Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Fluorescencia Basado Técnica de extensión del cebador para determinar la transcripción puntos de partida y los sitios de escisión de RNasas
En este artículo
Resumen
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
Resumen
Fluorescencia de extensión del cebador basado (FPE) es un método molecular para determinar los puntos de partida de la transcripción o sitios de procesamiento de moléculas de ARN. Esto se consigue mediante la transcripción inversa del ARN de interés usando cebadores marcados con fluorescencia específicos y el análisis posterior de los fragmentos de ADNc resultantes mediante electroforesis en gel desnaturalizante de poliacrilamida. Simultáneamente, una reacción tradicional de secuenciación de Sanger se ejecuta en el gel para mapear los extremos de los fragmentos de ADNc a sus bases correspondientes exactas. En contraste con 5'-RACE (amplificación rápida de extremos de ADNc), donde el producto debe ser clonado y secuenciado múltiples candidatos, la mayor parte de fragmentos de ADNc generados por la extensión del cebador se puede detectar simultáneamente en una carrera gel. Además, todo el procedimiento (de la transcripción inversa para el análisis final de los resultados) se puede completar en un día laborable. Mediante el uso de cebadores marcados con fluorescencia, el uso de isótopos radiactivos peligrosos reactivos marcadosse pueden evitar y los tiempos de procesamiento se reducen como productos pueden ser detectados durante el procedimiento de electroforesis.
En el siguiente protocolo, se describe un método de extensión de cebador fluorescente in vivo para detectar de forma fiable y rápida los extremos 5 'de los ARN para deducir puntos de partida de la transcripción y sitios de procesamiento del ARN (por ejemplo, por componentes del sistema toxina-antitoxina) en S. aureus, E. coli y otras bacterias.
Introducción
La extensión del cebador 1 es un método molecular para determinar los extremos 5 'de moléculas de ARN específicas hasta una resolución una base. La ventaja de otros métodos tales como 5'-RACE (amplificación rápida de extremos de ADNc) es el tiempo de respuesta rápido y la capacidad de analizar fácilmente una mezcla de diferentes longitudes de moléculas de ARN.
Este método funciona sometiendo moléculas de ARN para revertir reacciones de transcripción utilizando cebadores fluorescentes específicas, generando fragmentos de ADNc de ciertas longitudes. Estas moléculas de ADNc se ejecutan junto tradicionales reacciones de secuenciación Sanger 2 en geles de poliacrilamida desnaturalizantes y pueden ser detectados por su fluorescencia debido a la utilización de cebadores marcados con fluorescencia. Las longitudes de los fragmentos de ADNc se evalúan por comparación con la escalera de secuenciación, lo que permite el mapeo de los extremos 5 'de ARN.
Tradicionalmente, las reacciones de extensión de cebadores se utilizan en combinacióncon isótopos radioactivos para detectar moléculas de ADNc de películas de rayos X. Debido a los peligros de salud, problemas de eliminación de residuos y la facilidad de manejo, nuevos protocolos utilizan la fluorescencia para la detección de la extensión del cebador con secuenciadores automatizados, aunque su sensibilidad es ligeramente inferior. El uso de cebadores marcados con fluorescencia, el procedimiento marcado radiactivo recurrentes se puede omitir, como cebadores fluorescentes son estables durante un largo tiempo (más de un año en nuestras manos).
El método se describe aquí utiliza un secuenciador automatizado de gel, pero con ligeras modificaciones, secuenciadores capilares puede también ser utilizado para la separación y detección de cDNA 3. La naturaleza paralela de análisis en gel hace que sea posible detectar incluso una pequeña cantidad de escisión de ARN o de procesamiento. Otra ventaja es la alta resolución de este método, como terminal de división o transformación de una sola base puede ser detectado.
En lo que se refiere a la detección de la escisión de ARN o el procesamiento, typically dos diferentes tipos de extensiones de imprimación se distinguen. En un caso, el tratamiento enzimático se realiza in vitro usando ARN purificado y enzima purificada, mientras que en el otro caso, el procesamiento se realiza in vivo y el ARN resultante se purifica. En ambos casos, el ARN se somete a una extensión del cebador lleva a cabo in vitro, sin embargo, dependiendo de la fuente de ARN, ya sea el método se llama una extensión del cebador vivo in vitro o in. En el protocolo que presentamos aquí, nos centramos únicamente en la extensión del cebador en vivo, debido a la facilidad de uso (no hay proteínas purificadas necesario) y la posibilidad de determinar los puntos de partida de la transcripción y procesamiento a la vez. Sin embargo, in vitro extensiones de cebadores son, en principio, establecer la misma manera y este protocolo pueden servir como punto de partida.
El método ilustrado aquí se puede aplicar a muchas especies bacterianas mientras que son susceptibles de altopreparación pureza y alto rendimiento de los ácidos nucleicos.
La investigación en nuestro laboratorio se centra en el alcance de la regulación de la toxina-antitoxina (TA) sistemas de 4,5, un campo en el que se utiliza ampliamente el método de extensión del cebador. TA-sistemas son pequeños elementos genéticos presentes en los genomas de procariotas que constan de una proteína tóxica estable y endógena activa y una proteína o ARN antitoxina en su mayoría inestables que contrarresta la toxicidad 6,7. Actividad de la toxina a veces se ejerce mediante la inhibición de la replicación, síntesis de la pared celular u otros mecanismos, pero más a menudo por la actividad de RNasa 8,9. Típicamente, la especificidad de RNasa se determina mediante la realización de diferentes pruebas, uno de los cuales es el método de extensión del cebador. Reacciones de extensión de cebador son muy adecuados para esta aplicación, como una mezcla de fragmentos de longitud escindidos y completo se puede analizar de forma simultánea para determinar sus extremos 5 '. Usando una mezcla de in vitro e in vivo extensiones de cebadores, latoxina RNasa escisión específica, por ejemplo, la especificidad de secuencia se puede determinar 10-13.

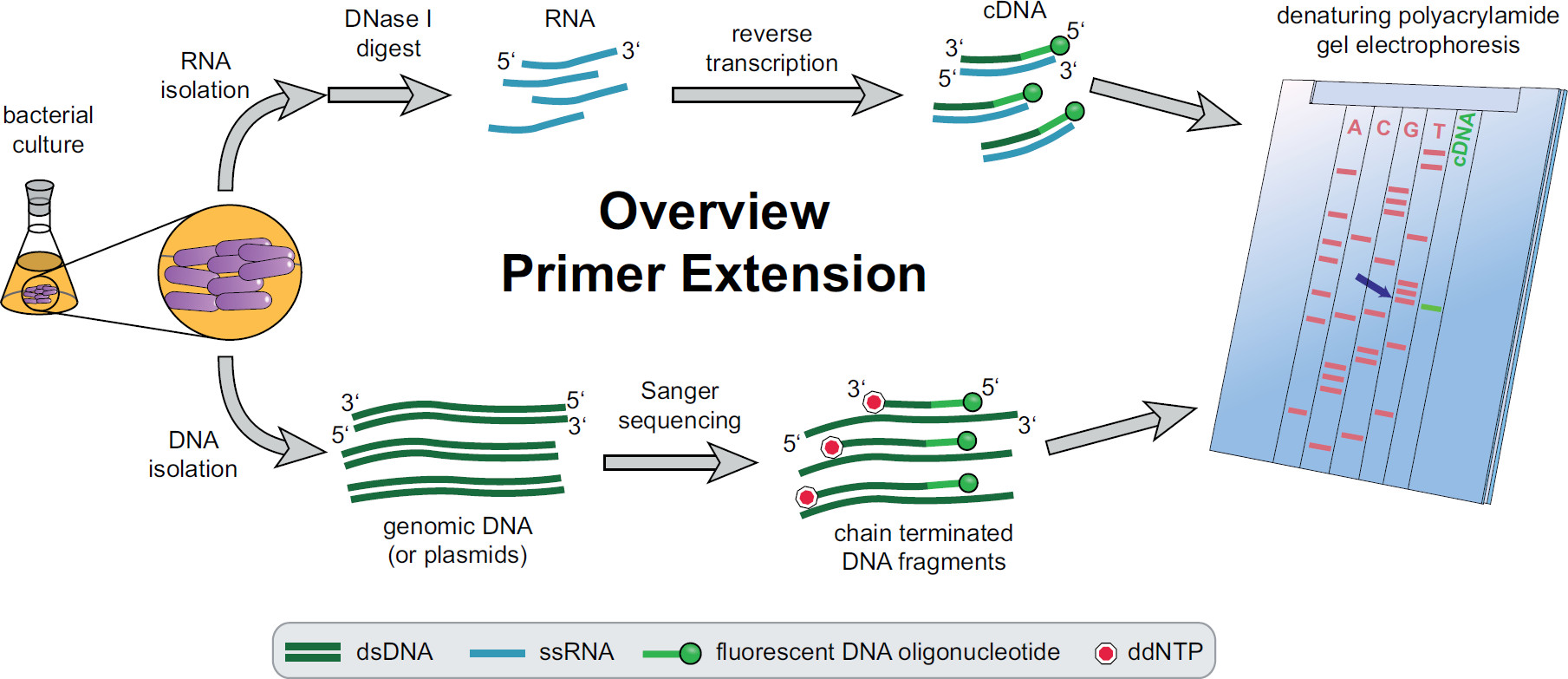
Figura 1. Descripción general del procedimiento de extensión del cebador. Los cultivos bacterianos se incubaron y tratados de acuerdo con las necesidades experimentales. El ARN total se extrajo de las células, se trató con DNasa I para eliminar las trazas de ADN y se sometieron a una reacción de transcripción inversa usando cebadores fluorescentes específicos diana de ADN rendimiento de ADNc. El ADN genómico o plásmidos se extrajeron y posteriormente utilizarse para reacciones de secuenciación Sanger fluorescentes para la comparación de tamaño con los fragmentos de ADNc. Productos de extensión del cebador se ejecutan junto a los productos de secuenciación de Sanger en un gel de poliacrilamida desnaturalizante de urea y se analizaron con un láser automatizado y el microscopio. La base de la secuencia que se alinea con la banda de ADNc es la last base del extremo 5 'de ADNc (flecha azul). Más información en Fekete, et al. 3 Haga clic aquí para ver una versión más grande de esta cifra.
{kind=link}
Una visión general de todo el procedimiento de extensión del cebador se puede encontrar en la Figura 1. Brevemente, las células bacterianas se cultivan, cosechado, el sedimento de células se lisaron y el ARN extraído. ARN purificado se trata a continuación con DNasa I para eliminar trazas de moléculas de ADN que podrían actuar como plantillas para la transcriptasa inversa. Cebadores fluorescentes específicos se añaden a la ARN, hibridado a la región de interés y posteriormente a transcripción inversa, lo que resulta en ADN complementario monocatenario (ADNc). Una escalera de secuenciación se crea mediante secuenciación Sanger tradicional empleando cebadores fluorescentes y se separó en un gel de poliacrilamida desnaturalizante junto de los fragmentos de ADNc de extensión del cebador. La resultantegel se analizó mediante la comparación de las bandas fluorescentes, lo que permite la identificación de los extremos 5 'de interés. Puntos de partida de la transcripción y sitios de procesamiento son luego evaluados individualmente por comparación de secuencias.
Protocolo
1. Alto Rendimiento Preparación de ARN
- Aislamiento de ARN
NOTA: Las altas concentraciones de ARN total se necesitan para la reacción de extensión del cebador. Kits columna de centrifugación por lo general no producen la cantidad de ARN necesaria (~ 5-16 g en 5 l de volumen). Por lo tanto se recomienda la purificación utilizando el método de extracción de tiocianato-fenol-cloroformo de guanidinio ácido, se describe a continuación.
NOTA: El fenol es cancerígeno, tóxico y corrosivo. Por favor, lea las hojas de datos de seguridad de los materiales y el uso bajo una campana de humos con la protección adecuada!- Crecer o tratar las células bacterianas (S. aureus o E. coli en este ejemplo) como se desee y la cosecha por 10 min de centrifugación a 4600 × g y 4 ° C. Nota: Por lo general se cosecha un total de 600 OD de 20 - 70. Los sedimentos celulares se pueden almacenar durante varias semanas a -20 ° C.
- Resuspender el sedimento celular en 1 ml de solución de tiocianato-fenol-cloroformo de guanidinio ácido y transferir a una2 ml de tornillo taza contiene 0,5 ml de 0,1 mm perlas de cristal de zirconio / sílice.
- Lisar las células tres veces en un batidor de preparación rápida / perlas de 6,5 m / s durante 30 s durante tres rondas, el enfriamiento de las muestras en hielo durante 5 minutos después de cada carrera. Nota: muestra homogeneizada se puede almacenar a -80 ° C durante varias semanas.
- Incubar lisado durante 5 min a RT y luego añadir 200 l de cloroformo.
- Agite vigorosamente o vórtice de la muestra durante 30 segundos para extraer ARN.
- Incubar a temperatura ambiente durante 3 min centrifugar durante 15 min a 13.000 - 15.000 × g y 4 ° C. Nota: Los disolventes se separan en una fase orgánica inferior (rosa, contiene proteínas), una interfase (blanco, contiene ADN) y una fase acuosa superior (claro, contiene ARN).
NOTA: A partir de este paso en el uso sólo RNasa reactivos libres y artículos de plástico! - Preparar fresco 1,5 ml tubos de reacción, etiqueta RNasa libre de manera adecuada y añadir unos 500 l de isopropanol 100% de RNasa libre de cada uno (utilizar aproximadamente la misma voLume como la fase acuosa en el tubo anterior).
- Sostenga el tubo en un ángulo y transferir la fase acuosa (aproximadamente 500 l) para los tubos preparados usando RNasa consejos gratis. No moleste a la interfase.
- Precipitar el RNA invirtiendo varias veces e incubando durante 10 min a TA.
- Centrifugar las muestras durante 15 min a 13.000 - 15.000 × g y 4 ° C y retirar el sobrenadante mediante pipeteado o aspiración (bomba de chorro de agua y la botella de alimentación). No perturbar el sedimento de ARN blanco transparente en la parte inferior.
- Añadir 1 ml de etanol 70-80% de RNasa libre (no hacer vórtice) para lavar. Nota: ARN en etanol se puede almacenar durante varias semanas a -20 ° C.
- Centrifugar durante 5 min a 7500 × g y 4 ° C y desechar el sobrenadante con la pipeta o preferiblemente la aspiración.
- Aire seco sedimento de ARN por 15 - 30 minutos bajo la campana de humos. No secar en exceso, de lo contrario gránulos pueden ser difíciles de disolver.
- Resuspender el precipitado en 50 l de RNasa libre de ddH 2 O o tampón de almacenamiento de ARN.
- Medir la concentración de ARN con una microvolumen UV-Vis o una cubeta de cuarzo (y fotómetro convencional) y proceder a la digestión DNasa I.
- DNasa I digestión de ARN para eliminar las trazas de DNA
NOTA: Dado que el ADN puede actuar como una plantilla espuria en la reacción de transcripción inversa (extensión del cebador), debe ser retirado de la muestra. Varios métodos para la eliminación de ADN a partir de soluciones de ARN están disponibles, que generalmente se basan en la digestión DNasa. Un método eficiente simple pero efectivo y el costo para la extracción de ADN se describe a continuación.- Precaliente el baño de agua a 37 ° C.
- Mezclar los compuestos enumerados en la Tabla 1 en un tubo de 1,5 ml de reacción.
- Incubar la mezcla durante 1 hora a 37 ° C en un baño de agua, y luego proceder directamente a la extracción con fenol / cloroformo. Nota: No se recomienda la inactivación por calor de la DNasa I, ya que esto podría degradar el ARN.
- Extracción con fenol / cloroformo de ARN después de DNasa I digestión
NOTA: El ARN debe ser purificada para eliminar los nucleótidos libres, fragmentos de ADN y los componentes del tampón de la DNasa I digestión. Extracción con fenol / cloroformo permite una alta recuperación y concentración de la muestra de ARN y por lo tanto se describe a continuación. Otros métodos para la purificación de ARN también pueden ser utilizados, si cumplen con estos requisitos.- Dividir la DNasa I digestión 500 l mezclar en dos muestras de 250 mu l en 2 ml tubos de reacción.
- Añadir 1 volumen (250 l) de solución ácida P / C / I (en agua saturada de fenol, cloroformo y isopentanol, relación de 25: 24: 1, pH 4,5-5).
NOTA: P solución / C / I es cancerígeno, tóxico y corrosivo. Por favor, lea las hojas de datos de seguridad de los materiales y el uso bajo una campana de humos con la protección adecuada! - Vigorosamente vórtice o lugar en una plataforma de agitación en vórtex durante 1 - 3 minutos.
- Centrifugar durante 30 min a 13.000 - 15.000 × g y 4 ° C.
- Recoger la fase superior (acuosa) y transferir a un tubo nuevo (250 l).
- Añadir 1/9 de volumen (28 l) de acetato de sodio 3 M pH 5,2.
- Añadir 2,5-3 volúmenes de etanol puro (700 l).
- Mezclar mediante agitación breve y lugar a -80 ° C durante 30 min o a -20 ° C por 2 - 3 horas. Si es necesario, guarde el ARN O / N a -20 ° C.
- Centrifugar durante 30 - 60 min a 13.000 - 15.000 × g y 4 ° C.
- Aspirar el sobrenadante con la pipeta o la aspiración.
- Lavar la pella mediante la adición de 1 ml de etanol al 70% en el sedimento. No te muestra vórtice.
- Centrifugar la muestra durante 5 min a 13.000 - 15.000 × g y 4 ° C.
- Aspirar el sobrenadante con la pipeta o la aspiración.
- Aire pellet seco bajo la campana de humos. Guarde el sedimento a -20 ° CO / N si es necesario.
- Disolver el precipitado en 30 l DEPC tratada con H 2 O por agitación durante 2 min y utilizar esta solución para disolver el PEllet del segundo tubo correspondiente por muestra (30 l solución por un par de extracción).
- Medir la concentración de ARN, y asegúrese de que sea superior a 1 mg / l para el uso en la extensión del cebador de ARNm expresadas medianamente.
- Si es necesario, almacenar el ARN a -20 ° C durante varias semanas hasta algunos meses.
2. cebador de extensión Reacción
- Primer diseño
NOTA: En el diseño de cebadores para un experimento de extensión del cebador, obedecer las directrices generales de diseño de cebadores de PCR (ver el manual que acompaña al secuenciador gel automatizado para obtener más información y discusión sección de este documento).- Específicamente, asegúrese de que los cebadores (i) no contienen tramos de bases, (ii) poseer una G o C en el extremo 3 ', (iii) tener un GC equilibrada: AT ratio, (iv) tienen una temperatura de recocido de alrededor de 55 - 60 ° C y (v) se unen al menos 50 pb, 100 pb mejor aguas abajo de la región de interés para recibir imágenes claras.
- Reacción de extensión del cebador
NOTA: La reacción de extensión del cebador (síntesis de ADNc) requiere grandes cantidades de ARN de plantilla. Si se eligen las cantidades de ARN que se utilizan para bajo, la señal puede ser demasiado baja para detectar! Por lo tanto recomendamos la purificación del ARN como se describe anteriormente.
NOTA: PRECAUCIÓN: El uso de RNasa reactivos libres y utensilios de plástico !!!- Precalentar el termociclador a una temperatura de 95 ° C y llevar a cabo todos los pasos adicionales de incubación en un termociclador para la facilidad de uso y reproducibilidad.
- Mezclar los compuestos de la Tabla 2 en un tubo de PCR para cada muestra de ARN.
- Desnaturalizar las muestras durante 1 min a 95 ° C.
- Colocar los tubos en el hielo y el frío durante 5 min para hibridar ARN y cebadores.
- Ajuste la máquina de PCR a 47 ° C.
- Mientras tanto preparar la mezcla maestra de transcripción inversa como se describe en la Tabla 3.
- Añadir 4 l de mezcla maestra de transcripción inversa para cada ARN hibridadomuestra.
- Incubar los tubos durante 1 hora a 47 ° C. Nota: La temperatura óptima para AMV RT es 42 ° C, las temperaturas más altas sin embargo ayudan a superar las estructuras secundarias de las moléculas de ARN.
- Detener la reacción por calentamiento de las muestras a 95 ° C durante 2 min.
NOTA: La formamida es corrosivo, tóxico y puede ser perjudicial para el feto. Por favor, lea las hojas de datos de seguridad de los materiales, manejar con cuidado y use protección adecuada! - Añadir 6 l de colorante de carga de formamida (95% (v / v) de formamida desionizada, EDTA 10 mM, 0,05% (w / v) de azul de bromofenol) y tienda para O / N dos semanas a -20 ° C en la oscuridad.
3. Preparación de la escalera de secuenciación
NOTA: La reacción escalera secuenciación requiere ya sea una cantidad moderada de plásmidos o grandes cantidades de ADN genómico. Siempre que sea posible, se recomienda el uso de plásmidos en la reacción de secuencia debido a la facilidad de aislamiento y de alta sigintensidad nal. En otros casos, se utiliza rutinariamente un método adoptado de Marmur 5,14 para preparar ADN genómico de E. coli y S. aureus células sin la necesidad de utilizar fenol. En principio cualquier método que produce grandes cantidades y pureza del ADN genómico se puede utilizar.
- Aislamiento de ADN genómico
- Crecer 10 ml de E. coli o S. células aureus O / N en LB, BM 5 o medio TSB.
- Cosecha células por centrifugación durante 10 min a 4600 × g en un tubo Falcon de 15 ml.
- Pellet se resuspendió en 2 ml de tampón P1 como se encuentra en algunos kits de preparación de los mini (Tris-HCl 50 mM pH 8,0, EDTA 10 mM, 100 mg / ml de RNasa A).
- Lisar las células durante 45 - 60 min con 20 - 40 l lisostafina (0,5 mg / ml, el almacenamiento a -20 ° C). Nota: Para E. células de E. coli el pretratamiento enzimático puede o bien omitirse o lisozima utilizado.
- Añadir 100 l de SDS-solución saturada (en 45% de etanol) a la suspensión y incubate durante 5 min a 37 ° C.
- Añadir 650 l 5 M NaClO 4 y las células de vórtice brevemente.
NOTA: El cloroformo es un carcinógeno potencial. Por favor, lea las hojas de datos de seguridad de los materiales y el uso bajo una campana de humos con la protección adecuada !!! - Añadir 3 ml de cloroformo / isopentanol (24: 1) a la mezcla y se agita durante al menos 60 segundos. Nota: El líquido debe convertirse en una emulsión blanca homogénea.
- Muestra Centrifugar durante 10 min a 4600 × g y RT para separar las fases.
- Transferir cuidadosamente la fase superior transparente (acuosa) a un tubo nuevo. Si la solución está turbia, repetir la extracción con cloroformo / isopentanol. Medir el volumen de la solución de ADN y prepare un tubo nuevo con 2 volúmenes de etanol (100%).
- Lentamente decantar o una pipeta la solución de ADN en el etanol que contiene el tubo. Nota: ADN debería precipitar como transparentes, bobinas densas en la parte inferior o cuando está completamente deshidratado como un racimo blanco flotante.
- Recuperar la DNA usando ganchos hechos de pipetas Pasteur de vidrio (Figura 2) y se lava dos veces cada muestra mediante inmersión en un tubo individual de 1 ml de etanol de 70%.
- Coloque los ganchos en posición vertical en un bastidor y secar al aire el sedimento durante 60 min. Si es necesario, almacenar el ADN se secó durante varios días a temperatura ambiente.
- Disolver el ADN por la ruptura de los ganchos de ADN cubierta de vidrio y colocar en un tubo de reacción de 2,0 ml que contiene 100 - 500 l ddH 2 O. Ajuste el volumen a una concentración final de ADN de 1.000 - 1.500 ng / l. Para una reacción de secuenciación, utilizar 10 - 18 g de ADN genómico.

Figura 2. Instrucción sobre cómo crear una barra de pesca de ADN. Sostenga la punta de una pipeta Pasteur de vidrio en la llama de un mechero Bunsen. Esto hace que el vidrio para iniciar la fusión después de varios segundos, creando un pequeño gancho en tél termina. Quite rápidamente del fuego y dejar enfriar durante 1 min.
- Aislamiento del plásmido
- Preparar plásmidos usando kits Mini Preparación estándar y disolver en tampón de elución (10 mM Tris-Cl, pH 8,5). Dependiendo del tamaño del plásmido, utilice 100 - 500 ng de plásmido para una escalera de secuenciación.
- Reacción de secuenciación de Sanger
NOTA: Encuentra debajo de un protocolo simple que utiliza un kit de secuenciación del cebador marcado con fluorescencia con 7-deaza-dGTP que funciona bien para el propósito de las extensiones de los cebadores. Consulte el manual del kit de secuenciación para obtener información detallada. Tenga en cuenta que la reacción de secuenciación debe utilizar el mismo cebador como la reacción de extensión del cebador para crear productos de la misma longitud.- Mezclar 12 l de ADN genómico (~ 10 - 15 g) con 1 l de DMSO y 1 l de cebador marcado con fluorescencia (2 pmol / l).
- A cada 1 l de las cuatro mezclas de reacción de secuenciación (A, C, G o T), añadir 3 l de la ADN / DMSO / mezcla de cebadores.
- Coloque las muestras en una máquina de PCR, y ejecute el siguiente programa de PCR: 95 ° C durante 2 min; 35 ciclos de 95 ° C durante 20 seg, 54 ° C durante 20 s, 70 ° C durante 30 s; mantener a 4 ° C para siempre.
- Después de la carrera, retirar las muestras de la máquina, añadir 6 l de colorante de carga y almacenar en hielo (a corto plazo) oa -20 ° C durante varios días o semanas.
4. Configuración del gel y Aparato Run
NOTA: La información detallada sobre cómo se monta el aparato de gel de secuenciación, el gel se prepara el gel y cómo se ejecuta se puede encontrar en el protocolo del fabricante.
- Preparativos
- Preparar 10x TBE como se indica en la Tabla 4.
- En el día de la carrera gel preparar 1 L de tampón 1x TBE con ultrapura ddH 2 O.
- Preparar 10% (w / v) APS. Nota: Puede ser almacenado en 200 ml de alícuotas a -20 ° C durante varios meses, pero la actividad puede disminuir con el tiempo <./ Li>
- Asamblea de cámara de colada de gel
- Evite el polvo y la pelusa entre las placas de vidrio. Superficies de trabajo tanto limpiar a fondo utilizando toallitas húmedas.
- Limpiar un par de placas de vidrio de 25 cm utilizando toallas de papel desechables y agua destilada en ambos lados y luego isopropanol para el lado interior de las placas de vidrio.
- Coloque 0.25 mm espaciadores en la placa de vidrio trasero y baje la placa de vidrio con muescas en la parte superior (Figura 3).
- Coloque los rieles de gel a ambos lados de las placas de vidrio con el extremo con muescas y los pilotos de entrada ferrocarril hacia arriba y apriete las perillas a la ligera.

Figura 3. Vista de las placas de vidrio de electroforesis en gel de despiece. Las placas de vidrio se deben utilizar direccionalmente. Tenga cuidado de hacer frente a la cara interior de las placas de vidrio hacia el interior y el exteriorlado hacia el exterior.

Figura 4. Vista de un aparato de gel montado. Después de inyectar la solución de gel, el bolsillo espaciador se coloca en la solución de entre las placas de vidrio. La placa de fundición se desliza entonces entre la placa de cristal frontal y los carriles de gel y se fija apretando las perillas de ferrocarril.
- Lanzar el gel
NOTA: acrilamida no polimerizada es neurotóxico! Por favor, lea las hojas de datos de seguridad de los materiales y su uso con la protección adecuada !!!- Añadir los compuestos enumerados en la Tabla 5 en un vaso de precipitados y mezclar usando una barra de agitación y un agitador magnético.
- Inmediatamente después de la adición de APS y TEMED, tomar la solución de gel en una jeringa de 50 ml y colocar un filtro de 0,45 nm en la punta.
- Cualquiera de sujetar el borde superior de la placa de vidrio con una mano o colocar la sandwich en una colada de gel de pie para crear una pendiente en ángulo 10-20 °.
- Lentamente dispensar la solución de gel entre las placas de vidrio mientras que se mueve continuamente la punta de la jeringa de un lado al otro y detener una vez que la solución de gel se encuentra con el extremo inferior.
- Mover a un lado o eliminar por completo las burbujas de forma usando un gancho de burbujas.
- Deslice el espaciador bolsillo gel (0,25 mm) entre las placas de vidrio en el extremo con muescas, se sumergen en la solución de gel y fijar por fijación de la placa de colada.
- Tornillos de los rieles superior Sujete ligeramente (Ver Figura 4 para el aparato completamente montado).
- Deje conjunto gel para 1 - 2 horas.
- Retire la placa y el bolsillo de fundición espaciador y limpiar el bolsillo de los residuos de sal y gel.
- Enjuague con ddH2O y limpie el exceso de solución con papeles de seda.
- Correr y visualizar el gel
NOTA: Los geles de secuenciación se sometieron a electroforesis directamente en el generador de imágenes de gel, mientrasla fluorescencia se detecta simultáneamente por un microscopio láser. En contraste a electroforesis en gel convencional, en el que el gel se ejecuta primero y luego teñidas y se visualizó, la unidad de detección se fija y escanea las bandas en tiempo real a medida que pasan el láser. A continuación un procedimiento para el software de recogida de datos ImagIR en OS / 2 se esboza, que puede ser adoptada para las versiones más recientes. Para obtener más información, consulte el manual de usuario.- Deslice el soporte de depósito de inercia en los carriles de gel sobre las placas de vidrio del frente y apriete las perillas.
- Coloque gel en el depósito de gel inferior del generador de imágenes de gel automatizado contra la placa de calentamiento y fijar deslizando el piloto de entrada ferroviaria en los soportes de aparatos.
- Rellene tampón TBE 1x en las cámaras de amortiguación de gel inferior y superior, cierre la cámara intermedia inferior y conecte la cámara de amortiguación superior a la corriente mediante el cable de alimentación.
- Si está presente, limpiar la bolsa de gel de salinas residuos pipeteando repetidamente búfer en el bolsillo.
- Cierre la cámara de la parte superior del tanque buffer usando la tapa de amortiguación superior.
- Cierre la puerta de la máquina y encienda el reproductor de imágenes y la computadora e iniciar el software de colección de datos Base ImagIR.
- Crear un nuevo archivo de proyecto (Archivo-> Nuevo ...), introduzca un nombre de archivo del proyecto, seleccione los rangos de láser apropiados (700 o 800 nm) y confirme con OK.
- Seleccione Opciones-> Auto ganancia ... en el menú de la imagen en la parte superior, haga clic en Auto para iniciar la medición de ganancia automática y aceptar la configuración haciendo clic en Aceptar.
- Enfoque el láser seleccionando Opciones-> Enfoque ... en el menú de control del escáner, haga clic en el botón Auto y aceptar la configuración haciendo clic en Aceptar.
- Repita el procedimiento automático de ganancia para ajustar a la región recién centrado.
- Configuración del control del escáner de acuerdo con estos valores: 2000 V, 35 mA, 45 W, 45 ° C, el filtro de exploración: 3, velocidad de escaneo: 3.
- Ejecución previa de la gel de vacío durante 20 minutos (SELECT en tensión y pulse ).
- Mientras tanto, calentar la escalera de secuenciación y los productos de extensión de cebadores en una máquina de PCR durante 2 min a 90 ° C, a continuación, enfriar en hielo.
- Detener la electroforesis, abrir el secuenciador automático de gel y retirar la tapa del tanque de amortiguación superior.
- Insertar el tiburón de dientes de peine entre las placas de vidrio y Pierce ligeramente el gel con los dientes de tiburón (véase la Figura 5).

Figura 5. Vista de primer plano de gel con peine de dientes de tiburón. Muestra (púrpura) se aplica entre los dientes de tiburón.
- Pipetear ya sea 1 - 2 l de los productos de extensión de escalera o reacciones de secuenciación del cebador en cada bolsillo de gel (formado por los dientes de tiburón).
- Si se necesitan no todos los bolsillos, llenar los bolsillos vacíos con colorante de carga para evitar que encomportamiento de marcha constante.
- Cerrar el depósito y la puerta del secuenciador gel buffer.
- Iniciar electroforesis y encienda láser (Seleccione Voltaje ON y Laser ON y pulse ).
- Deje de electroforesis vez región de interés ha pasado el láser.
Resultados
Como se representa en la figura 6, una reacción de extensión del cebador se puede utilizar para determinar los puntos de partida de la transcripción de las transcripciones de interés y puede ayudar a deducir regiones promotoras (normalmente identificado por -10 y -35 elementos). El fragmento de ADNc más alta (más larga) representa el extremo 5 'del ARNm y por lo tanto se puede asignar fácilmente cuando se compara con la escalera de secuenciación.
Discusión
Extensión del cebador fluorescente es un método sencillo y rápido para determinar los extremos 5 'de los ARN, ya sea para TSP- o identificación procesamiento del ARN secundaria. Debido a la utilización de cebadores fluorescentes, las reacciones se pueden configurar y ejecutar sin las precauciones de seguridad adicionales (a diferencia de en el caso de cebadores marcados radiactivamente). Como las muestras son detectados por fluorescencia, que pueden obtenerse imágenes, mientras que la electroforesis está en p...
Divulgaciones
The authors have nothing to disclose that would present a conflict of interest.
Agradecimientos
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
Materiales
| Name | Company | Catalog Number | Comments |
| Name | Supplier | Catalog Number(s) | Comment |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
Referencias
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados