
Method Article
Rapid Subtractive Patterning of Live Cell Layers with a Microfluidic Probe
In This Article
Summary
We present a protocol to perform subtractive patterning of live cell monolayers on a surface. This is achieved by local and selective lysis of adherent cells using a microfluidic probe (MFP). The cell lysate retrieved from local regions can be used for downstream analysis, enabling molecular profiling studies.
Abstract
The microfluidic probe (MFP) facilitates performing local chemistry on biological substrates by confining nanoliter volumes of liquids. Using one particular implementation of the MFP, the hierarchical hydrodynamic flow confinement (hHFC), multiple liquids are simultaneously brought in contact with a substrate. Local chemical action and liquid shaping using the hHFC, is exploited to create cell patterns by locally lysing and removing cells. By utilizing the scanning ability of the MFP, user-defined patterns of cell monolayers are created. This protocol enables rapid, real-time and spatially controlled cell patterning, which can allow selective cell-cell and cell-matrix interaction studies.
Introduction
In their native environment, cells in biological tissues perceive a range of biochemical and physical cues directing their growth, organization, and development. Understanding these cues requires selective investigation of cell-cell and cell-matrix interactions. This necessitates the development of methods for patterning cell monolayers. Methods to geometrically separate different cell types in culture (patterning) enable broad studies of physical and chemical cues in cell biology. Most of the current approaches for patterning cell layers1-4 depend on depositing cell-adhesion proteins on surfaces or using microfabricated stencils for selective growth on substrates. In contrast, here we present a method to rapidly pattern cell monolayers in situ, i.e., cells in culture, by removing cells in selected regions of the monolayer. Methods that perform such subtractive patterning5-9 usually require specialized substrates, surface treatment, complex operation, physical contact or ablation using a laser, inadvertently affecting the live cells. We here use a microfluidic probe (MFP)10,11, a non-contact scanning microfluidic technology that hydrodynamically confines a liquid on a substrate. One important component of the MFP is the microfabricated head containing microchannels (Figure 1). The associated platform consists of syringe pumps for liquid control, stages for scanning control and an inverted microscope for visualization and feedback (Figure 2). In its basic configuration, the MFP head comprises two microchannels with apertures at the apex, one for injecting a processing liquid and the other for aspirating the injected processing liquid together with some immersion liquid (Figure 3A). During MFP operation, the apex is at a fixed distance from the substrate. When the aspiration flow rate (Qa) is sufficiently higher than the injection flow rate (Qi), i.e., Qa:Qi≥ 2.5, the processing liquid is confined on the substrate. This results in a hydrodynamic flow confinement (HFC). The region in which the processing liquid is in direct contact with the substrate is termed the footprint. For typical operating conditions, the flow of the processing liquid within the HFC is characterized by a low Reynolds number (Re ≈ 10-2) and a high Péclet number (Pe ≈ 102). This implies liquid flows in laminar regime with convection being the primary mode of mass transfer of chemical species. The numerical and analytical models for flow confinement are described elsewhere12-14.
In this paper, we use the approach of simultaneously confining multiple processing liquids, which is called hierarchical HFC (hHFC)14. To implement hHFC with the MFP, two additional apertures are needed to provide a secondary source of injection and aspiration. This enables us to confine one liquid within a second liquid. The inner (processing) liquid benefits from being shielded from stray debris on the substrate by the outer (shielding) liquid. In addition, hHFC allows operation in two modes: (i) the nested mode, in which the processing liquid in the inner HFC contacts the surface (Figure 3B), and (ii) the pinched mode, in which the inner liquid loses contact and only the outer liquid is in contact with the substrate (Figure 3C). Switching between the two modes allows users to effect or stop processing the substrate and is achieved by controlling the head-to-surface gap or by changing the ratio between the injection flows (Qi2/Qi1). For a given channel geometry, the footprint of the processing liquid on the substrate can be controlled by modulating flow conditions (Figure 3D). Sodium hydroxide (NaOH) is used as the inner processing liquid to lyse the cells, and the lysate is continuously aspirated from the surface. Because the chemical effects of the processing liquid are localized to the inner HFC footprint, the adjacent cells remain unperturbed, which allows spatiotemporal studies of cell-cell interactions. The scanning functionality of the MFP allows the creation of user-defined geometries of cell patterns (Figure 4). Further, the choice of NaOH as processing liquid accommodates downstream DNA analysis (Figure 7).
Protocol
1. MFP Head and Platform Cleaning and Preparation
NOTE: This protocol uses vertically-oriented silicon-glass hybrid MFP heads15,16. The silicon component of the head contains the microchannels, which are etched to a depth of 100 µm. The etched silicon is bonded to glass using anodic bonding. The channel design comprises a pattern consisting of six channels, two each for injection and aspiration, and two for injecting the immersion liquid (Figure 1). The channels used for injecting immersion liquid replenish the media surrounding the biological sample, thus avoiding losses due to aspiration and evaporation. The inner channels and outer channels used in the current work are 100 × 100 µm and 200 × 100 µm respectively. Post fabrication and processing, MFP heads with clear channels and polished apexes are obtained.
- Preparation of the pumping station and fluid-handling apparatus
- Use capillaries with a 1/16'' (1.59 mm) outer diameter and 0.02'' (0.51 mm) inner diameter for all tubings connected to the syringes. Vary capillary size based on application and obtain the appropriate connectors and fittings to interface the MFP head with the syringes. Use ultrapure water wherever dilutions are necessary.
- Clean syringes (250 - 500 µl volume) and syringe plungers by sonication in 0.5% bleach solution in ultrapure water prior to- and post- live cell experiments. Rinse them thoroughly with water. Fill them with water by immersing their tips completely in a water bath and aspirating using the plunger. Purge the liquid while within the water bath with the syringe shaft contacting the seal at the exit of the syringe. Repeat aspirate and purge until no air bubbles are seen in the syringe columns.
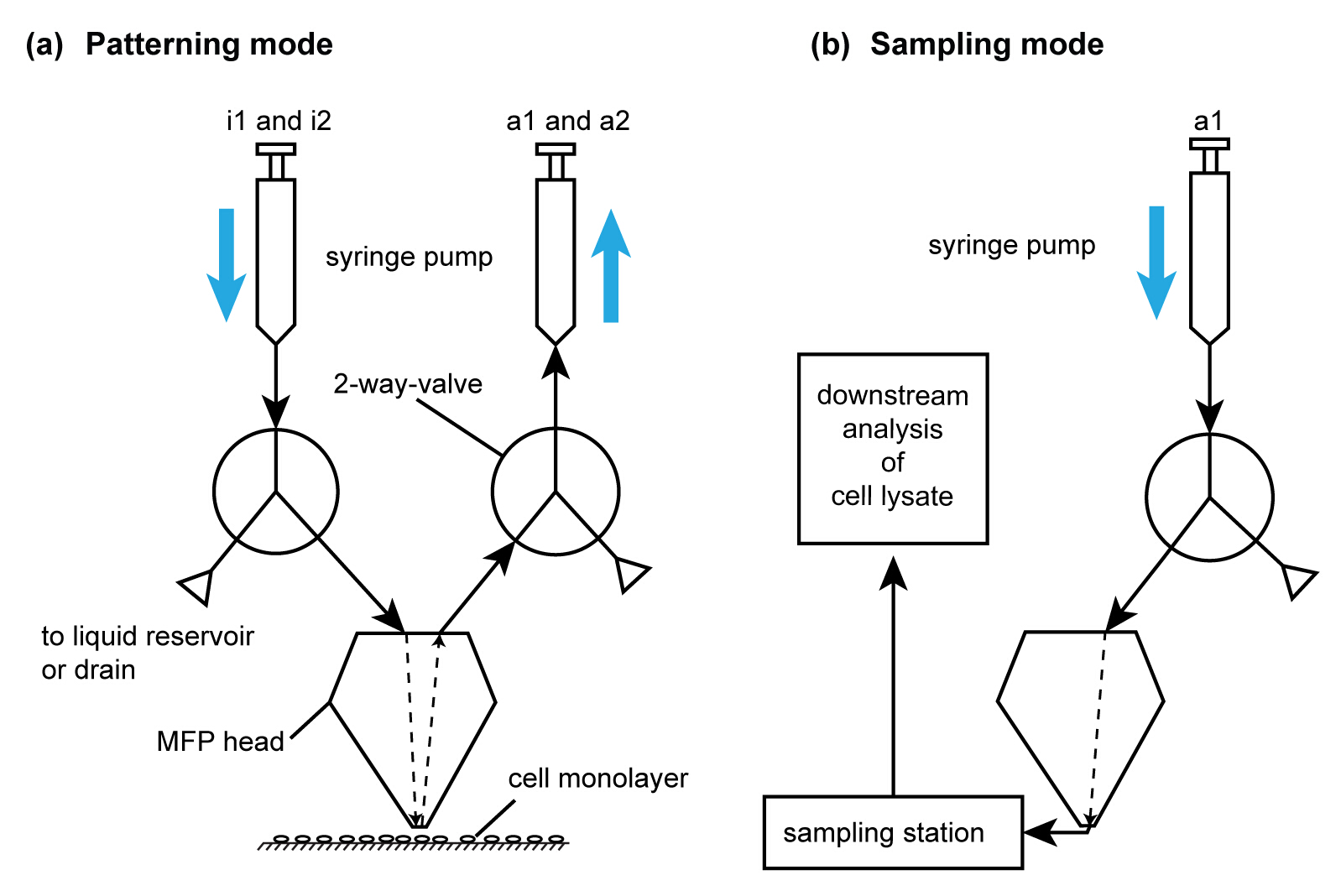
- Connect filled syringes to the syringe pumps using Luer-lock connectors. Use a switch valve mounted on the pumps to direct the liquid from the syringe to one of two capillaries leading either to the MFP head or to the liquid reservoirs (Figure 6). (If no switch valve is available see section 1.4.4). Purge both capillaries with water from the syringes at a flow rate of about 10 - 50 µl/min, depending on the size of the syringe until about 10 µl of water is left in the syringes.
- Pre-insert the purged capillaries into an appropriate microfluidic connector/adaptor which interfaces with the channel vias in the head (e.g., M1 connector — see materials).
- MFP head preparation
- Clean the MFP head by sonication using glassware detergent for standard cleaning or 0.5% bleach for stringent cleaning, for 5 min. Purge channels with water by immersing the apex in water and applying vacuum to the vias.
- Inspect channels under a stereomicroscope for potential obstructions (clogging) and repeat the previous step if necessary.
- Mount the clean head on the head holder and screw the connector with the pre-inserted and purged tubing onto the head. Screw the head holder to the Z-stage, which is used for the control of gap distance between the head and the cell monolayer.
- Calibrating the scanning stages of the MFP platform
- Perform endpoint calibration of the X-, Y- and Z-stages prior to connecting the head to the platform, according to manufacturer's protocol with an appropriate software interface. Calibrated stages ensure accuracy in positioning the MFP head.
- Obtain a crude zero gap distance (zeroing) by bringing the MFP head over a chamber slide without cells and slowly descend in 5 µm steps. Upon probe contact with the substrate, Newton's rings should be observed. This is a crude estimate. An accurate position is to be obtained after adjusting coplanarity of the probe apex to the substrate.
- To ensure coplanarity of the probe apex, adjust the tilt of the head using a goniometer (at the interface of the head and the Z stage). When formed ensure that the Newton's rings are symmetric (Figure 1). Move the MFP head 20 µm away from the substrate and adjust tilt using the goniometer. Repeat descend, zeroing and tilt adjustment until the Newton's rings are symmetric on contact. With tilt adjusted, set the z position which produces symmetric Newton rings as zero.
NOTE: The Z-stage controls the head-to-substrate distance, whereas the X-Y stage controls the scanning of the substrate (Figure 2). A detailed explanation can be found in our earlier work14.
- Chemical preparations for local removal of cell layers
Caution: Where appropriate, use safety equipment (e.g., nitrile gloves, safety glasses) for the chemicals being prepared. Use a fume hood if necessary for preparing solutions. Prepare all chemicals and buffers with ultrapure water as the diluent.- Prepare 50 mM NaOH solution as the processing liquid for the inner injection channel (i2 with flow QI2 in Figure 1).
- Prepare the extraction buffer solution required for the outer injection (i1 with flow QI1 in Figure 1), composed of 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Tween 20, and 10 µM rhodamine B in 50 mM Tris(hydroxymethyl)aminomethane at pH 8. For purging, use water in the aspiration syringes.
- Filter all solutions using a 0.2 µm syringe filter. Degas all filtered solutions using a desiccator until the dissolved air stop bubbling to the surface.
- Using the drain line connected to the syringe pumps, purge the remnant water and aspirate the degassed solutions into the injection syringes at 40 µl/min until the syringes are full (250 or 500 µl). This ensures bubble-free filling of the syringes, connectors, and capillaries. The syringe–switch valve–two-capillary system allows filling of the syringes mid-experiment. In the absence of such a switch valve, disconnect the capillaries from the head and fill the capillaries and the syringe by aspirating the degassed solutions before reconnecting them to the head.
- Aspirating the processing and shielding liquids through the capillaries allows for the use of small volumes during operation. If the chemicals can be prepared in large volumes, fill syringes with the required solutions directly. For example, the aspiration syringes that contain water can be filled using this approach.
- Purge the capillaries to the MFP head with the liquids allocated for each channel as defined in 1.4.1 and 1.4.2.
NOTE: The extraction buffer solution shields the NaOH in the inner confinement and the rhodamine component in the buffer facilitates the visualization of the hHFC during operation. - If the cells are over-confluent, with cell aggregates appearing on the cultured surface, supplement the extraction buffer with 10% Proteinase K. This supplements the action of NaOH on these aggregates by dissolving denatured proteins as the lysate enters the aspiration channels. This prevents clogging of the channels during operation.
- Preparation of cell monolayers on chamber slides
Caution: Use a cell culture hood for culturing the cells and handle equipment as per regulations set by the biosafety officer of the laboratory. Note special requirements of specific cell lines and adapt protocol and equipment accordingly.- Use cell culture incubators (at 5% CO2 and at 37 ºC) for culture and expansion of cells to be patterned. Perform the expansion using standard cell culture protocols17,18 in T flasks. Use culturing media as per requirements of the specific cell line (e.g., serum and antibiotic supplemented DMEM for culturing MCF7 and MDAMB 231).
- On reaching cell confluence in the culture flasks, trypsinize and collect cells and seed 2×105 cells/cm2 in each of the 2-chamber slides for patterning and culture over 48 hr. Use the cells in one of the chambers as a control for cell growth and viability.
- On reaching cell confluence on the chamber slides, incubate the cells for 45 min with 500 µl cell-tracker dye solution (e.g., green - CMFDA or orange - CMRA) at 10 µM concentration prepared in serum-free medium. This is done for cell visualization during patterning. Wash the labelled cells with PBS by gently flushing each chamber using a pipette, and subsequently culture the cells in serum-supplemented media for the patterning experiments.
- Obtain a reference image of the cell-tracker-stained cell surface for the purpose of cell counting. This is done to ensure confluency of the cell layer. In addition, it serves as an aid for quantification experiments if sample recovery and DNA analysis are objectives.
NOTE: In case that cell viability is to be assessed during patterning, the cells can be stained with a Live/Dead cytotoxicity kit in accordance to the manufacturer's instructions.
2. Creating the Hierarchical Hydrodynamic Flow Confinement (hHFC)
- Move the MFP over the cell monolayer to a gap distance of 50 µm from the glass slide. This gap distance while ensuring contact of the hHFC also accounts for monolayer surface and thickness variation.
- Inject NaOH at 6 or 8 µl/min through i2. Evaluate other flow rates (i.e., QI1, QA1 and QA2) using the flow rules in Figure 3.
- Modulate the size of the inner HFC by changing the ratio of QI2/QI1 using the injection syringes. For example, use a QI1 between 1.3 µl/min and 4 µl/min, with the QI2 fixed at 8 µl/min, which results in a NaOH footprint of 150 - 300 cells (100 - 200 µm2/cell) (Figure 3D).
- Inject complete medium from the outer-most apertures on the MFP head at a flow rate of 20 µl/min to account for evaporation of the media and aspiration during the operation of the hHFC.
3. Patterning Cell Monolayers Using hHFC
Note: The scanning pattern determines the areas of the cell monolayer where the cells are extracted (subtractive patterning), leaving the remaining cells to study specific biological questions. This pattern can be straight lines or an array of spots, for example. Complex patterns require design of a suitable scanning trajectory. For example, a checkered scanning trajectory provides a grid of cell areas (shown for example in Figure 4A), which would enable studying the effect of different stimuli on cells in different squares while being in close proximity. These patterns can be created using control over the X-Y stages of the platform, where the control software allows scripting of scan trajectories for the MFP head over the cell monolayer.
- Set the stage software to scan the probe head over the cell monolayer in user-defined patterns (by setting X, Y and Z coordinates) at a scan velocity of 10 µm/s at a gap distance of 50 µm.
- With the nested hHFC in operation and in contact with the monolayer, scan the MFP with a trajectory of the desired pattern to effect patterned cell removal.
- For a co-culture after the first cell type removal, seed a different cell line to fill the gaps, using methods described in section 1.5. Move between nested and pinched modes (by increasing gap distance, for example) to control cell removal.
NOTE: The scan velocity may have to be investigated for other cell lines. The scan velocity used in this demonstration effects complete cell removal over the scanned regions for the used cell lines.
4. Downstream Processing for Sampling and DNA Amplification
- Prepare the sampling station for downstream analysis of the lysate. For the sampling station, use a 3D printed 8-strip PCR tube holder. Alternatively, choose an appropriate tube holder to serve as this adaptor, which is capable of being mounted within the scanning range of the head outside the substrate. Wipe the tube holder with 70% ethanol or other surface decontaminants based on the stringency desired for the application. Use a magnetic clip on the tube holder to attach it onto the substrate holder.
- After preparing the sampling station, position the MFP head 100 µm from the monolayer. Begin operating the hHFC with 50 mM NaOH solution as the processing liquid for the inner injection channel with flow rates of 1, 6, -7 and -17.5 µl/min for QI1, QI2, QA1 and QA2 respectively.
- Once the flow confinement has stabilized (in approximately 10 sec), descend the probe head to a gap distance of 50 µm to perform NaOH based local lysis of the chosen sub-population of cells with the hHFC. Once lysis of the sub-population is complete (approximately 30 sec per footprint), direct the head towards the tubes in the sampling station. Eject the collected lysate into the PCR tubes directly (Figure 6).
- For downstream processing of the lysate, first neutralize the pH of the solution by mixing the lysate with Tris buffer (1:1). After neutralization, heat the lysate to 95 ºC for 30 min. Then directly load the lysate into a standard qPCR workflow as set by supplier of the instrument.
Results
The described protocol for rapid subtractive patterning of cell monolayers is demonstrated using a multi component MFP platform (Figures 1 and 2). The protocol employs the hierarchical hydrodynamic flow confinement (hHFC; Figure 3) to locally treat and remove cells from cell monolayers, using NaOH as the processing liquid. The hHFC configuration comprises an inner HFC and an outer HFC. The confined NaOH in the inner hHFC introduces chemical action and shear on the cells in contact. Within this footprint, the cells are homogeneously exposed to the chemical action of NaOH, owing to convection driven mass transfer within and with negligible diffusion to the region outside the confinement. The shear on the cells on the other hand, can be altered by varying the flow rate of NaOH. To simplify operational parameters, we chose to make chemical action the dominant mechanism of cell removal in comparison to the shear. To estimate the shear force applied by the hHFC on the surface, a finite-element model was built using Comsol Multiphysics 5.0. Simulations were run using the CFD module for laminar flows. Two inlet boundary conditions to the injection apertures of the geometry and two outlet boundary conditions to the aspiration apertures (Figure 5) were applied for the range of flow rates and aperture dimensions used in the current demonstration. In the model obtained, the flow rules dictated the shear profile, whereas the flow rates defined the magnitude of the shear on the surface. Practically, a combination of both determines if the hHFC contacts the surface. Keeping these factors in mind, we set out to find a range of flow rates for operation in order to obtain chemically dominant cell removal. To create an hHFC, we use the flow rule of a total aspiration to injection flow rate ratio of 3.5. The other flow rules used were defined to minimize clogging within the aspiration channels, which can be caused by denatured proteins sticking to the channel surfaces. Using the developed model, we found flow rates from 5 to 10 µl/min translating to a shear stress between 1 and 3 N/m2. Without the chemical effect of NaOH, for instance in the case of the extraction buffer, the shear stress would not be high enough to remove the cells19. Within the observed range, we note that operation at higher flow rates is more practical owing to perturbations in the flow path at low aspiration flow rates (i.e., QI2 < 4 µl/min) due to cell debris in the channels.
Considering the studied shear profile and practical considerations, NaOH flow rates (QI2) of 6 and 8 µl/min are used for the patterning experiments and QI1, QA1 and QA2 according to the flow rules shown in Figure 3. The ratio of injection flows (QI2/QI1) allows us to further modulate the size of the hHFC footprint (Figure 3D and Figure 4B), with the underlying principle elaborated by Autebert et al.14. Using the liquid-shaping ability of the hHFC coupled with high-resolution scanning ability of MFP platform, we demonstrate live-cell grid generation at multiple scales and furthermore show the application of the given protocol in developing co-cultures (Figure 4C).
The platform also allows us to perform sample lysate retrieval for downstream analysis. To show the quality of the obtained lysate, we sampled locally lysed cells from one and five footprints in two independent experiments, showing the variation in quantities of DNA obtained from the lysate (Figure 7). Here, we amplified DNA contained in the lysate using β-actin primers (forward: GGATGCAGAAGGAGATCACT and reverse: CGATCCACACGGAGTACTTG) using 4 µl of the neutralized lysate in each PCR reaction.

Figure 1. Modules of the MFP platform and the head. (A) The operational modules of the platform comprise motorized syringes, motorized stages and a controller. The MFP is connected to a motorized Z-stage to control the gap distance between the head and the substrate, and the substrate holder is attached to the X- and Y-motorized stages constituting the scanning system. (B) The MFP head has fluidic vias to connect the pumping station, mounting holes to mount the head onto the Z-stage, and channels that exits the polished apex. The apex is set coplanar to the cell culture substrate. Symmetric Newton's rings can be observed when the apex and the substrate are coplanar and in contact. Please click here to view a larger version of this figure.
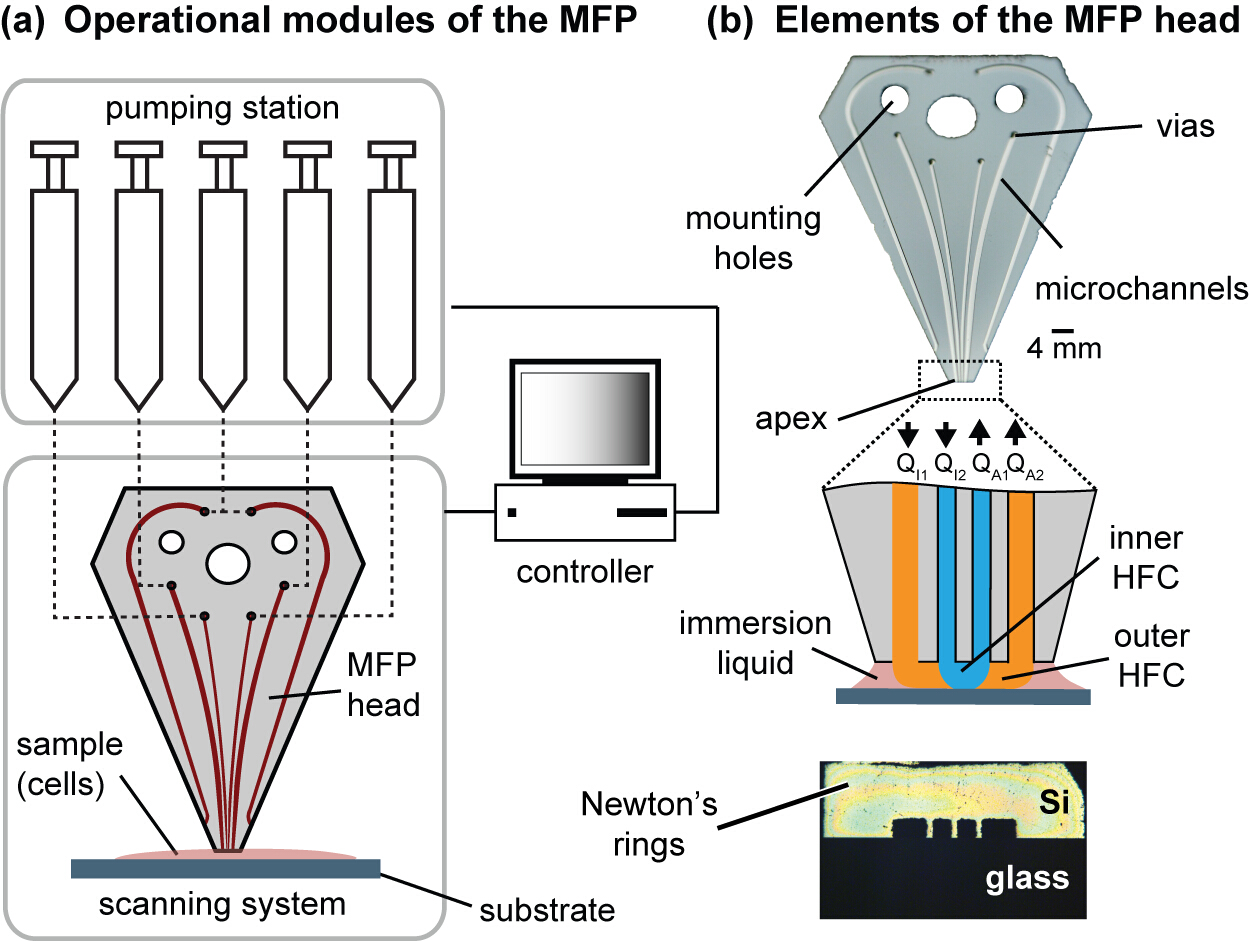{kind=link}

Figure 2. MFP platform. The high-resolution scanning platform is equipped with a machined head holder interfacing with the high-precision motorized Z-stage. The substrate holder is connected to the X-Y stage for scanning purposes. For recovery of the lysate for downstream analysis, a 3D printed sampling station is clipped magnetically to the side of the substrate holder (shown in inset). Syringe pumps, an inverted microscope, controllers and displays are located around the platform. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Hierarchical hydrodynamic flow confinement (hHFC) for spatial and temporal control of cell removal. (A) Schematic of a single HFC. (B) Nested and (C) pinched mode of hHFC operation. (D) Image of the footprint for two different injection/aspiration flow ratios. Please click here to view a larger version of this figure.
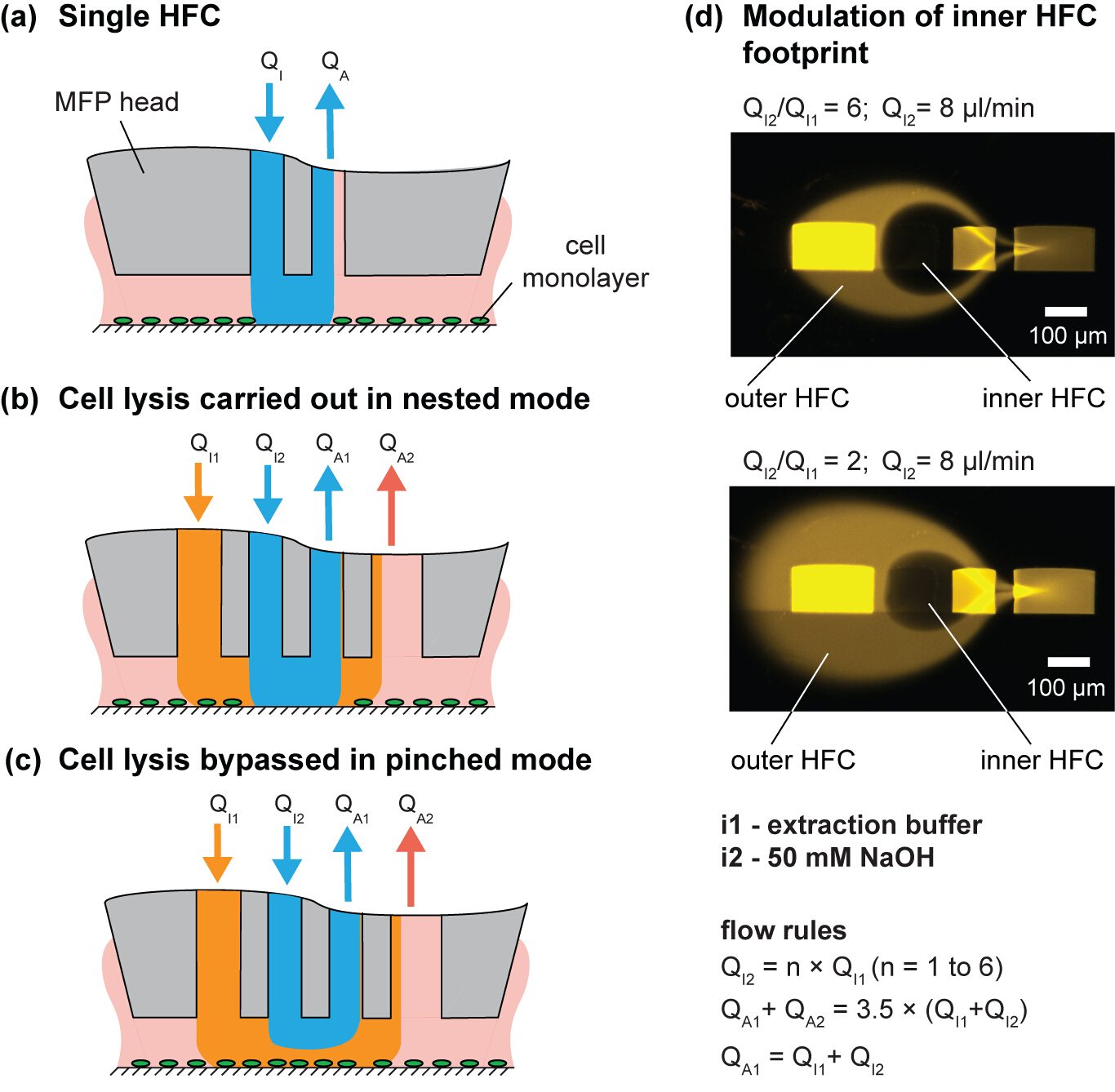{kind=link}

Figure 4. Patterns of cell monolayers using the MFP. (A) Cell-grid generation by programmed scanning of the MFP on a MDA-MB-231 cell monolayer. Cells were stained with green cell-tracker dye. (B) The footprint for the different injection ratios (n) on an MCF7 cell monolayer. The schematic shows the expected variation in the shape of the inner HFC with a change in n. (C) Patterned co-culture by subtractive patterning of MCF7 monolayer followed by seeding MDA-MB-231 cells in the subtracted regions. Please click here to view a larger version of this figure.
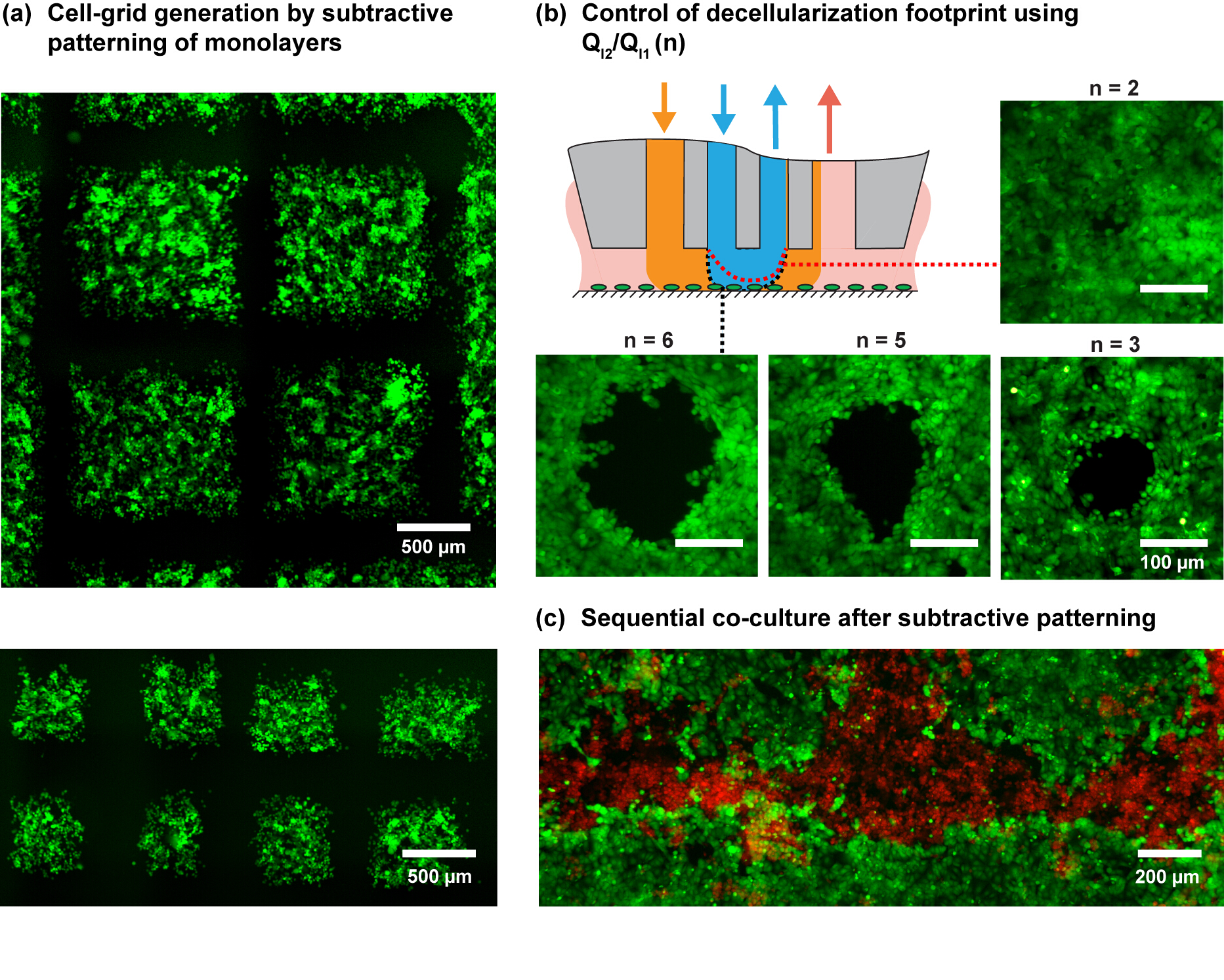{kind=link}

Figure 5. Shear stress on the surface when applying hHFC. The shear stress on the surface increases linearly with the inner injection flow rate. The highest shear point is found between the two inner apertures (bottom right inset), where the processing liquid is confined (red flow lines, top left inset). The aperture dimensions used in the finite-element model are 200, 100, 100 and 200 µm for i1, i2, a1 and a2, respectively. Please click here to view a larger version of this figure.
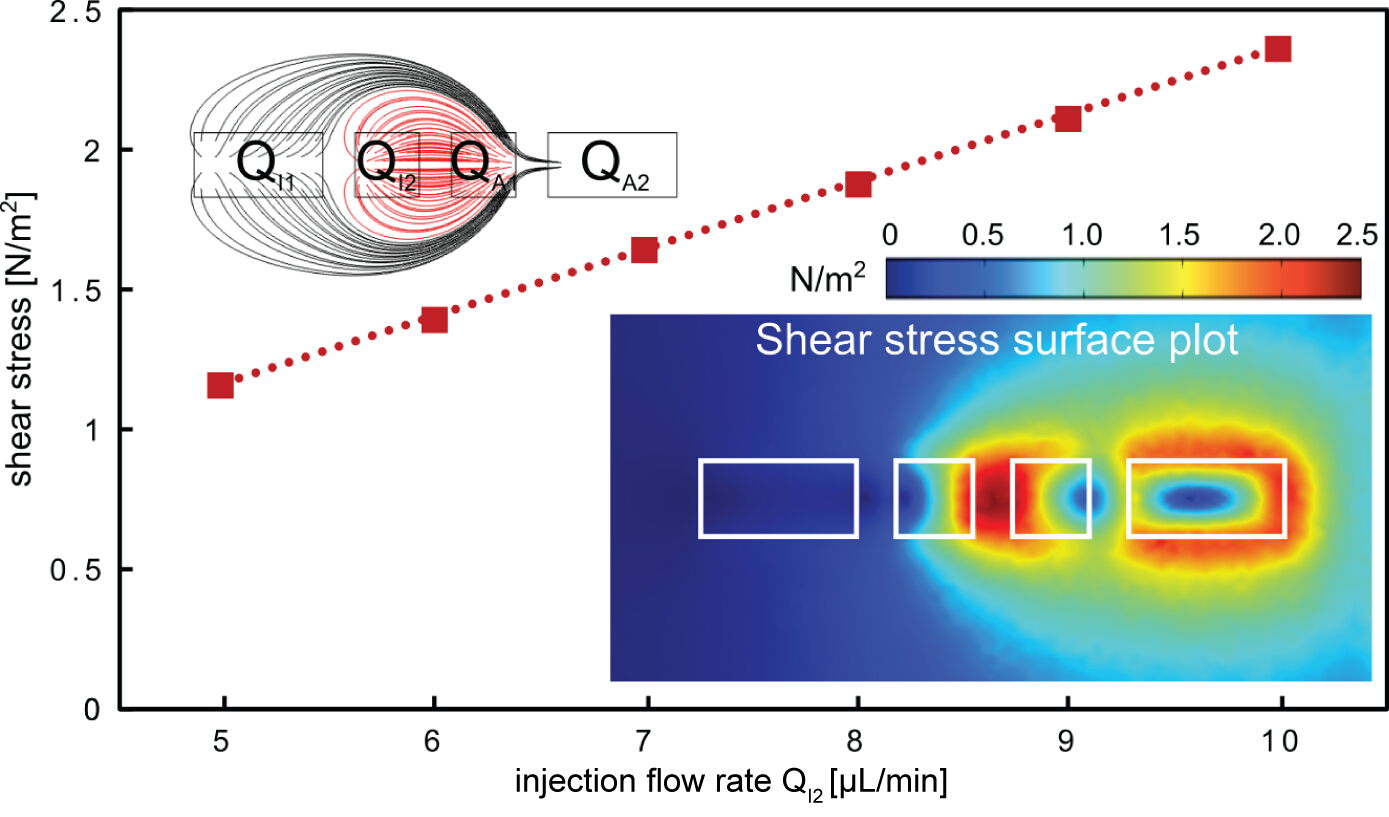{kind=link}

Figure 6. Operating modes of the MFP platform to perform cell removal and patterning. (A) Schematic of the flow path for subtractive patterning by cell lysis. For simplicity, only one of each injection and aspiration flow paths are shown. The syringes are filled using the drain valve in the pumps. i1 and i2 are used for injecting extraction buffer and NaOH, respectively. (B) Schematic of flow path for lysate recovery. This flow path is activated after collection of cell lysate for analysis using the flow path in (A). Please click here to view a larger version of this figure.
{kind=link}

Figure 7. Downstream analysis of DNA from cell lysate using qPCR. (A) Amplification plots of DNA in lysate extracted from 5 footprints (5 fp) and 1 footprint (1 fp). Controls were extracted after lysate collection for both cases. (B) Melt curves of DNA amplified from the lysate showing the quality of the extracted DNA. qPCR was performed (N = 2, n = 3) to amplify the β-actin gene. Please click here to view a larger version of this figure.
{kind=link}

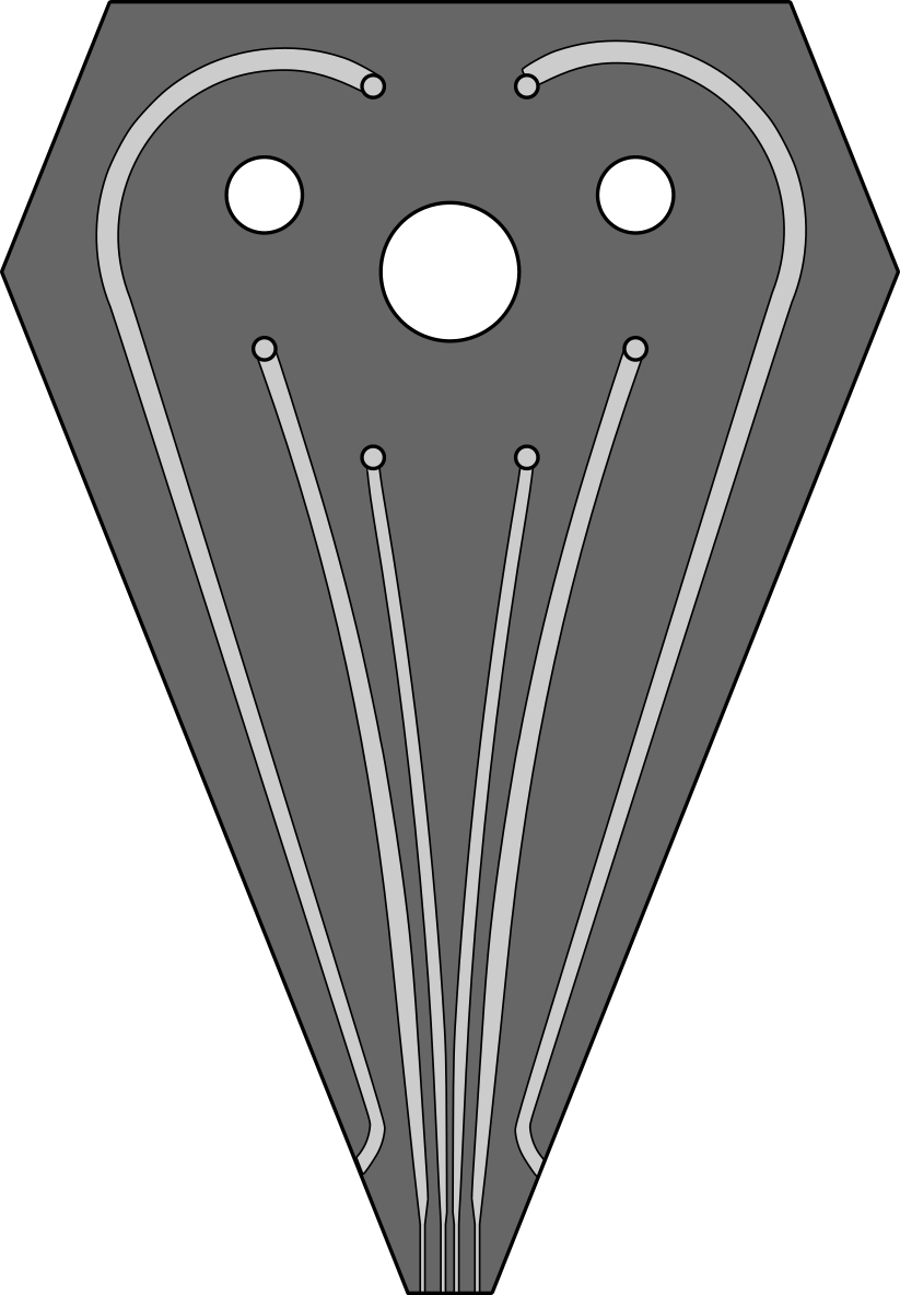
Figure S1. A scaled image of channel design for the 6-channel MFP head used for patterning experiments. Channels performing the hHFC are 200, 100, 100, 200 µm wide, and 100 µm deep. The two outermost channels, which replenish immersion liquid are 500 µm wide and 100 µm deep. A GDS file for the same design has been provided as a supplementary to this article. Please click here to view a larger version of this figure.
{kind=link}
Discussion
We present a versatile protocol for the subtractive patterning of cell monolayers that enables rapid generation of spatially defined cell patterns. Selective lysis of cell monolayers is performed using NaOH as the processing liquid in the hHFC. The NaOH instantaneously denatures the proteins contained in the cell membrane. We recommend the use of 50 mM concentration of NaOH to ensure downstream processing of the lysate. This concentration can be increased for more rapid cell removal, provided lysate downstream processing is not an objective. The hHFC ensures that the unprocessed surrounding areas of the cell monolayer remain unaffected and are available for either expanding the pattern or probing other properties of the cells.
The rate of cell removal is a function of both the chemistry and shear presented by the flow. We choose an operating range of flow rates to have chemistry dominant cell removal. Scanning velocities of 10 - 20 µm/sec using a NaOH injection flow rate of 6 - 8 µl/min are used to facilitate 'rapid' subtractive patterning. The rapid rate of cell removal addresses some challenges faced by surface printing based patterning approaches20,21. These methods require a mechanism to limit growth of cells in spatially defined areas, for example, by selective deposition of cell adhesion proteins via micro-contact printing and subsequent seeding of cells to obtain the pattern. They are limited by low throughput and require several sequential steps for pattern generation as well as a new stencil/stamp for each pattern. The described method does not require selective growth of cells on defined areas, and overcomes several limitations by performing subtractive patterning in contrast to printing, while not requiring any additional treatment of the cell culture substrate.
With the protocol outlined in this paper, the throughput of patterning cell monolayers is increased, while obtaining immediate visual feedback of the pattern generated. This facilitates real-time modification of the patterns. Such control might be crucial in scenarios in which cell viability on a given surface is not homogeneous. Another advantage of the method is that the same head and chemistry can be used to generate multiple patterns, thereby reducing the number of steps involved in generating a patterned cell monolayer.
The dimensions of the channels in the head and the head geometry can be scaled according to the requirements of the application, thus allowing control over the resolution of the patterning. All experiments in the current work were performed using an MFP head with fixed aperture dimensions, specifically with inner apertures at 100 × 100 µm and outer apertures at 200 × 100 µm. This design with larger outer apertures was chosen to ensure continuous operation of the hHFC without clogging of the apertures by stray cell debris. We have tested heads with inner aperture dimensions 50 × 50 µm and 50 µm spacing between them, with successful results in cell removal. Use of smaller apertures, down to 10 × 10 µm with 10 µm spacing, would permit scaling to a smaller footprint size (~ 30 × 30 µm), thus enabling a higher resolution in patterning. We observed operational issues with aperture sizes smaller than 10 µm due to clogging from particulates. Because of such operational difficulties, 100 µm is the current limit of resolution and thus a limitation of the described technique. However, we can address this using the liquid shaping ability of the hHFC. We have demonstrated that the resolution of cell removal can be tuned for a given set of aperture dimensions by controlling QI2/QI1 (Figure 4b) and the apex-to-substrate gap10,14. The stages used in the current work has a minimum step size of 100 nm. A combination of these controllable parameters can improve the spatial resolution of cell removal in terms of the footprint size and scanning distance.
If patterned co-cultures are desired to study specific cell-cell interactions between different cell types, sequential seeding and patterning can be performed using the protocol described. Finally, amplifiable DNA of high quality can be obtained from the lysate, as evident by the singular peak in the DNA melt curve (see Figure 7). We evaluated a DNA quantity of around 1.6 ng from a single footprint (about 300 cells) using qPCR, which is close to theoretical expectation (6-8 pg/cell). This indicates a quantity of the extracted DNA suitable for several downstream processes while obviating use of any DNA isolation methods. This opens up avenues for DNA-analysis-based local probing of cultured cells. The capability of hHFC to shape liquids can also be used to deposit live cells and proteins on activated surfaces, which in combination with the subtractive patterning and sequential co-culturing presented in this protocol enables the creation of complex cell and cell-matrix patterns on culture substrates. The versatility and the control provided by hHFC-based subtractive patterning of cell monolayers and the possibility of performing DNA analysis on the extracted cells, provides a powerful new tool set to biologists to perform spatially resolved studies involving cell interactions.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the European Research Council (ERC) Starting Grant, under the 7th Framework Program (Project No. 311122, BioProbe). We thank Dr. Julien Autebert and Marcel Buerge for technical assistance and discussions during the development of the protocol and the platform. Prof. Petra Dittrich (ETH Zurich), Prof. Bradley Nelson (ETH Zurich), Dr. Bruno Michel and Dr. Walter Riess are acknowledged for their continuous support.
Materials
| Name | Company | Catalog Number | Comments |
| Materials | |||
| Microfluidic Probe (MFP) | Silicon/glass hybrid probe head with channels patterned in silicon. Fabrication done in-house at IBM Research - Zürich | ||
| Lab Tek II Chamber slides with 2 chambers | ThermoFisher scientific, USA | 154461 | 2 Chamber chamber slides for cell culture with each chamber providing an area of 4 cm2 and capable of holding up to 3 ml of media volume |
| Chemicals and cell lines | |||
| Sodium Hydroxide, Tris-Cl, Ethylenediaminetetraaceticacid, Rhodamine B | SigmaAldrich chemicals, USA | S5881, 252859, E9884, R6626 | Chemicals used for processing and shielding liquids |
| Proteinase K | Qiagen, Germany | 19131 | protease to digest denatuired proteins |
| Deconex 16 Plus | Bohrer Chemie, Switzerland | Universal cleaning agent for labaratory consumables. Used for non stringent cleaning. | |
| DNA away | ThermoFisher scientific, USA | 7010 | Surface decontaminant that denatures DNA and DNAases. |
| DMEM, high glucose, GLUTAMAX supplement | ThermoFisher scientific, USA | 10566-016 | Culturing medium for epithelial cells. |
| CellTracker Green CMFDA dye | ThermoFisher scientific, USA | C7025 | Membrane permeant live cell labeling dye. Dye active for 72 hours. |
| CellTracker Orange CMRA dye | ThermoFisher scientific, USA | C34551 | |
| β-actin genomic primers for qPCR | Integrated DNA technologies, USA | Custom oligos used for DNA quality validation and qPCR. | |
| MCF7 breast carcinoma cells | ATCC, USA | ATCC HTB-22 | Cell lines used to produce co-cultures. |
| MDA-MB-231 breast carcinoma cells | ATCC, USA | ATCC HTB-26 | |
| Equipment and fluidic connections | |||
| Motorized high precision stages | Custom machined components. Linear axis motors from LANG GmBH, Germany | Customized linear axis stages from LT series | 3 × LT for 3 axes. LSTEP controller used for interfacing stages and PC through RS 232 port. |
| Syringes | Hamilton, Switzerland | 1700 TLLX series | Interchangeable with syringes provided by other manufacturers with a 250-500 µl range. |
| Nemesys low pressure syringe pumps | cetoni GmbH, Germany | Component of pumping station. | |
| Circular M1-connector | Dolomite microfluidics, United Kingdom | 3000051 | Interface between vias in MFP head and tubing |
| Tilt/rotation stage Goniometer | OptoSigma, USA | KKD-25C | Goniometer to adjust coplanarity of MFP apex |
| DS Fi2 HD color camera (CCD) | Nikon, Switzerland | Controlled using DS-U3 controller unit | |
| Software | |||
| Win - Commander | LANG GmBH, Germany | Stage control software. | |
| Qmix Elements | cetoni GmbH, Germany | Pump control software. | |
| NIS Elements | Nikon, Switzerland | Basic research module for image acquisition and analysis. | |
References
- Théry, M. Micropatterning as a tool to decipher cell morphogenesis and functions. J. Cell Sci. 123 (Pt 24), 4201-4213 (2010).
- Goubko, C. A., Cao, X. Patterning multiple cell types in co-cultures: A review. Mater. Sci. Eng. C. 29 (6), 1855-1868 (2009).
- Kaji, H., Camci-Unal, G., Langer, R., Khademhosseini, A. Engineering systems for the generation of patterned co-cultures for controlling cell-cell interactions. Biochim. Biophys. Acta. 1810 (3), 239-250 (2011).
- Cho, C. H., Park, J., Tilles, A. W., Berthiaume, F., Toner, M., Yarmush, M. L. Layered patterning of hepatocytes in co-culture systems using microfabricated stencils. Biotechniques. 48 (1), 47-52 (2010).
- Schütze, K., Lahr, G. Identification of expressed genes by laser-mediated manipulation of single cells. Nat. Biotechnol. 16 (8), 737-742 (1998).
- Salazar, G. T. A., Wang, Y., et al. Micropallet arrays for the separation of single, adherent cells. Anal. Chem. 79 (2), 682-687 (2007).
- Guillaume-Gentil, O., Zambelli, T., Vorholt, J. A. Isolation of single mammalian cells from adherent cultures by fluidic force microscopy. Lab Chip. 14 (2), 402-414 (2014).
- Iwata, F., Adachi, M., Hashimoto, S. A single-cell scraper based on an atomic force microscope for detaching a living cell from a substrate. J. Appl. Phys. 118 (13), (2015).
- Riahi, R., Yang, Y., Zhang, D. D., Wong, P. K. Advances in wound-healing assays for probing collective cell migration. J. Lab. Autom. 17 (1), 59-65 (2012).
- Juncker, D., Schmid, H., Delamarche, E. Multipurpose microfluidic probe. Nat. Mater. 4 (8), 622-628 (2005).
- Kaigala, G. V., Lovchik, R. D., Delamarche, E. Microfluidics in the "open space" for performing localized chemistry on biological interfaces. Angew. Chem. Int. Ed. Engl. 51 (45), 11224-11240 (2012).
- Qasaimeh, M. A., Gervais, T., Juncker, D. Microfluidic quadrupole and floating concentration gradient. Nat. Commun. 2 (May), 464 (2011).
- Christ, K. V., Turner, K. T. Design of hydrodynamically confined microfluidics: controlling flow envelope and pressure. Lab Chip. 11 (8), 1491-1501 (2011).
- Autebert, J., Kashyap, A., Lovchik, R. D., Delamarche, E., Kaigala, G. V. Hierarchical hydrodynamic flow confinement: efficient use and retrieval of chemicals for microscale chemistry on surfaces. Langmuir. 30 (12), 3640-3645 (2014).
- Kaigala, G. V., Lovchik, R. D., Drechsler, U., Delamarche, E. A Vertical Microfluidic Probe. Langmuir. 27 (9), 5686-5693 (2011).
- Cors, J. F., Lovchik, R. D., Delamarche, E., Kaigala, G. V. A compact and versatile microfluidic probe for local processing of tissue sections and biological specimens. Rev. Sci. Instrum. 85 (3), 034301 (2014).
- Pollard, J. W., Walker, J. M. . Basic Cell Culture Protocols. , (1997).
- Mitry, R. R., Hughes, D. R. Human cell culture protocols. Methods Mol. Biol. Springer. 531 (1), (2012).
- Sakai, K., Ushida, T., Nagase, T., Nakamigawa, H. Quantitative analysis of cell detachment by shear stress. Mater. Sci. Eng. C. 17 (1-2), 55-58 (2001).
- Rodriguez, N. M., Desai, R. A., Trappmann, B., Baker, B. M., Chen, C. S. Micropatterned multicolor dynamically adhesive substrates to control cell adhesion and multicellular organization. Langmuir. 30 (5), 1327-1335 (2014).
- Tasoglu, S., Demirci, U. Bioprinting for stem cell research. Trends Biotechnol. 31 (1), 10-19 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved