 1 мин на RT. Будьте крайне осторожны, чтобы избежать нарушения слоев при передаче трубки на стойку.
ПРИМЕЧАНИЕ: Трубка должна теперь содержать три фазы: 1) Верхний слой (т.е. супернатант), смесь воды и метанола; 2) Средний слой (т.е. межфаза), белый осаждаемый белок; и 3) Нижний слой (т.е. нижняя фаза), хлороформ. - Аккуратно удалите супернатант.
- Добавьте 300 л метанола к оставшейся межфазной и нижней фазе. Вихрь энергично.
- Центрифуга при 9000 х г в течение 2 мин на RT. Будьте крайне осторожны, чтобы избежать нарушения слоев при передаче трубки на стойку.
- Аккуратно удалите супернатант.
- Аккуратно аспирировать как можно больше жидкости, как это возможно под потоком воздуха (например, с помощью вакуумного концентратора) на RT, пока гранулы только немного влажным (10 мин). Поскольку необходимое время может быть различным для каждого образца, проверьте каждые 2 мин, чтобы оценить. Храните гранулы при -80 градусах по Цельсию до дальнейшей обработки.
6. Пищеварение белка
- Притязание осажденного протеинового пеллета в 100 л буфера л.
ПРИМЕЧАНИЕ: Необязательно измерять концентрацию белка на этом этапе. - Непосредственно перед использованием, подготовить 1 мкг / Л трипсина, добавив 100 злитрораствор атрипсина (50 мм уксусной кислоты) на дно 100 мкг трипсина стеклянный флакон и инкубировать в течение 5 минут на RT. Store оставшийся реагент в одноразовых дозах при -80 градусов по Цельсию.
- Добавьте 2,5 л трипсина на 100 мкг белка. Дайджест образца на ночь при 37 градусах Цельсия. Этот шаг имеет решающее значение для полной растворимостизации белка; не изменяйте эти условия. После пищеварения, это необязательно для измерения концентрации белка с помощью стандартных белковых анализов.
7. Пептидная маркировка
- Непосредственно перед использованием, уравновесить TMT этикетки комплект реагентов rt.
- Растворите каждый из 0,8 мг TMT тегов флаконы путем добавления 41 л ангидроус ацетонитрила к каждой трубке. Инкубировать реагент в течение 5 минут на RT с случайным вихрем. Кратко центрифуга труб.
ПРИМЕЧАНИЕ: Концентрация 0,8 мг тега TMT, как правило, достаточно, чтобы пометить два набора. Другие исследователи, однако, показали, что эта концентрация может быть снижена дальше и по-прежнему дают надежные данные15. - Тщательно добавляйте 41 зл реагента метки TMT к каждому образцу 100 л.
- Инкубировать реакцию на 1 ч на RT.
- Добавьте в образец 8 зЛ 5% гидроксиламина и инкубируйте в течение 15 мин, чтобы утолить реакцию.
- Разделите образцы на равные количества в новой центрифуге трубки и храните при -80 градусов по Цельсию.
ПРИМЕЧАНИЕ: На этом этапе образцы стабильны и могут быть представлены для массовой спектроскопии. Необязательно измерять концентрацию в этой точке с помощью стандартных белковых анализов.
8. Масс-спектроскопия
- Отправить образцы на объект протеомики (это исследование используется UF ICBR Proteomics Core объекта), где все образцы объединены и очищены с помощью C18 спин столбцов.
ПРИМЕЧАНИЕ: Обсудите с основным объектом, как представить образцы до их подготовки, чтобы подтвердить точные шаги, которые они предпочитают для представлений. - Запросите следующие процедуры на комбинированный образец мультиплекса: твердая фаза экстракции, HPLC (SCX, SE), цип-код, и LC-MS/MS (2 ч градиент для белка ID, если зgt; 10'E Plus).
- После сбора данных основной объект будет обрабатывать RAW-файлы с использованием поставляемого поставщиком программного обеспечения для идентификации белка.
9. Анализ данных
- Данные обычно доставляются из ядра обратно пользователю в формате 7z, что может потребовать примерно 16 ГБ дискового пространства на каждый набор данных (в данном случае 11 образцов). Для обработки данных убедитесь, что компьютер доступен, по крайней мере 3,4 ГГц.
- Извлекайте файлы с помощью 7-цип-файл-менеджера. Эти извлеченные файлы содержат данные RAW, файл формата pdStudy и файл формата pdResultView. Сохраните все файлы для дальнейшего анализа.
- Открытый файл с помощью программного обеспечения Proteome Discovery 2.2.
ПРИМЕЧАНИЕ: Формат файла "File name.pdStudy". При открытии "File name.pdResultView" невозможно выбрать контрольный образец. - Выберите контрольные образцы на панели"Образцы".
- Открытый результат, выбрав id на панели"Результаты анализа".
- Экспорт программного обеспечения для электронных таблиц.
- Сохранить необработанные данные (все белки определены).
- Откройте файл программного обеспечения электронной таблицы. Это будет содержать все белки, которые были определены.
- В файле программного обеспечения электронной таблицы используйте функцию«Фильтр»для экранирования «Protein FDR Confidence: Combined« в высоком (колонка B), « #Uniqueпептиды» выше 2 (колонка K),и либо один из «Коэффициент изобилия» пустой исключительно(колонка S до W).
- Вставьте столбец для расчета"p-value"с функцией
«TTEST»(контрольная группа, экспериментальная группа, хвосты, тип) - Вставьте столбец для "Статистическое значение" с функцией
ЗИФ (p-value-lt;0.05, "Значение","NS") - Используйте функцию«Фильтр»для проверки «Статистическое значение» показывая«Значимость». Результат показывает проанализированные белки со статистической значимостью в контрольной и экспериментальной группе.
- Определите значительно более высокое или более низкое обилие выражения протеина в экспериментальной группе сравненный к контрольной группе, вставьте колонку для «Регулирования« с функцией
(AVERAGE (оценка) гт;СРЕДНИй (экспериментальная группа),"Upregulated","Downregulated")
10. Методы оценки значительных попаданий
- Для выявления белково-белковых взаимодействий между значительными хитами, выявленными в исследованиях TMT, используйте инструмент поиска взаимодействующих генов/белков (STRING) версии 11.021: https://string-db.org/
- Для классификации по группам (т.е. молекулярной функции, биологическим процессам и классам белка) используйте анализ белка с помощью эволюционного отношения (PANTHER) онтологии классификации программного обеспечения22: http://www.pantherdb.org/
- Для выявления белковых взаимодействий в различных путях, используйте программное обеспечение для анализа путей23.
11. Протеомные данные, загруженные в банк-репозиторий
- Для представления протеомических данных в базу данных Proteomics IDEntificantions Database (PRIDE) или Mass Spectrometry Interactive Virtual Environment (MassIVE) содержится следующая информация: файлы пикового списка (обработанные файлы массового спектра в стандартном формате, таких как mzXML, mzML, или MGF), файлы результатов (идентификация спектра в стандартном формате, таких как mzIdentML или mzTab) и файлы исходного спектра (файлы некачественного массового спектра в нестандартном или инструмента-специфическом формате, таких как . RAW файлы или . ФАЙЛы WIFF).
- Чтобы отправить, создать учетную запись и включить информацию, такую как принадлежность и детали проекта. Затем выберите файлы, перечисленные в шаге 11.1, и загрузите их.
- Чтобы создать официальный набор данных, запустите рабочий процесс представления этих загруженных файлов.
ПРИМЕЧАНИЕ: После представления, набор данных будет частным в банке репозитория. С помощью частного варианта данные доступны только авторизованным пользователям. Есть два дополнительных варианта: 1) Общий набор данных, который дает доступ к рецензентам и сотрудникам журналов; или 2) Общедоступный набор данных, который будет отображаться в общедоступных поисках наборов данных. Другой важной особенностью этих репозиторий является возможность обновления загруженных данных и ассоциации последующих публикаций с существующим набором данных.
Результаты
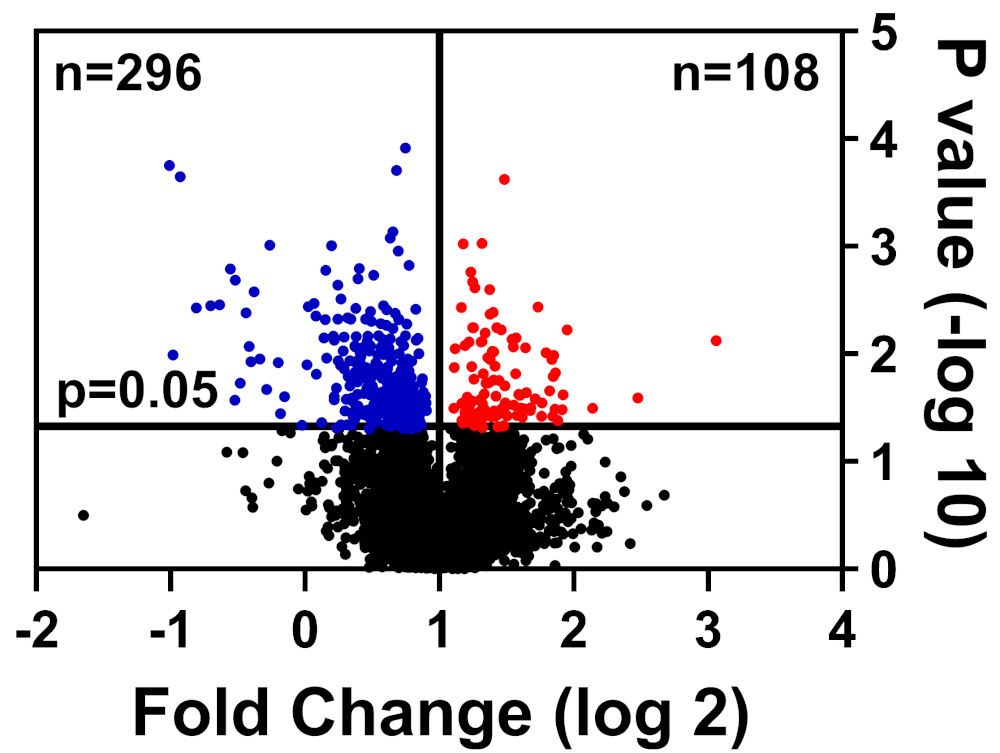
Здоровые и больные клетки были лисицированы в буфере CHAPS, подготовлены, как подробно описано в нашем методе маркировки TMT, и представлены в Университете Флориды Междисциплинарный центр биотехнологических исследований (UF-ICBR) Протеомика ядро для жидкой хроматографии с тандемной масс-спектрометрии. После получения и доставки данных из ядра, набор данных был открыт в программном обеспечении, поставляемом поставщиком, и были применены следующие фильтры отсечения: 2 уникальных пептида, ионы репортеров для каждого образца белка, присутствуют во всех каналах, и включают в себя только значительно измененные белки (p 0.05). В таблице 1 приводятся данные: 39 653 пептида, из которых 7211 имеют равные или более двух уникальных пептидов, а 3829 содержат ионы репортеров для всех каналов. Значения p для этих 3829 пептидов были рассчитаны по тесту студента т, и р 0,05 считались значительными. Кроме того, для определения относительного распределения белков от больных по сравнению с здоровыми клетками использовалось сокращение от изменения складок: регулируемых (синий) или upregulated (красный)(рисунок 1).
Список значительно дисрегулируемой экспрессии белка был оценен с помощью системы классификации онтологии PANTHER и анализа STRING. Анализы пантер ы показали категоризированный список белков, основанных на значительно более низком(рисунок 2A)или более высоком изобилии больных клеток на основе молекулярной функции(рисунок 2B). Струнный анализ белков значительно ниже(рисунок 2С)и выше(Рисунок 2D) изобилие определило несколько взаимодействий и сильные ассоциации между белками.

Рисунок 1: Вулкан участок отображения белков, изобилие которых не было значительно изменено (черный), значительно снижена (синий), или значительно увеличилось (красный) в больных по сравнению со здоровыми контрольными клетками. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 2: Репрезентативные оценки значительно дисрегулируемых хитов, выявленных PANTHER (A, B) и String (C, D) значительно более низких или более высоких белков изобилия. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
| Всего пептиды | Всего идентифицировано | 2 уникальных пептида | Количественные белки | Значительно измененные белки | |
| Низкой | Высокой | ||||
| 39653 | 7211 | 4457 | 3829 | 296 | 108 |
Таблица 1: Представительная таблица количественных белков на анализ набора данных.
Обсуждение
Для успешной подготовки образцов для протеомического анализа с использованием TMT основе изобарических изобарических методологий маркировки изотопов, очень важно выполнять извлечения белка очень тщательно при 4 КС и использовать буфер лиза, который содержит коктейль-ингибитор протеазы24,25. Коктейль ингибитора протеазы является важным реагентом, чтобы избежать неожиданной деградации белка во время переваривания белка. Одним из ключевых различий между нашим протоколом и текущим, предоставленным поставщиком, является то, что мы настоятельно рекомендуем использовать буфер лиза CHAPS на основе нашего опыта с клетками млекопитающих и тканями. Мы также предлагаем использовать метанол / хлороформ белка осадков подход как для клеточных гранул и тканей.
В идеале, экстракция белка, измерение, уменьшение /алкилирующие реагенты лечения, и метанол / хлороформ ные те же осадки выполняются в тот же день. После этой рекомендации приведет к более точной концентрации белка для последующей маркировки. Шаг выпадения белка имеет важное значение для удаления реагентов, которые будут мешать тандемной масс-спектрометрии. В том числе шаг осадков значительно повышает разрешение TMT26. В целом, основными преимуществами нашего протокола TMT являются высокая эффективность маркировки различных типов образцов, его воспроизводимость и достоверные данные.
Поскольку мультиплексный характер этой стратегии нецелевой протеомики TMT продолжает расширяться, она будет постепенно повышать способность исследователей в самых различных областях делать новые открытия. В частности, в биомедицинской области, мы и другие обнаружили, что эта технология становится все более информативным в исследованиях, исследуя новые механизмы действия в болезни и относительные последствия различных терапевтических. По всем этим причинам эта мощная технология дополняет репертуар других подходов OMICS, используемых в современных исследованиях, и предоставляет ключевую информацию, которая может служить для дальнейшего терапевтического развития.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Мы хотели бы отметить протеомику UF-ICBR за их обработку наших образцов. Эта работа была частично поддержана Национальными институтами здравоохранения R01 HL136759-01A1 (CAP).
Материалы
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium bicarbonate (TEAB), 50 mL | Thermo Fisher | 90114 | Reagent for protein labeling |
| 50% Hydroxylamine, 5 mL | Thermo Fisher | 90115 | Reagent for protein labeling |
| Acetic acid | Sigma | A6283 | Reagent for protein digestion |
| Anhydrous acetonitrile, LC-MS Grade | Thermo Fisher | 51101 | Reagent for protein labeling |
| Benzonaze nuclease | Sigma-Aldrich | E1014 | DNA shearing |
| Bond-Breaker TCEP solution, 5 mL | Thermo Fisher | 77720 | Reagent for protein labeling |
| BSA standard | Thermo | 23209 | Reagent for protein measurement |
| CHAPS | Thermo Fisher | 28300 | Reagent for protein extraction |
| Chloroform | Fisher | BP1145-1 | Reagent for protein precipitation |
| cOmplete, EDTA-free Protease Inhibitor Cocktail Tablet | Roche | 4693132001 | Reagent for protein extraction |
| DC Protein Assay | BioRad | 500-0116 | Reagent for protein measurement |
| Excel | Microsoft Office | Software for data analyses | |
| Heat block | VWR analog | 12621-104 | Equipment for protein digestion incubation |
| HEPES | Sigma | RDD002 | Reagent for protein extraction |
| Methanol | Fisher | A452-4 | Reagent for protein precipitation |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | 90058 | Reagent for protein digestion |
| Potassium chloride | Sigma | 46436 | Reagent for protein extraction |
| Sigma Plot 14.0 | Sigma Plot 14.0 | Software for data analyses | |
| Sonicator | Fisher Scientific | FB120 | DNA shearing |
| Spectra Max i3x Multi-Mode Detection Platform | Molecular Devices | Plate reader for protein measurement | |
| Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | 23275 | Reagent for protein measurement |
| Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay | Thermo Fisher | 23290 | Reagent for protein measurement |
| Thermo Scientific Proteome Discoverer Software | Thermo Fisher | OPTON-30945 | Software for data analyses |
| TMT 10plex Isobaric Label Reagent Set 0.8 mg, sufficient reagents for one 10plex isobaric experiment | Thermo Fisher | 90110 | Reagent for protein labeling |
| TMT11-131C Label Reagent 5 mg | Thermo Fisher | A34807 | Reagent for protein labeling |
| Water, LC-MS Grade | Thermo Fisher | 51140 | Reagent for protein extraction |
Ссылки
- Graves, P. R., Haystead, T. A. Molecular biologist's guide to proteomics. Microbiology and Molecular Biology Reviews. 66 (1), 39-63 (2002).
- Erdjument-Bromage, H., Huang, F. K., Neubert, T. A. Sample Preparation for Relative Quantitation of Proteins Using Tandem Mass Tags (TMT) and Mass Spectrometry (MS). Methods in Molecular Biology. 1741, 135-149 (2018).
- O'Farrell, P. H. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 250 (10), 4007-4021 (1975).
- Rabilloud, T., Lelong, C. Two-dimensional gel electrophoresis in proteomics: a tutorial. Journal of Proteomics. 74 (10), 1829-1841 (2011).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 1 (5), 376-386 (2002).
- Anderson, N. G., Anderson, N. L. Twenty years of two-dimensional electrophoresis: Past, present and future. Electrophoresis. 17 (3), 443-453 (1996).
- Haynes, P. A., Yates, J. R. Proteome profiling-pitfalls and progress. Yeast. 17 (2), 81-87 (2000).
- Bunai, K., Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 815 (1-2), 227-236 (2005).
- Sury, M. D., Chen, J. X., Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Molecular & Cellular Proteomics. 9 (10), 2173-2183 (2010).
- Zhang, G., Neubert, T. A. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods in Molecular Biology. 527, 79-92 (2009).
- Wang, X., et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Scientific Reports. 8 (1), 8441 (2018).
- Thompson, A., et al. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 75, 1895-1904 (2003).
- Cheng, L., Pisitkun, T., Knepper, M. A., Hoffert, J. D. Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods in Molecular Biology. 1355, 53-70 (2016).
- Navarrete-Perea, J., Yu, Q., Gygi, S. P., Paulo, J. A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research. 17 (6), 2226-2236 (2018).
- Zecha, J., et al. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Bachor, R., Waliczek, M., Stefanowicz, P., Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 24 (4), E701 (2019).
- Suzuki-Hatano, S., et al. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Molecular Therapy - Methods & Clinical Development. 13, 167-179 (2019).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. (40), e1954 (2010).
- Perumal, N., et al. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. Journal of Visualized Experiments. (144), e59140 (2019).
- Wessel, D., Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 138, 141-143 (1984).
- Jensen, L. J., et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, D412-D416 (2009).
- Mi, H., Muruganujan, A., Casagrande, J. T., Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 8 (8), 1551-1566 (2013).
- Cirillo, E., Parnell, L. D., Evelo, C. T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Frontiers in Genetics. 8, 174 (2017).
- Plaxton, W. C. Avoiding Proteolysis during the Extraction and Purification of Active Plant Enzymes. Plant and Cell Physiology. 60 (4), 715-724 (2019).
- Ryan, B. J., Henehan, G. T., Walls, D., Loughran, S. T. . Protein Chromatography: Methods and Protocols. , 53-69 (2017).
- Fic, E., Kedracka-Krok, S., Jankowska, U., Pirog, A., Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 31 (21), 3573-3579 (2010).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены