
Method Article
TMT Sample Preparation for Proteomics Facility Submission and Subsequent Data Analysis
In This Article
Summary
We present an optimized tandem mass tag (TMT) labeling protocol that includes detailed information for each of the following steps: protein extraction, quantification, precipitation, digestion, labeling, submission to a proteomics facility, and data analyses.
Abstract
Proteomic technologies are powerful methodologies that can aid our understanding of mechanisms of action in biological systems by providing a global view of the impact of a disease, treatment, or other condition on the proteome as a whole. This report provides a detailed protocol for the extraction, quantification, precipitation, digestion, labeling, and subsequent data analysis of protein samples. Our optimized TMT labeling protocol requires a lower tag-label concentration and achieves consistently reliable data. We have used this protocol to evaluate protein expression profiles in a variety of mouse tissues (i.e., heart, skeletal muscle, and brain) as well as cells cultured in vitro. In addition, we demonstrate how to evaluate thousands of proteins from the resulting dataset.
Introduction
The term "proteomics" was first defined as the large-scale characterization of the entire protein complement of a cell, tissue, or organism1. Proteomic analyses enable the investigation of mechanisms and cellular processes involved in disease development, therapeutic pathways, and healthy systems using techniques to perform relative quantitation of protein expression levels2. The initial descriptions of such studies were published in 1975 and demonstrated the use of two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) for this purpose1,3. The 2D method separates proteins based on charge (isoelectric focusing, IEF) and molecular mass (sodium dodecyl sulfate polyacrylamide gel electrophoresis, or SDS-PAGE)4. For years, the combination of 2D-PAGE and subsequent tandem mass spectrometry performed on each gel component was the most common untargeted protein expression analysis technique performed and identified numerous previously unknown protein expression profiles5,6. General disadvantages to the 2D-PAGE approach are that it is time-consuming, does not work well for hydrophobic proteins, and there are limitations in the total number of proteins assessed due to low sensitivity7,8.
The stable isotope labeling by amino acids in cell culture (SILAC) method became the next popular approach to identify and quantify protein abundance in samples9. It consists of metabolic labeling of cells that are incubated in medium lacking a standard essential amino acid and supplemented with an isotope-labeled version of that specific amino acid10. The advantage of this technique is its efficiency and precise labeling9. The main limitation to the SILAC approach is primarily the reduced cell growth rate caused by isotope label incorporation, which can be particularly challenging in relatively sensitive cell lines modeling human diseases11.
In 2003, a novel and robust proteomics technique involving tandem mass tag (TMT) isobaric labels was introduced to the field12. TMT labeling is a powerful method due to its increased sensitivity to detect relative protein expression levels and posttranslational modifications13. As of this publication date, TMT kits have been developed that can simultaneously label 6, 10, 11, or 16 samples. As a result, it is possible to measure peptide abundance in multiple conditions with biological replicates at the same time14,15,16. We recently used TMT to characterize the cardiac proteomic profile of a mouse model of Barth Syndrome (BTHS)17. In so doing, we were able to demonstrate widespread improvement in the cardiac profiles of BTHS mice treated with gene therapy and identify novel proteins impacted by BTHS that revealed novel therapeutic pathways involved in cardiomyopathies.
Here, we describe a detailed method to perform multiplex TMT quantitative proteomics analyses using tissue samples or cell pellets. It can be beneficial to perform the sample preparation and labeling prior to submission to a core because the labeled tryptic peptides are more stable than raw frozen samples, not all cores have experience handling all sample types, and preparing samples in a laboratory can save time for cores, which often have long backlogs. For detailed descriptions of the mass spectroscopy portion of this process please see Kirshenbaum et al. and Perumal et al.18,19.
The sample preparation protocol consists of the following major steps: extraction, quantification, precipitation, digestion, and labeling. The major benefits of this optimized protocol are that it reduces the costs of labeling, improves protein extraction, and consistently generates high-quality data. In addition, we describe how to analyze TMT data to screen thousands of proteins in a short amount of time. We hope that this protocol encourages other research groups to consider incorporating this powerful methodology into their studies.
Protocol
The Institutional Animal Care and Use Committee from University of Florida approved all animal studies.
1. Preparation of reagents
- Prepare CHAPS Lysis Buffer (150 mM KCl, 50 mM HEPES pH = 7.4, 0.1% CHAPS, and 1 protease inhibitor cocktail tablet per 50 mL of buffer). Buffer without protease inhibitors can be stored at 4 °C for up to 6 months or buffer with protease inhibitor stored at -20 °C up to 1 year.
- Prepare 100 mM triethylammonium bicarbonate (TEAB): Add 500 µL of 1 M TEAB to 4.5 mL of ultrapure water.
- Prepare 200 mM tris (2-carboxyethyl) phosphine hydrochloride (TCEP): Add 70 µL of 0.5 M TCEP, a denaturing reagent, to 70 µL of ultrapure water. Then add 35 µL of the 1 M TEAB.
- Prepare 5% hydroxylamine: Add 50 µL of 50% hydroxylamine to 450 µL of 100 mM TEAB.
2. Protein extraction
- Isolate quadriceps muscle from a euthanized mouse according to an IACUC approved protocol. Freeze and maintain at -80 °C or continue with the protocol for immediate use.
- Cut to isolate approximately 10 mg of fresh or frozen quadriceps mouse tissue. Separate fibers using tweezers when working with skeletal muscle. Alternatively, if working with cell cultures, resuspend ~3 x 106 cells in 300 µL of CHAPS lysis buffer and skip to step 2.4.
- Homogenize tissue using a bead disrupter using 2 mL tubes filled with approximately 200 µL of 1 mm zirconia/silica beads and 500 µL of CHAPS lysis buffer. Scale up or down as appropriate (e.g., 5 mg of tissue in 250 µL of CHAPS lysis buffer).
- Perform sonication (10x for 10 s each with 50% amplitude and 30 s intervals on ice) to release protein bound to DNA. The same DNA degradation results can be achieved either with syringe lysis by passing the lysate 10x through a 21 G needle attached to a 1 mL syringe, or by benzonase incubation (44 U/mL) at 37 °C for 30 min.
- Centrifuge the lysate at 16,000 x g for 10 min at 4 °C and transfer the supernatant to a new centrifuge tube.
3. Protein measurement
- Determine the protein concentration of the supernatant using established protocols (see Table of Materials).
NOTE: It is best to use samples at ≥2 µg/µL, but less concentrated samples may also be used. If a less concentrated sample is used it will be necessary to appropriately adjust the volumes of the reducing/alkylating reagents in step 5.1. - Prepare a BSA standard curve dilution using CHAPS lysis buffer.
- Follow the manufacturer's instructions, and after 15 min, read the absorbance at 750 nm.
4. Reducing/alkylating reagent treatment
- Transfer 200 µg of protein per condition into a new centrifuge tube and adjust to a final volume of 100 µL using CHAPS lysis buffer. It is possible to scale up to 200 µL when the protein concentration is too low, but do not forget to appropriately adjust the volume of reducing/alkylating reagent.
- Add 5 µL of the 200 mM TCEP and incubate samples at 55 °C for 1 h.
- Immediately prior to use, prepare 375 mM iodoacetamide by dissolving one tube of iodoacetamide (i.e., 9 mg) into 132 µL of 100 mM TEAB. Protect this solution from light.
- Add 5 µL of 375 mM iodoacetamide to the sample and incubate for 30 min at room temperature (RT) protected from light.
5. Methanol/chloroform precipitation20
- Add 400 µL of methanol to each 100 µL of protein and briefly vortex samples.
- Centrifuge at 9,000 x g for 10 s at RT. This is to incorporate liquids deposited on the sides of the sample tube.
- Add 100 µL of chloroform to the mixture and briefly vortex. Use 200 µL of chloroform if the sample has a high concentration of phospholipids.
- Centrifuge at 9,000 x g for 10 s at RT. This is to incorporate liquids deposited on the sides of the sample tube.
- Add 300 µL of water and vortex vigorously. It is important to obtain a homogenous solution.
- Centrifuge at 9,000 x g for 1 min at RT. Be extremely careful to avoid disturbing the layers when transferring the tube to a rack.
NOTE: The tube should now contain three phases: 1) Top layer (i.e., supernatant), a mixture of water and methanol; 2) Middle layer (i.e., interphase), white precipitated protein; and 3) Bottom layer (i.e., bottom phase), chloroform. - Carefully remove the supernatant.
- Add 300 µL of methanol to the remaining interphase and bottom phase. Vortex vigorously.
- Centrifuge at 9,000 x g for 2 min at RT. Be extremely careful to avoid disturbing the layers when transferring the tube to a rack.
- Carefully remove the supernatant.
- Gently aspirate as much liquid as possible under a stream of air (e.g., using a vacuum concentrator) at RT until the pellet is just a bit moist (~10 min). As the necessary time may be different for each sample, check every 2 min to assess. Store the pellet at -80 °C until further processing.
6. Protein digestion
- Resuspend the precipitated protein pellet in 100 µL of TEAB lysis buffer.
NOTE: It is optional to measure the protein concentration at this step. - Immediately prior to use, prepare 1 µg/µL trypsin by adding 100 µL of the trypsin storage solution (50 mM acetic acid) to the bottom of the 100 µg trypsin glass vial and incubate for 5 min at RT. Store remaining reagent in single-use doses at -80 °C.
- Add 2.5 µL of trypsin per 100 µg of protein. Digest the sample overnight at 37 °C. This step is crucial for complete solubilization of the protein; do not modify these conditions. Following the digestion, it is optional to measure the protein concentration using standard protein assays.
7. Peptide labeling
- Immediately prior to use, equilibrate the TMT label kit reagents to RT.
- Dissolve each of the 0.8 mg TMT tag vials through the addition of 41 µL of anhydrous acetonitrile to each tube. Incubate the reagent for 5 min at RT with occasional vortexing. Briefly centrifuge the tubes.
NOTE: A concentration of 0.8 mg of TMT tag is usually enough to label two sets. Other investigators, however, have demonstrated that this concentration can be reduced further and still yield reliable data15. - Carefully add 41 µL of the TMT label reagent to each 100 µL sample.
- Incubate the reaction for 1 h at RT.
- Add 8 µL of 5% hydroxylamine to the sample and incubate for 15 min to quench the reaction.
- Split the samples into equal amounts in a new centrifuge tube and store at -80 °C.
NOTE: In this step the samples are stable and can be submitted for mass spectroscopy. It is optional to measure concentration at this point using standard protein assays.
8. Mass spectroscopy
- Submit the samples to a proteomics facility (this study used the UF ICBR Proteomics Core facility), where all samples are combined and purified using C18 spin columns.
NOTE: Discuss with the core facility how to submit the samples prior to preparing them to confirm the exact steps they prefer for submissions. - Request the following procedures per combined multiplex sample: solid phase extraction, HPLC (SCX, SE), zip tip, and LC-MS/MS (2 h gradient for protein ID, if >10QE Plus).
- Once the data are collected, the core facility will process RAW files using vendor-supplied software for protein identification.
9. Data analysis
- Data are typically delivered from the core back to the user in the 7z format, which can require approximately 16 GB of disk space per each dataset (in this case 11 samples). For data processing, make sure a computer is available that is at least 3.4 GHz.
- Extract files using 7-Zip File Manager. These extracted files contain RAW data, pdStudy format file, and pdResultView format file. Save all files for further analyses.
- Open file using Proteome Discovery 2.2 Software.
NOTE: The file format is "File name.pdStudy". If the "File name.pdResultView" is opened it is not possible to select the control sample. - Select control samples on "Samples" panel.
- Open Result by selecting ID on "Analysis Results" panel.
- Export to spreadsheet software.
- Save raw data (all proteins identified).
- Open the spreadsheet software file. This will contain all proteins that have been identified.
- In the spreadsheet software file use the "Filter" function to screen "Protein FDR Confidence: Combined" in high (column B), "#Unique Peptides" higher than 2 (column K), and either one of "Abundance Ratio" blank exclusively (column S up to W).
- Insert a column for "p-value" calculation with the function
=TTEST(control group,experimental group, tails, type) - Insert a column for "Statistical Significance" with the function
=IF(p-value<0.05, "Significance","NS") - Use the "Filter" function to screen "Statistical Significance" showing "Significance". The result shows the analyzed proteins with the statistical significance in the control group and experimental group.
- Determine significantly higher or lower protein expression abundance in the experimental group compared to the control group, insert a column for "Regulation" with the function
=IF(AVERAGE(controlgroup)>AVERAGE(experimentalgroup),"Upregulated","Downregulated")
10. Methods to evaluate significant hits
- To identify protein-protein interactions between the significant hits identified in the TMT studies, use Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) version 11.021: https://string-db.org/
- To classify by groups (i.e., molecular function, biological processes, and protein classes) use Protein Analysis through Evolutionary Relationships (PANTHER) ontology classification software22: http://www.pantherdb.org/
- To identify protein interactions in a variety of pathways, use pathway analysis software23.
11. Proteomic data upload to a repository bank
- To submit proteomic data to the Proteomics IDEntificantions Database (PRIDE) or Mass Spectrometry Interactive Virtual Environment (MassIVE) include the following information: the peak list files (processed mass spectrum files in a standard format such as mzXML, mzML, or MGF), result files (spectrum identifications in a standard format such as mzIdentML or mzTab), and raw spectrum files (raw mass spectrum files in a nonstandard or instrument-specific format such as .RAW files or .WIFF files).
- To submit, create an account and include information such as affiliation and project details. Then, select the files listed in step 11.1 and upload them.
- To create an official dataset, run a submission workflow on these uploaded files.
NOTE: Following submission, the dataset will be private in the repository bank. With the private option, the data are only available to authorized users. There are two additional options: 1) Shared dataset, which gives access to journal reviewers and collaborators; or 2) Public dataset, which will show up in public dataset searches. Another important feature of these repositories is the ability to update the uploaded data and associate subsequent publications with the existing dataset.
Results
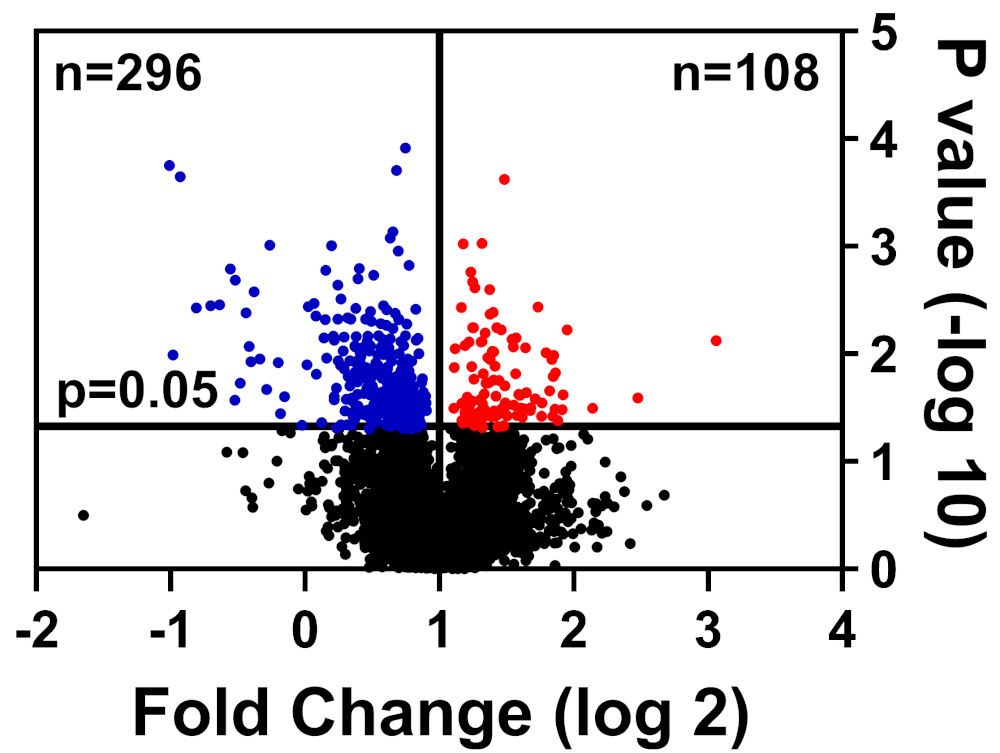
Healthy and diseased cells were lysed in CHAPS buffer, prepared as detailed in our TMT labeling method, and submitted to the University of Florida Interdisciplinary Center for Biotechnology Research (UF-ICBR) Proteomics Core for Liquid Chromatography with tandem mass spectrometry. Following data acquisition and delivery from the core, the dataset was opened in vendor-supplied software and the following cutoff filters were applied: ≥2 unique peptides, reporter ions for each protein sample present in all channels, and include only significantly altered proteins (p ≤ 0.05). Table 1 summarizes the data: 39,653 total peptides, of which 7,211 have equal or greater than two unique peptides, and 3,829 include reporter ions for all channels. The p values for these 3,829 peptides were calculated by Student's t test and p ≤ 0.05 was considered significant. In addition, a fold-change cutoff was used to determine the relative distribution of proteins from diseased compared to healthy cells: downregulated (blue) or upregulated (red) (Figure 1).
The list of significantly dysregulated protein expression was assessed using the PANTHER ontology classification system and STRING analyses. Panther analyses showed a categorized list of proteins based upon significantly lower (Figure 2A) or higher abundance in diseased cells based upon molecular function (Figure 2B). String analyses of proteins of significantly lower (Figure 2C) and higher (Figure 2D) abundance identified multiple interactions and strong associations between proteins.

Figure 1: Volcano plot displaying proteins whose abundance was not significantly altered (black), significantly lowered (blue), or significantly increased (red) in diseased vs. healthy control cells. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative evaluations of significantly dysregulated hits identified by PANTHER (A, B) and String (C, D) of significantly lower or higher abundance proteins. Please click here to view a larger version of this figure.
{kind=link}
| Total peptides | Total identified | ≥ 2 unique peptides | Quantified proteins | Significantly altered proteins | |
| Low | High | ||||
| 39653 | 7211 | 4457 | 3829 | 296 | 108 |
Table 1: Representative table of quantified proteins per dataset analysis.
Discussion
To successfully prepare samples for proteomic analysis using TMT-based isobaric stable isotope labeling methodologies, it is crucial to perform protein extractions very carefully at 4 °C and to use a lysis buffer that contains a protease inhibitor cocktail24,25. The protease inhibitor cocktail is a crucial reagent to avoid unexpected protein degradation during protein digestion. One key difference between our protocol and the current one provided by the vendor is that we strongly recommend the use of CHAPS lysis buffer based upon our experience with mammalian cells and tissues. We also suggest using a methanol/chloroform protein precipitation approach for both cell pellets and tissues.
Ideally, protein extraction, measurement, reducing/alkylating reagent treatments, and methanol/chloroform precipitations are all performed on the same day. Following this recommendation will result in more accurate protein concentrations for subsequent labeling. The protein precipitation step is important for the removal of reagents that will interfere with tandem mass spectrometry. Including the precipitation step significantly enhances the resolution of TMT26. In sum, the major advantages of our TMT protocol are the high labeling efficiencies for different types of samples, its reproducibility, and the reliable data acquired.
As the multiplex nature of this TMT untargeted proteomics strategy continues to expand, it will progressively enhance the ability of researchers across a wide variety of fields to make novel discoveries. Specifically in the biomedical field, we and others have found this technology increasingly informative in studies exploring novel mechanisms of action in disease and relative impacts of various therapeutics. For all of these reasons, this powerful technology complements the repertoire of other OMICS approaches used in modern research studies and provides key information that can guide further therapeutic development.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to acknowledge the UF-ICBR proteomics facility for their processing of our samples. This work was supported in part by the National Institutes of Health R01 HL136759-01A1 (CAP).
Materials
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium bicarbonate (TEAB), 50 mL | Thermo Fisher | 90114 | Reagent for protein labeling |
| 50% Hydroxylamine, 5 mL | Thermo Fisher | 90115 | Reagent for protein labeling |
| Acetic acid | Sigma | A6283 | Reagent for protein digestion |
| Anhydrous acetonitrile, LC-MS Grade | Thermo Fisher | 51101 | Reagent for protein labeling |
| Benzonaze nuclease | Sigma-Aldrich | E1014 | DNA shearing |
| Bond-Breaker TCEP solution, 5 mL | Thermo Fisher | 77720 | Reagent for protein labeling |
| BSA standard | Thermo | 23209 | Reagent for protein measurement |
| CHAPS | Thermo Fisher | 28300 | Reagent for protein extraction |
| Chloroform | Fisher | BP1145-1 | Reagent for protein precipitation |
| cOmplete, EDTA-free Protease Inhibitor Cocktail Tablet | Roche | 4693132001 | Reagent for protein extraction |
| DC Protein Assay | BioRad | 500-0116 | Reagent for protein measurement |
| Excel | Microsoft Office | Software for data analyses | |
| Heat block | VWR analog | 12621-104 | Equipment for protein digestion incubation |
| HEPES | Sigma | RDD002 | Reagent for protein extraction |
| Methanol | Fisher | A452-4 | Reagent for protein precipitation |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | 90058 | Reagent for protein digestion |
| Potassium chloride | Sigma | 46436 | Reagent for protein extraction |
| Sigma Plot 14.0 | Sigma Plot 14.0 | Software for data analyses | |
| Sonicator | Fisher Scientific | FB120 | DNA shearing |
| Spectra Max i3x Multi-Mode Detection Platform | Molecular Devices | Plate reader for protein measurement | |
| Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | 23275 | Reagent for protein measurement |
| Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay | Thermo Fisher | 23290 | Reagent for protein measurement |
| Thermo Scientific Proteome Discoverer Software | Thermo Fisher | OPTON-30945 | Software for data analyses |
| TMT 10plex Isobaric Label Reagent Set 0.8 mg, sufficient reagents for one 10plex isobaric experiment | Thermo Fisher | 90110 | Reagent for protein labeling |
| TMT11-131C Label Reagent 5 mg | Thermo Fisher | A34807 | Reagent for protein labeling |
| Water, LC-MS Grade | Thermo Fisher | 51140 | Reagent for protein extraction |
References
- Graves, P. R., Haystead, T. A. Molecular biologist's guide to proteomics. Microbiology and Molecular Biology Reviews. 66 (1), 39-63 (2002).
- Erdjument-Bromage, H., Huang, F. K., Neubert, T. A. Sample Preparation for Relative Quantitation of Proteins Using Tandem Mass Tags (TMT) and Mass Spectrometry (MS). Methods in Molecular Biology. 1741, 135-149 (2018).
- O'Farrell, P. H. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 250 (10), 4007-4021 (1975).
- Rabilloud, T., Lelong, C. Two-dimensional gel electrophoresis in proteomics: a tutorial. Journal of Proteomics. 74 (10), 1829-1841 (2011).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 1 (5), 376-386 (2002).
- Anderson, N. G., Anderson, N. L. Twenty years of two-dimensional electrophoresis: Past, present and future. Electrophoresis. 17 (3), 443-453 (1996).
- Haynes, P. A., Yates, J. R. Proteome profiling-pitfalls and progress. Yeast. 17 (2), 81-87 (2000).
- Bunai, K., Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 815 (1-2), 227-236 (2005).
- Sury, M. D., Chen, J. X., Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Molecular & Cellular Proteomics. 9 (10), 2173-2183 (2010).
- Zhang, G., Neubert, T. A. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods in Molecular Biology. 527, 79-92 (2009).
- Wang, X., et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Scientific Reports. 8 (1), 8441(2018).
- Thompson, A., et al. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 75, 1895-1904 (2003).
- Cheng, L., Pisitkun, T., Knepper, M. A., Hoffert, J. D. Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods in Molecular Biology. 1355, 53-70 (2016).
- Navarrete-Perea, J., Yu, Q., Gygi, S. P., Paulo, J. A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research. 17 (6), 2226-2236 (2018).
- Zecha, J., et al. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Bachor, R., Waliczek, M., Stefanowicz, P., Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 24 (4), E701(2019).
- Suzuki-Hatano, S., et al. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Molecular Therapy - Methods & Clinical Development. 13, 167-179 (2019).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. (40), e1954(2010).
- Perumal, N., et al. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. Journal of Visualized Experiments. (144), e59140(2019).
- Wessel, D., Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 138, 141-143 (1984).
- Jensen, L. J., et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, D412-D416 (2009).
- Mi, H., Muruganujan, A., Casagrande, J. T., Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 8 (8), 1551-1566 (2013).
- Cirillo, E., Parnell, L. D., Evelo, C. T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Frontiers in Genetics. 8, 174(2017).
- Plaxton, W. C. Avoiding Proteolysis during the Extraction and Purification of Active Plant Enzymes. Plant and Cell Physiology. 60 (4), 715-724 (2019).
- Ryan, B. J., Henehan, G. T. Protein Chromatography: Methods and Protocols. Walls, D., Loughran, S. T. , Springer. New York. 53-69 (2017).
- Fic, E., Kedracka-Krok, S., Jankowska, U., Pirog, A., Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 31 (21), 3573-3579 (2010).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved