
Method Article
Split Point Analysis and Uncertainty Quantification of Thermal-Optical Organic/Elemental Carbon Measurements
In This Article
Summary
This article presents a protocol and software tool for the quantification of uncertainties in the calibration and data analysis of a semi-continuous thermal-optical organic/elemental carbon analyzer.
Abstract
Researchers from myriad fields frequently seek to quantify and classify concentrations of carbonaceous aerosols as organic carbon (OC) or elemental carbon (EC). This is commonly accomplished using thermal-optical OC/EC analyzers (TOAs), which enable measurement via controlled thermal pyrolysis and oxidation under specific temperature protocols and within constrained atmospheres. Several commercial TOAs exist, including a semi-continuous instrument that enables on-line analyses in the field. This instrument employs an in-test calibration procedure that requires relatively frequent calibration. This article details a calibration protocol for this semi-continuous TOA and presents an open-source software tool for data analysis and rigorous Monte Carlo quantification of uncertainties. Notably, the software tool includes novel means to correct for instrument drift and identify and quantify the uncertainty in the OC/EC split point. This is a significant improvement on the uncertainty estimation in the manufacturer’s software, which ignores split point uncertainty and otherwise uses fixed equations for relative and absolute errors (generally leading to under-estimated uncertainties and often yielding non-physical results as demonstrated in several example data sets). The demonstrated calibration protocol and new software tool enabling accurate quantification of combined uncertainties from calibration, repeatability, and OC/EC split point are shared with the intent of assisting other researchers in achieving better measurements of OC, EC, and total carbon mass in aerosol samples.
Introduction
The ability to accurately measure atmospheric concentrations of carbonaceous species is exceedingly important to many researchers. Carbonaceous species in ambient particulate matter (PM, the largest environmental risk factor for early death1) have been suggested to be the key component of PM responsible for adverse health effects and outcomes2,3,4. Particulate carbon in the atmosphere is a critical climate pollutant, where different carbonaceous species are known to have variable, even opposite, impacts. Black carbon is potentially the second strongest direct radiative forcer in the earth’s atmosphere5,6,7,8. When deposited on snow and ice, black carbon also reduces the reflectivity of the arctic landscape, enhancing the absorption of sunlight, and increasing the rate of melt9,10,11,12. Contrastingly, hygroscopic organic carbon particles act as cloud condensation nuclei, increasing the mean reflectivity of earth, and causing a cooling effect13. Accurate classification of sampled carbonaceous material and concurrent quantification of measurement uncertainties are thus essential aspects of particulate matter measurements.
Differentiating between organic and elemental carbon in a particulate-laden sample can be achieved using a thermal-optical analysis14. Commercial, laboratory-based systems for thermal-optical carbon analyses have been created15,16,17 including an on-line, semi-continuous analyzer18 that enables the execution of thermal-optical analyses in the field. The present work describes a detailed procedure for calibrating this latter OCEC instrument (see Table of Materials) and shares an open-source software tool for the rigorous quantification of calibration and analysis uncertainties. Although the initial release of the open-source software is designed for the output file format of the semi-continuous instrument, the software tool could readily be extended by others in future to work with outputs generated by other instruments.
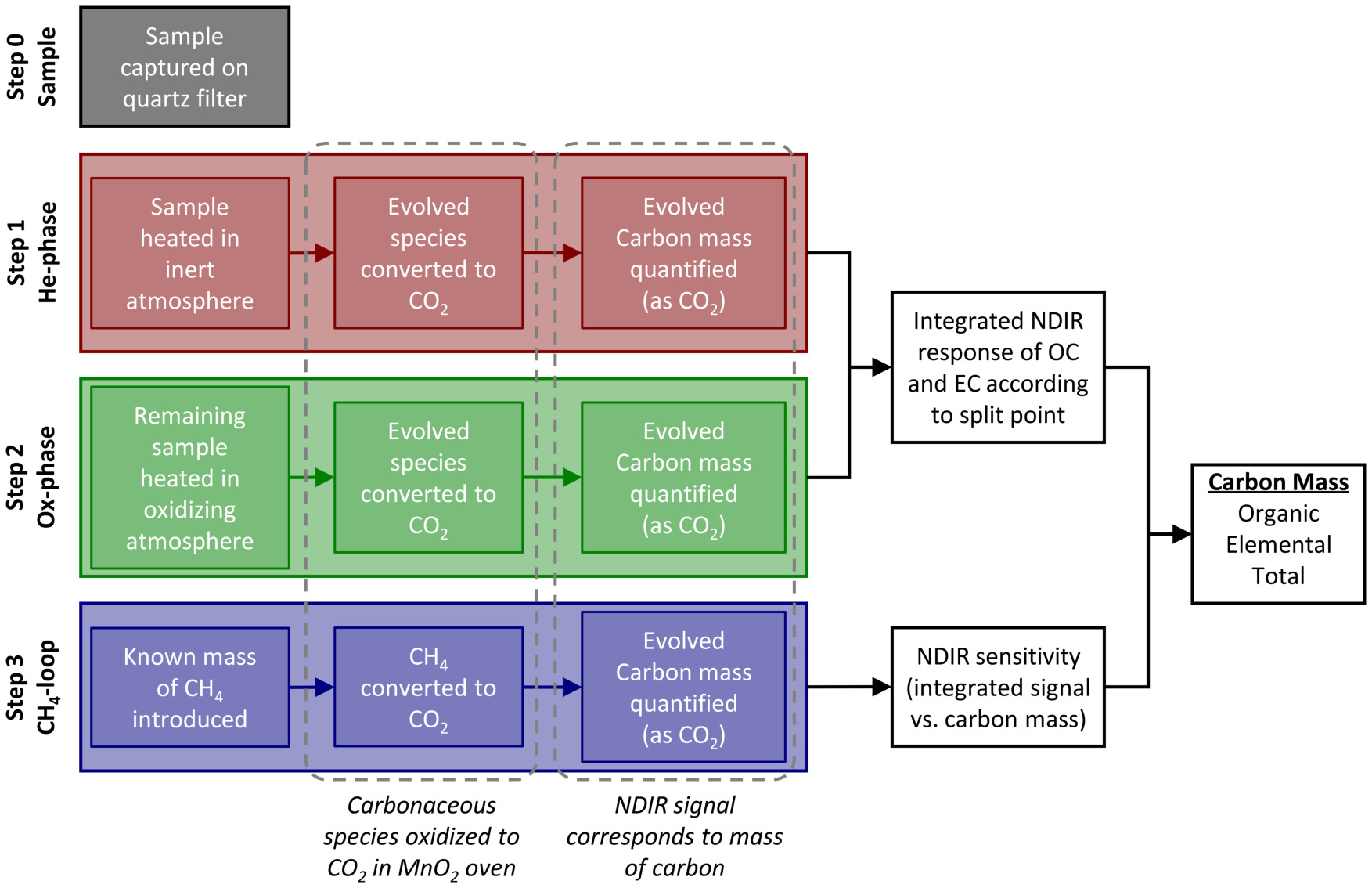
The semi-continuous thermal-optical organic/elemental carbon analyzer (OCEC) quantifies organic carbon (OC) and elemental carbon (EC) in a sample volume. The analysis procedure contains four phases outlined in Figure 1. Firstly, a sample volume is pulled through the instrument, where particulate matter is deposited onto, and gas-phase organics are adsorbed by, a quartz filter. At the termination of sampling, the quartz filter is heated through a prescribed temperature protocol in an inert, helium (He) atmosphere. During this procedure, a portion of the carbonaceous material is thermally pyrolyzed from the quartz filter. Gaseous exhaust is forwarded to a fixed-temperature, Manganese Oxide (MnO2) oven that converts pyrolyzed carbonaceous species into carbon dioxide (CO2). The time-resolved concentration of released CO2 is subsequently measured by a non-dispersive infrared (NDIR) detector. Following initial heating in the He environment, the sample is heated through a similar protocol in an oxidizing (Ox) environment. In the presence of oxygen, refractory carbonaceous species remaining on the quartz filter are oxidized and then forwarded through the MnO2 oven and NDIR detector in the same manner. Once sampled carbonaceous species have been fully evolved from the quartz filter, a final in-test calibration procedure is performed. A fixed quantity (nominally 0.8 mL) of a 5% methane (CH4)-helium mixture is introduced into the instrument, oxidized in the MnO2 oven, converted to CO2, and subsequently measured by the NDIR. The integrated NDIR signal during this in-test calibration phase (termed the CH4-loop) corresponds to the known carbon mass (introduced as CH4) and therefore quantifies the sensitivity of the NDIR, which can drift in time. This measure of NDIR sensitivity is then used to infer carbon masses from the NDIR signal during the prior He- and Ox-phases of the analysis.

Figure 1: Thermal-optical analysis procedure. Analysis procedure of the thermal-optical OCEC instrument. Following the acquisition of a sample onto the quartz filter (step 0), three main analysis steps are performed. Two thermal protocols first in a helium atmosphere (He-phase, step 1) and then in an oxidizing atmosphere (Ox-phase, step 2) are executed, where carbonaceous components are pyrolyzed/oxidized from the quartz filter, further oxidized into CO2 within a secondary catalyzing oven, and subsequently measured by an NDIR detector. A final in-test calibration procedure (CH4-loop, step 3) is performed, where the oxidation of a known mass of methane provides a measure of sensitivity of the NDIR detector. Detector sensitivity is coupled with NDIR-measured CO2 during the He- and Ox-phases to quantify carbon mass loading on the quartz filter. Please click here to view a larger version of this figure.
{kind=link}
The mass of injected carbon during the CH4-loop can be sensitive to operational conditions such that intermittent calibration is required. This calibration uses an aqueous sucrose solution of known concentration (approximately 0.99%m/m) as an external standard. Repeated tests are performed where different known volumes of the sucrose solution are introduced in random order into the instrument and the thermal-optical analysis is performed. Each repeated test (i.e., results from each injection and subsequent analysis) yields an integrated NDIR signal during the CH4-loop (“calibration area”) and an integrated NDIR signal for total carbon (i.e., signal during the He- and Ox-phases; referred to as “total area”), which corresponds to the known mass of carbon in sucrose. Linear regression of the known carbon mass with instrument-reported “total area” provides a measure of mean NDIR sensitivity. This sensitivity is then coupled with the mean “calibration area” to yield calibrated knowledge of the carbon mass injected during the CH4-loop.
Beyond calibration, a key challenge in the interpretation of results from the OCEC instrument is the determination of the relative fractions of OC and EC in the measured sample. As OC pyrolyzes during the He-phase of the temperature protocol, a fraction chars on the filter rather than being released, oxidized in the MnO2 oven, and detected by the NDIR. In theory, this charred refractory OC (termed pyrolyzed carbon, PC) remains on the filter until the Ox-phase, when it is oxidized alongside EC. Consequently, naively labelling all evolved carbon during the He-phase as OC and evolved carbon in the Ox-phase as EC leads to biased estimates of the true fraction of OC and EC. A common means to define the split point (i.e., the moment in time when all prior carbon evolution is considered as OC and all subsequent carbon evolution as EC) is the thermal/optical-transmittance (TOT) method19. Here, a laser is directed through the quartz filter during the thermal analysis and its power (optically downstream of the quartz filter) is detected by a photodetector. By assuming that OC is not optically active at the laser wavelength (i.e., OC negligibly absorbs light) and PC shares optical properties with EC, the split point can be estimated. The premise is to first measure the attenuated laser power at the start of the analysis. As OC evolves (partly into light-absorbing PC), the attenuation of the laser is enhanced such that the photodetector signal drops. As the Ox-phase is entered and EC/PC are co-evolved, attenuation is reduced, and the photodetector signal begins to increase. The split point is defined as the instance in time when the measured laser power returns to its initial value. While the logic of this approach is robust, the result relies upon the above-noted assumptions. As such, it is common to declare that reported OC and EC results are “operationally-defined” ― i.e., they are specific to the technique employed to evaluate the split point14,20,21.
Although it is true in theory that OC is evolved in the He-phase and PC/EC are evolved in the Ox-phase, it has been observed that evolution of PC/EC can indeed occur during the He-phase due to various mechanisms22,23,24,25, such that the true split point may occur prior to the introduction of oxygen. This ambiguity in the prediction of where the split point should lie, coupled with uncertainty in the assumptions of the optical properties of OC, PC, and EC, suggests that uncertainty in the split point could be a dominant source of uncertainty in measured carbon masses. Fortunately, the methodical estimation of the split point via the TOT method enables an objective estimate of split point uncertainty. To the authors’ knowledge however, there is no direct estimation (and subsequent propagation) of split point uncertainty in the manufacturer’s software; reported total uncertainties are instead computed with fixed relative and absolute components26. As part of this work, a novel technique to estimate split point uncertainty is presented − the “attenuation decline” technique. In this technique, uncertainty in the split point is quantified as the difference between evolved carbon mass at the nominal split (via the TOT method) and evolved carbon mass at a subsequent point where laser attenuation has decreased beyond some critical quantity, a prescribed fraction of the initial attenuation. A critical attenuation decline is estimated based on the uncertainty in laser attenuation relative to its initial value; in theory, this approach captures uncertainty in the key attenuation-matching principle of the TOT method. Furthermore, to consider (at least in part) split point uncertainty due to assumed optical properties of PC and EC, the suggested critical attenuation decline is expanded by a factor of two.
This article presents a detailed protocol for calibrating the OCEC instrument alongside a software tool for rigorously quantifying calibration and analysis uncertainties. Firstly, sections 1 to 3 of the protocol outline instructions for creating the aqueous sucrose solution, preparing the instrument for calibration, and acquiring calibration data. Section 4 uses the novel, open-source, software tool (see Table of Materials) to analyze calibration data via the software’s graphical user interface. Section 5 specifies considerations for acquiring a sample using the OCEC instrument and section 6 describes use of the above-noted software for calculating carbon masses and associated uncertainties, including contributions from the estimation of the split point. Novel techniques to improve the processing of OCEC data ― including the “attenuation decline” technique introduced above ― are described in the software’s online documentation.
Within the presented software tool, calibration constants, measured carbon masses, and associated uncertainties are computed using a Monte Carlo (MC) method. This procedure propagates errors that are not, to the authors’ knowledge, currently considered in the manufacturer’s proprietary software. For calibration, these sources of error include uncertainty in the concentration of sucrose in the aqueous solution, accuracy in the volume of applied sucrose solution (instrument accuracy, inter-user reproducibility, and intra-user repeatability), and uncertainty in the linear regression. With respect to data analysis, considered sources of error include the calibration uncertainty and repeatability and, importantly, the estimated uncertainty of the split point. Ultimately, the software enables a user to accurately quantify uncertainty in the calibration of the instrument and propagate this uncertainty alongside that of split point estimation into the calculation of carbon masses. This represents a notable improvement upon the manufacturer’s protocol, by directly considering key sources of error in the measurement, rather than estimating partial uncertainties using a fixed equation.
Protocol
NOTE: This instrument contains a visible, Class 1 laser. While exposure to this low-power laser is unlikely to result in harm, the instrument includes an interlock device in the form of a laser shroud that blocks the user from the optical path when the access panel is open. Removal of the laser shroud disables the laser, such that exposure to the laser throughout the presented protocol should not occur. The instrument’s ovens may heat to ~900 °C during normal operation of the instrument and components manipulated throughout the presented protocol may become hot. Prior to performing work within the instrument’s access panel, ensure that the instrument software reads a “Status” of “Idle” and that the “Front Oven” temperature is <90 °C. Use caution when manipulating components near the oven unit immediately following high-temperature operations.
1. Prepare aqueous sucrose solution.
- On a precision balance, place a clean, sealable, glass vessel of minimum 1 L in volume. Add 10 g of high-purity sucrose and 1000 g of distilled, de-ionized (DDi) water to the vessel.
- Seal the vessel and mix thoroughly by shaking until the sucrose is fully dissolved. Transfer a portion of the solution to a small, clean, glass jar (Table of Materials) for pipetting.
- Store both solutions in a refrigerator for up to six months.
2. Prepare instrument for calibration.
- Energize the instrument and allow the rear oven to come to temperature.
- Run a minimum of five CH4-loops to allow startup transients to dissipate.
- In the Sample ID # field of the instrument’s software, type text to indicate startup, such as Instrument Warm-Up. In the PAR FILE field, click the … button to browse and select the OCECgo_WarmUp.par protocol. In the Output raw data file field, click the … button to browse and select or create a suitable .txt file such as yyyy-mm-dd_WarmUp.txt.
NOTE: The parameter file OCECgo_WarmUp.par is available in the on-line software repository for the OCECgo software tool (Table of Materials). - Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select 0. Ensure the Cycle checkbox is checked. Click the Start Analysis button. Allow the instrument to operate for ~20 min.
NOTE: Interaction with the Sample Minutes dropdown menu must be by mouse. The instrument will not recognize manual text input. - During the final analysis, uncheck Cycle checkbox and allow the current analysis to finish.
- In the Sample ID # field of the instrument’s software, type text to indicate startup, such as Instrument Warm-Up. In the PAR FILE field, click the … button to browse and select the OCECgo_WarmUp.par protocol. In the Output raw data file field, click the … button to browse and select or create a suitable .txt file such as yyyy-mm-dd_WarmUp.txt.
- Replace quartz filters (optional).
NOTE: It is recommended by the manufacturer of the instrument18 that filters are replaced weekly (assuming consistent use of the instrument).- Remove the quartz (tube) insert.
- Open the access panel and remove the laser shroud (de-energizing the laser). Remove the photodetector (no tools required) by loosening the white polyoxymethylene (POM) nut behind the photodetector, disconnecting the metal tube fitting on the left side of the photodetector, and sliding the photodetector housing off the quartz insert. Place the photodetector in the bottom of the instrument.
- Remove the quartz insert by loosening the white POM nut holding the quartz insert in place and — while wearing non-powdered, disposable, plastic gloves — sliding the quartz insert out of the POM fitting.
CAUTION: The quartz insert is very fragile, place it stably on a lint-free tissue on a flat surface.
- Using the filter removal tool, remove and dispose of the existing quartz filters. Install new quartz filters.
- Place a large quartz filter on a lint-free tissue on a flat surface. Using the filter punch tool, punch out one filter.
- Using clean tweezers, remove the filter from the punch and place against the POM housing for the quartz insert such that the textured surface of the filter is facing away from the oven. Wearing non-powdered, disposable, plastic gloves, use the quartz insert to slide the quartz filter until fully seated against the oven.
- Repeat steps 2.3.2.1 and 2.3.2.2 so that two quartz filters are installed.
- Remove the quartz (tube) insert.
- Install a third quartz filter (referred to as the quartz “boat”) for introduction of the sucrose standard to the instrument. Repeat step 2.3.2.1. Using clean tweezers, remove the filter from the punch and place the quartz filter “boat” into the end of the quartz insert such that the cross-sections of the filter and insert are perpendicular.
- Replace components and close the instrument.
- Wearing non-powdered, disposable, plastic gloves, re-introduce the quartz insert into the instrument. Loosely hand-tighten the white POM nut that secures the quartz insert in place.
- Replace photodetector head (no tools required). Slide the photodetector housing onto the end of the quartz insert. Loosely re-connect the metal tube fitting on the left side of the photodetector to ensure proper alignment of the photodetector and quartz insert. Loosely hand-tighten the white POM nut securing the photodetector head onto the quartz insert. Fully hand-tighten the metal tube fitting on the left side of the photodetector.
- Ensure all POM nuts are fully hand-tight and secure.
NOTE: It is critical that the instrument’s oven is well-sealed from atmosphere. That is, although tools should not be used, hand-tighten all POM nuts as much as possible. In the authors’ experience, while delicate, the quartz insert is strong in the circumferential direction — significant tightening of the POM nuts, which ensures good seals, is unlikely to damage the quartz insert. - Replace the laser shroud (re-energizing the laser) and close the access panel.
- Clean newly-installed quartz filters by running a minimum of one oven clean cycle. Click the Run dropdown menu in the instrument software’s toolbar and select Clean Oven, which raises the front oven temperature to above ~835 °C.
- Ensure residual carbon has been removed from the filter using blank analytical cycle(s).
- In the PAR FILE field of the instrument’s software, click the … to browse and select the desired thermal protocol’s .par file. Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select 0. Ensure the Cycle checkbox is unchecked. Click the Start Analysis button, confirm that only one analysis/cycle is desired, and allow the thermal analysis to execute.
NOTE: Interaction with the Sample Minutes dropdown menu must be by mouse. The instrument will not recognize manual text input.
- In the PAR FILE field of the instrument’s software, click the … to browse and select the desired thermal protocol’s .par file. Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select 0. Ensure the Cycle checkbox is unchecked. Click the Start Analysis button, confirm that only one analysis/cycle is desired, and allow the thermal analysis to execute.
- Repeat steps 2.6 and 2.7 until the instrument-reported total carbon mass is statistically equal to zero.
3. Acquire calibration data.
- Obtain one calibration point.
- Remove the quartz insert via step 2.3.1 and following manufacturer-recommended pipetting procedures, aspirate 5 μL or 10 μL of sucrose solution. Carefully deposit the sample onto the quartz boat, as close as possible to the end of the quartz insert, ensuring a blowout procedure is executed such that the entire volume is ejected onto the quartz boat.
- Re-introduce the quartz insert, close the instrument via step 2.5, and dry the wet filter. Click the Run dropdown menu in the instrument software’s toolbar and select dry wet filter, which raises the front oven temperature to 110 °C.
- Once the front oven has cooled, run the instrument protocol to be used in post-calibration measurements (selected in step 5.3.1).
- In the Sample ID # field of the instrument’s software, type text to indicate applied sucrose volume, such as 5 uL. In the PAR FILE field, click the … button to browse and select the desired thermal protocol’s .par file. In the Output raw data file field, click … button to browse and select or create a suitable .txt file such as yyyy-mm-dd_Calibration.txt.
- Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select 0. Ensure the Cycle checkbox is unchecked. Click the Start Analysis button, confirm that only one analysis/cycle is desired, and allow the thermal analysis to execute.
NOTE: Interaction with the Sample Minutes dropdown menu must be by mouse. The instrument will not recognize manual text input.
- Obtain one blank/background calibration point. Perform step 3.1 without depositing a sucrose sample onto the quartz boat.
NOTE: To obtain an accurate blank/background, ensure that the quartz boat is exposed to ambient air as though the user were depositing a sample of sucrose. - Repeat step 3.1 such that one calibration point each at 5 and 10 μL are obtained. Further repeat steps 3.1 and 3.2 as required to achieve desired calibration uncertainty as computed in step 4.
NOTE: Section 4 may be executed following each iteration of section 3.3 to support the user in determining satisfactory convergence of the calibration. - Remove the quartz boat. Remove the quartz insert via step 2.3.1. Using clean tweezers, remove the quartz boat from the quartz insert. Re-introduce the quartz insert and close the instrument via step 2.5.
- To ensure the instrument is prepared for post-calibration measurements, run a minimum of one oven clean cycle as in step 2.6.
4. Compute calibration constant with uncertainty.
NOTE: The OCECgo software tool employs mouseover utilities to aid the user with the input of data and selection of analysis parameters. Further information, including default and permissible ranges for user-editable fields are listed in the tool’s online documentation.
- Load the software tool (OCECgo) and click to migrate to the Calibration Tool tab.
- Input calibration data. In section (1) of the graphical user interface (GUI), input calibration data: nominal volume of the applied sucrose solution, instrument-reported integrated NDIR signal corresponding to total carbon (“total area”), instrument-reported integrated NDIR signal during the CH4-loop (“calibration area”), and a Boolean to indicate whether specific points should be used in calibration (“1” for yes; “0” for no). Repeat for each data point, adding and deleting rows to the table, as necessary, by clicking the “+ Row” and “- Row” buttons.
NOTE: The user can alternatively click the Import Calibration button to upload previous calibration data and inputs to the software tool. If this option is exercised, go to step 4.4 to re-create plots in GUI section (3) or go directly to section 6 to analyze instrument data. - Define uncertainty data for use in the Monte Carlo analysis.
- In GUI section (2)(a), input data pertaining to the aqueous sucrose solution. Input masses of sucrose and DDi water measured during step 1.1 and the absolute 2σ bias of the scale [g] used to measure DDi and sucrose masses — absolute bias is equivalent to the scale’s reported accuracy. Input the nominal minimum purity of sucrose [%m/m] listed on the sucrose vessel’s label and insert the range of ambient temperatures [°C] observed during the acquisition of calibration data.
NOTE: 2σ corresponds to two times the standard deviation, which in the context of a normal (Gaussian) distribution is a conservative estimate of the 95% confidence interval (CI). - In section (2)(b), provide data pertaining to pipette uncertainty. Input relative 2σ equipment-reported accuracy (bias error), equipment reported repeatability (precision error), precision error corresponding to intra-user repeatability, and bias error corresponding to inter-user reproducibility for aspirated volumes of 5 μL and 10 μL.
NOTE: Default pipette uncertainties correspond to the instrument listed in the Table of Materials. Default 2σ human-errors were estimated based on the pooled variances of studies of intra-user repeatability and inter-user reproducibility at each volume. - In section (2)(c), input the desired number of Monte Carlo draws for the computation of calibration metrics.
NOTE: The number of Monte Carlo draws corresponds to the number of random computations of the mass calibration constant under the Monte Carlo framework. Larger numbers yield more consistent results but take longer to process (more computational time). The default value in OCECgo is 106 while permitted values are [102, 108].
- In GUI section (2)(a), input data pertaining to the aqueous sucrose solution. Input masses of sucrose and DDi water measured during step 1.1 and the absolute 2σ bias of the scale [g] used to measure DDi and sucrose masses — absolute bias is equivalent to the scale’s reported accuracy. Input the nominal minimum purity of sucrose [%m/m] listed on the sucrose vessel’s label and insert the range of ambient temperatures [°C] observed during the acquisition of calibration data.
- Run analysis. In GUI section (3), press
 to run the Monte Carlo analysis to process calibration data.
to run the Monte Carlo analysis to process calibration data. - Update the instrument calibration file with the results presented in section (4). Open the instrument’s parameter file: SCInstrumentParameters.txt. Find the line of text containing the existing calibration data — this line of text includes a comment on the righthand side reading “Calibration Constant…”. Replace the numerical data with the reported “Calibrated Carbon Mass” and “Mean Calibration (CH4-loop) Area”. Save and close the parameter file and re-start the instrument’s software.
- Save and/or export calibration results (optional).
- Click the Save as Default Calibration button to store the calibration result for default use by the software.
NOTE: Calibration results are stored in an initialization file that, upon rebooting of the software, reloads the latest calibration. The user is warned if the current date is more than 30 days from the latest calibration. - Click the Export Calibration Results button to export the calibration data.
NOTE: Numerical data are exported to a pre-formatted .xlsx file and visualization of the Monte Carlo results are exported as a .png file. This saved calibration file is useful if results are to be reanalyzed/imported at a later date using the applicable calibration.
- Click the Save as Default Calibration button to store the calibration result for default use by the software.
- Once calibration is complete, remove the quartz boat. Following step 2.3.1, remove the quartz insert from the instrument. Using forceps or tweezers, remove the quartz boat used for calibration. Following step 2.5, replace the quartz insert and close the instrument.
5. Obtain measurement data.
- Set the desired sample flow rate (optional). Open the instrument’s valve control file: valve_table.txt. Set the “collect” parameter of “Valve A”, which represents the target sample flow rate in liters per minute, to an integer value between 2 and 8 (inclusive).
NOTE: The instrument's software must be restarted following changes to the valve control file. - Set the desired sampling period.
- If an immediate sample is desired. Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select the desired sample period in minutes. If multiple, consecutive examples are desired, ensure the Cycle checkbox is checked. Otherwise, ensure the Cycle checkbox unchecked.
NOTE: Interaction with the Sample Minutes dropdown menu must be by mouse. The instrument will not recognize manual text input. - If it is desired to delay sample acquisition. Open the instrument’s sample time control file: SamTimePar1.txt. Each row of this file contains a comma-separated pair of the sample time start and duration. Edit this file as desired, then save and close the file and re-start the instrument’s software.
- If an immediate sample is desired. Ensure the Use Sample File Times checkbox is unchecked. In the Sample Minutes dropdown menu, select the desired sample period in minutes. If multiple, consecutive examples are desired, ensure the Cycle checkbox is checked. Otherwise, ensure the Cycle checkbox unchecked.
- Execute the thermal analysis.
- In the Sample ID # field of the instrument’s software, type text to define the sample, such as Sample_01. In the PAR FILE field, click the … button to browse and select the desired thermal protocol’s .par file. In the Output raw data file field, click the … button to browse and select or create a suitable .txt file such as yyyy-mm-dd_Samples.txt.
- Click the Start Analysis button. If required, confirm that only one analysis/cycle is desired.
NOTE: Experience has identified, with two unique instruments, that the internal measurement of sample volume disagrees with measurements using an external high-accuracy mass flow meter, with errors exceeding 10% in some cases. Furthermore, errors in the instrument-reported sample volume have been noted to be sensitive to both sample flow rate and sample duration. It is therefore recommended to externally measure the sampled volume at the outlet of the sample pump with a high-accuracy mass flow meter such as that listed in the Table of Materials.
6. Compute carbon masses and uncertainties.
NOTE: The OCECgo software tool exploits mouseover utilities to aid the user with the input of data and selection of analysis parameters. Further information, including default and permissible ranges for user-editable fields are listed in the tool’s online documentation.
- Load the software tool (OCECgo) and click to migrate to the Data Analysis – Inputs tab.
- Load time-resolved instrument data — GUI section (1). In sub-section (a), click the Browse… button and, in the file selection dialog, select the .txt results file defined in step 5.3.1. In sub-section (b), review the Sample IDs (as defined in step 5.3.1), and click to select the analysis of interest. In sub-section (c), review the analysis metadata, particularly the analysis’ Sample Start timestamp.
- Define data-processing options — GUI section (2).
- In sub-section (a), select the desired laser correction procedure: a quadratic- or linear-dependence on oven temperature.
NOTE: In the experience of the authors, the laser correction procedure typically has a negligible effect ― as such, the quadratic correction is recommended and is loaded as the default value. - In sub-section (b), select the desired NDIR correction procedure: correction via a convex hull to the raw NDIR data or a linear correction using the instrument-reported NDIR areas (From results file).
NOTE: The novel convex hull technique (briefly described in the software’s online documentation) corrects the NDIR signal by fitting a convex hull as a lower-bound to the NDIR time series; this technique allows for a non-linear (piecewise) correction to the NDIR signal. In the experience of the authors, linear correction of the NDIR detector can, in some circumstances, yield non-physical results ― as such, the “Convex Hull” procedure is recommended and is loaded as the default value. - In sub-section (c), if desired, adjust the parameters of the generalized t-distribution reported for the mass calibration constant (calculated in step 4.4) and the estimated calibration repeatability error.
NOTE: Execution of step 4.4 or import of prior calibration results (see step 4.6.2) automatically updates the generalized t-distribution parameters. Repeatability in instrument calibration (“Rep. [%]”) is set to a default value of 7.90%, based on repeatability testing by the authors27. - In sub-section (d), press
 to create/update the analysis’ thermogram and AVEC (laser attenuation vs. evolved carbon21) plots.
to create/update the analysis’ thermogram and AVEC (laser attenuation vs. evolved carbon21) plots.
NOTE: If the From results file button is selected (step 6.3.2), in the file selection dialog, select the .xlsx results file created by the instrument.
- In sub-section (a), select the desired laser correction procedure: a quadratic- or linear-dependence on oven temperature.
- Define the split point determination procedure — GUI section (3) and (4).
- In sub-section (3)(a), select the desired procedure to calculate the split point and associated uncertainty: the novel “Attenuation decline” procedure described in the introduction section, a manually-defined split point and uncertainty (“Manual selection”), or the default TOT procedure of the manufacturer (“Manufacturer”).
NOTE: The width of the split point range when using the manufacturer’s procedure is set to zero (i.e., the manufacturer’s procedure does not consider split point uncertainty). - In sub-section (3)(b), depending on the selected procedure to calculate the split point and uncertainty, define the nominal (mean) split point, split point uncertainty, initial laser attenuation, and/or critical attenuation decline.
NOTE: The user inputs the initial laser attenuation and a threshold of attenuation decline for the “Attenuation decline” procedure and inputs the split mean and split uncertainty for the “Manual selection” procedure. Initial laser attenuation is not used in the “Manual selection” procedure but can be tuned to support the manual selection of the split point. - In section (4), review the accuracy and uncertainty of the split point. Leveraging the AVEC plot, repeat steps 6.4.1 and 6.4.2 as required, until a satisfactory split point and reasonable split point uncertainty are achieved. Use the zoom in (
 ), zoom out (
), zoom out ( ), and pan (
), and pan ( ) utilities as required to manipulate the AVEC plot and support the selection of the split point and its uncertainty.
) utilities as required to manipulate the AVEC plot and support the selection of the split point and its uncertainty.
- In sub-section (3)(a), select the desired procedure to calculate the split point and associated uncertainty: the novel “Attenuation decline” procedure described in the introduction section, a manually-defined split point and uncertainty (“Manual selection”), or the default TOT procedure of the manufacturer (“Manufacturer”).
- Run Monte Carlo analysis — GUI section (5).
- In sub-section (a), insert the estimated precision of the instrument as a whole.
NOTE: Instrument precision (repeatability) in units of µg. The default value in OCECgo (0.031 μg) is based on estimates by the authors via replicate blank analyses. - In sub-section (b), insert the desired number of Monte Carlo draws for the computation of carbon masses.
NOTE: The number of Monte Carlo draws corresponds to the number of random computations of the carbon masses under the Monte Carlo framework. Larger numbers yield more accurate and consistent results, at the cost of computational time. The default value in OCECgo is 106 while permitted values are [102, 108]. - In sub-section (c), press to run the Monte Carlo analysis to compute carbon masses and associated uncertainties.
NOTE: Following execution of the Monte Carlo analysis, the user is migrated to the Data Analysis Tool – Results tab.
- In sub-section (a), insert the estimated precision of the instrument as a whole.
- Review results. The Data Analysis Tool – Results tab reports statistics of the measured OC, EC, and total carbon (TC); histograms of the Monte Carlo results; and the best-fitting posterior probability distribution of carbon masses for use in subsequent Monte Carlo procedures, chosen by the Akaike information criterion28.
- Press the Export Analysis Results button to export the Monte Carlo results.
NOTE: Numerical data are exported to a pre-formatted .xlsx file and visualization of the Monte Carlo results are exported as a .png file.
- Press the Export Analysis Results button to export the Monte Carlo results.
Results
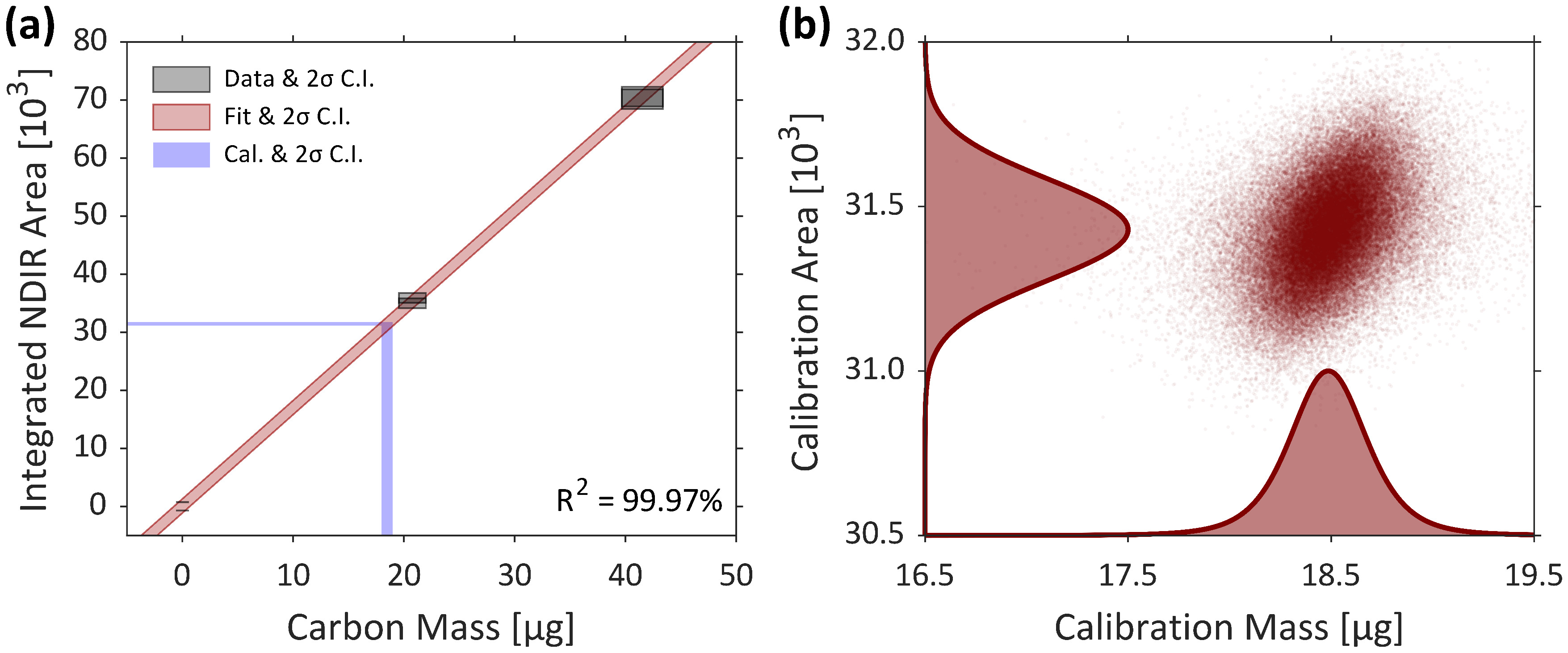
Representative results for the calibration of the OCEC instrument are presented using example calibration data acquired by the authors, shown in Table 1. Here, a six-point calibration is used, obtained using the developed software and following the manufacturer’s example in the instrument manual18. Results are shown in Figure 2. Figure 2a presents the results of linear regression on the calibration data under an MC framework. Black points correspond to the 2σ confidence interval of each of the six calibration points — i.e., carbon mass in the deposited sucrose is on the horizontal axis and instrument-reported total area (column 2 of Table 1) is on the vertical axis. The red shaded region represents the 2σ CI of the linear regression based on these six uncertain calibration data points ― two each for 0, 5, and 10 μL of the sucrose solution (column 1 of Table 1). The mean carbon mass injected during the CH4-loop is determined within the MC method. For each MC draw (2×107 in this example), a randomized calibration area (based on the distribution of the mean of calibration areas from column 3 of Table 1) is coupled with the uncertain linear model to obtain a MC-estimate of CH4-loop carbon mass. The horizontal and vertical blue shaded regions correspond to the 2σ CIs of the mean calibration area and mass calibration constant from the MC analysis. The MC-estimates of these calibration data are shown in the scatterplot-histogram in Figure 2b. This example dataset yielded a calibration constant of 18.49 μgC ± 2.78%.
| Calibration Data | ||
| Volume of Sucrose Solutiona [μL] | "Total" NDIR Area [-] | "Calibration" NDIR Area [-] (CH4-loop) |
| 0 | 3041 | 31297 |
| 5 | 38229 | 31281 |
| 5 | 37321 | 32056 |
| 10 | 72472 | 31435 |
| 0 | 1589 | 31583 |
| 10 | 72914 | 30926 |
| a Nominal concentration of 4.148 ± 0.022 gC/L at 25 °C | ||
Table 1: Representative calibration data. Representative calibration data containing two blanks and two measurements each at 5 μL and 10 μL of sucrose solution, consistent with the manufacturer’s example in the instrument manual18.

Figure 2: Representative calibration results. (a) Results of the MC-regression of a six-point calibration dataset obtained by the authors. Calibration data with 2σ uncertainties are represented by black boxes and the 2σ CI of the linear regression is shown in the red shaded region. The horizontal blue bar corresponds to the 2σ CI of the mean NDIR area during the CH4-loops (“calibration areas”), which are coupled with the uncertain linear regression to yield the 2σ CI of the mass calibration constant (represented by the vertical blue bar). (b) A scatterplot-histogram representing the MC-computed calibration data; mass calibration constant on the horizontal axis and mean calibration area on the vertical axis. In this example uncertainty in the mass calibration constant was approximately 2.78%. Please click here to view a larger version of this figure.
{kind=link}
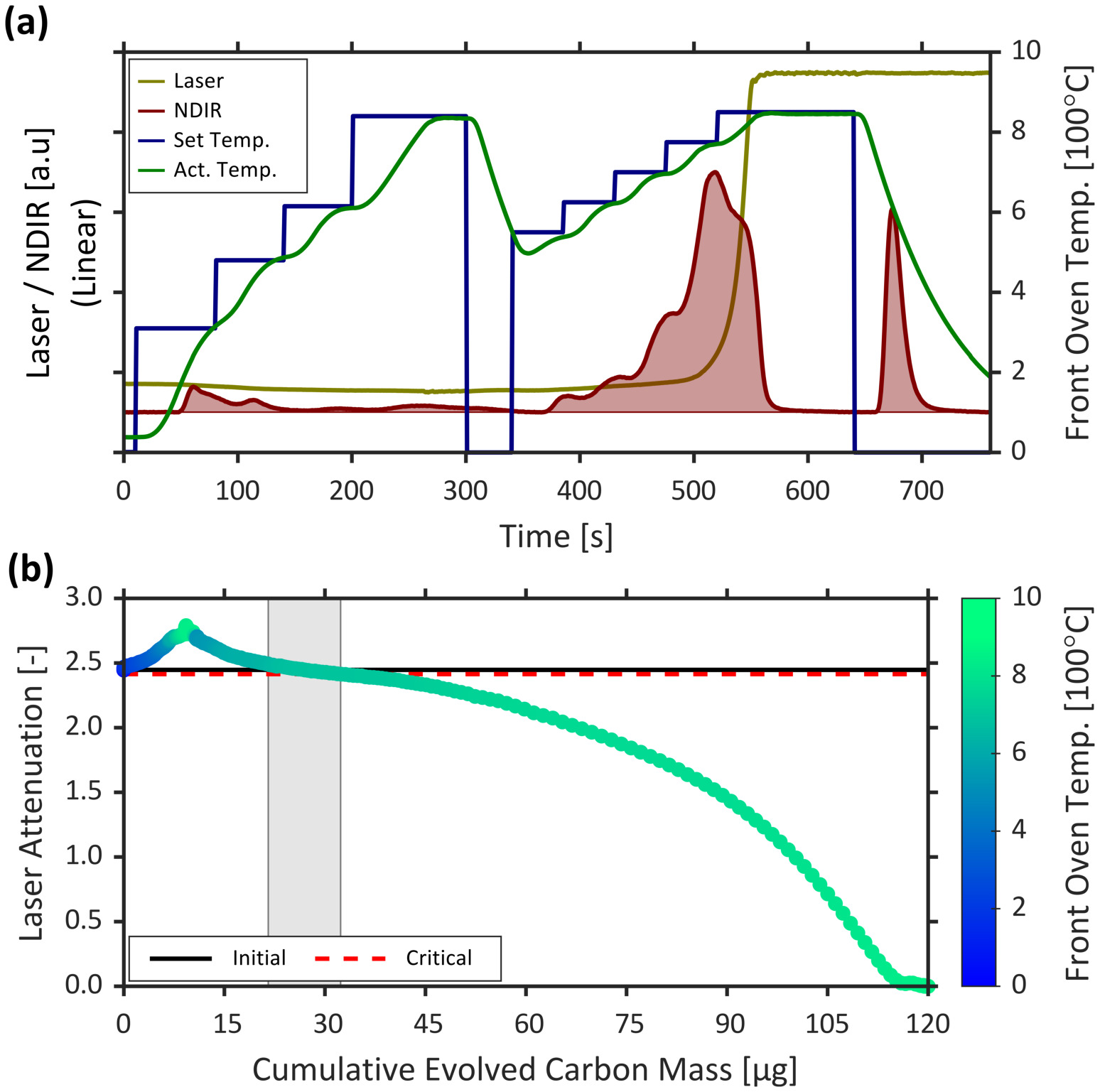
A representative data analysis using the developed software is provided for a measurement of carbonaceous emissions from a laboratory soot generator (MiniCAST 5201 Type C) burning nitrogen-diluted propane fuel29. Summaries of the sample data in the form of an OCEC thermogram (a plot displaying laser power, NDIR, and oven temperature during an OCEC analysis) and an AVEC21 plot — created during protocol step 6.3.4 — are shown in Figure 3a and Figure 3b, respectively. In this example, uncertainty in the split point is estimated using the above-described “Attenuation decline” technique developed by the authors. The critical decline in attenuation was quantified as 1.342% yielding a split point uncertainty of 4.50% of TC mass. Key results of this analysis ― carbon mass statistics and best-fitting posterior distributions ― are summarized in Table 2.

Figure 3: Representative analysis data. (a) A thermogram of OCEC-measured laser power, NDIR signal, desired (set), and measured (actual, act.) oven temperature. (b) The attenuation vs. evolved carbon (AVEC) plot corresponding to the presented histogram. The points correspond to instantaneous data reported at 1 Hz, with point colouring representing the instantaneous front oven (filter) temperature. The horizontal black line corresponds to the initial laser attenuation (following sample acquisition and prior to thermal analysis) used in the determination of the split point, while the horizontal red line corresponds to a 1.342% decline in initial laser attenuation used to estimate uncertainty in the split point. The grey shaded region represents the 2σ CI of the split point for this analysis, which is approximately 4.50% of TC mass. Please click here to view a larger version of this figure.
{kind=link}
| Carbon Type | Mean Mass [μgC] | 2σ Uncertainty of MC Data | Best-fitting Distributiona |
| Organic Carbon (OC) | 26.94 | -21.3% +22.2% | N(26.94, 2.925) |
| Elemental Carbon (EC) | 93.11 | -9.98% +10.4% | Γ(385.7, 0.2414) |
| Total Carbon (TC) | 120.05 | -8.32% +8.40% | N(120.1, 5.014) |
| a Normal: N(μ, σ); Gamma: Γ(a, b[scale]) | |||
Table 2: Representative carbon mass results. MC-computed OC, EC, and TC masses for the example data plotted in Figure 3 with a 2σ uncertainty in the split point of 4.50%, corresponding to a 1.342% decline in initial laser attenuation. Listed are the mean and 2σ uncertainties of the MC data, in addition to the best-fitting posterior distributions.
Discussion
Table 3 shows the contribution of the specific sources of uncertainty to the mass calibration constant for the example case described in Table 1 and Figure 2. Cumulative calibration uncertainty resulting from bias error in the NDIR detector, bias error in the concentration of sucrose, and precision and bias error in pipetted volumes are listed. Bias error in the NDIR detector (i.e., variance in the “calibration area”) tends to dominate, with bias in the pipetting procedure being second-most important (although quite small in the representative example). Proper estimation of pipetting error is thus critical to ensuring accurate quantification of overall calibration uncertainty; referring to protocol step 4.3.2, it is therefore suggested that intra-user repeatability and inter-user reproducibility be assessed for each group of users and pipette. In contrast, uncertainty due to the concentration of sucrose in the external standard is negligibly small. Furthermore, there appears to be negligible contribution from regression uncertainty, a likely consequence of the good linearity of the instrument − the coefficient of determination (R2) for a linear fit of the calibration data generally exceeds 99.95%. If calibration data are not sufficiently linear, the software automatically warns the user, who is then able to troubleshoot the dataset via the Boolean control noted in protocol step 4.2; the user can then modify their calibration dataset by acquiring replacement data as needed.
| Considered Uncertainties | Full (6-point) Calibration |
| NDIR bias | ± 2.61% |
| + Sucrose solution | ± 2.61% |
| + Pipette | ± 2.78% |
| Nominal Result [μgC] | 18.49 |
Table 3: Uncertainties in mass calibration constant. Representative contribution of uncertainties in the calibration of the OCEC instrument for the example six-point calibration (see Table 1). Overall calibration uncertainty is dominated by bias in the NDIR detector with error due to accuracy in the pipetting of the sucrose solution, including human-errors (inter-user reproducibility and intra-user repeatability), being second-most important, followed by uncertainty in linear regression and sucrose concentration (which are both negligible).
Calibration of the OCEC instrument is a time-consuming procedure, typically requiring 2−3 hours to complete depending upon the length of the thermal protocol employed. A more rapid calibration procedure is desirable. To this end, the efficacy of a modified, truncated calibration protocol was analyzed with the presented software tool. The developed MC procedure was executed using all possible subsets of the example calibration data listed in Table 1 — limited to cases with three or more data and a minimum of one blank measurement. All resulting mass calibration constants from this analysis are plotted in Figure 4 as a function of number of calibration data used, where calibration constants have been normalized by the full (6-point) calibration result. Unsurprisingly, uncertainty in the calibration constant increases as available calibration data are reduced. Importantly however, the means of all truncated calibrations fall within the 2σ CI of the full calibration result, which is a consequence of the above-noted linearity of the instrument. This consistency in the MC-average suggests that a modified, faster calibration consisting of few calibration data can be employed as a “bump test” check of the OCEC instrument’s calibration. That is, if the MC-average of a 3-point calibration data set is within the 2σ CI of the existing calibration, it is likely that the OCEC instrument does not require re-calibration. It is also apparent in Figure 4 that calibration uncertainty is reduced with more calibration data, but the reduction in uncertainty suffers from diminishing returns. Referring to Table 3 and its discussion above, since calibration uncertainties are dominated by NDIR bias (quantified with the standard error of the “calibration areas”), the marginal decrease in calibration uncertainty by including the nth data point can be estimated with the factor √(1−1/n). Consequently, in the representative example, the marginal reduction in uncertainty is larger when moving from a three- to a four-point calibration than when moving from a five- to a six-point calibration. The developed software tool, which can be executed following acquisition of each calibration data point (i.e., following each repetition of protocol step 3.3), allows the user to quantify calibration uncertainty throughout the acquisition of data. Critically, this ability enables the user to not only decide upon adequate convergence of the calibration in the context of its uncertainty, but also detect the presence of spurious data — that is, an incremental decrease in calibration uncertainty that is notably different than expected highlights to the user that the most recently acquired calibration data point could be flawed.

Figure 4: Assessment of sample size on calibration uncertainty. Computed mass calibration constant for all possible combinations of calibration data listed in Table 1 (requiring a minimum of three data, including at least one blank) normalized by the result using all six data. Relative uncertainty in the calibration constant decreases with an increase in the number of calibration data. The blue shaded area in the figure corresponds to the 2σ CI of the calibration constant computed using all calibration data. It is apparent that all nominal results are within this CI suggesting that — although uncertain — a truncated calibration procedure of just three calibration data points might be used as a “bump test” check of the instrument’s calibration. Please click here to view a larger version of this figure.
{kind=link}
Calculated carbon masses and uncertainties for four example datasets are detailed in Table 4; these data are sourced from measurements of carbonaceous emissions from hydrocarbon flames27,29, gas turbines29, and fine-mode (< 2 μm) carbonaceous particles obtained from sub-ocean sediment samples30. Computed (MC-average) OC, EC, and TC masses are shown in Table 4 alongside the computed critical attenuation decline and the EC/TC ratio for each dataset, showing the breadth of the example data in the context of sampled carbon composition. Summarized in Table 4 are the sources and overall uncertainties in carbon mass using the presented software tool compared to those reported by the instrument. Within the software tool, the combination (product distribution) of calibration uncertainty and repeatability yields the overall uncertainty in MC-computed TC mass (quantified as −8.32/+8.40% in the present work), which is independent of uncertainty in the split point and therefore acts as a lower limit in the uncertainty of OC and EC masses. These representative calibration uncertainties are applied to each example dataset while maintaining the nominal mass calibration constant used in the original analyses.

Table 4: Uncertainties in data analysis. Contribution to uncertainty in OCEC-measured carbon masses for four example datasets from a broad range of sources and performed by different laboratories27,29,30. (a) Key numerical results from the example datasets: OC, EC, and TC masses, critical attenuation decline for the quantification of split point uncertainty, and elemental-to-total carbon ratio. (b) A summary of uncertainties in the calculated carbon masses. Contributing sources of uncertainty include that of the mass calibration constant, repeatability of the calibration procedure, and uncertainty in the split point (relative to TC mass) corresponding to the critical attenuation decline listed in (a). Uncertainties in the carbon masses using the fixed equations (Eq. (1)) employed by the instrument are also shown in (b). Red and yellow highlighting of the data correspond to under- and over-estimation of uncertainty, respectively, when using the fixed equations relative to the present method. In most cases, the instrument under-estimates carbon mass uncertainty however, if measured OC or EC mass is small, the instrument may over-estimate uncertainty as compared to the present software. Please click here to download this file.
For these examples, the attenuation decline technique for quantifying the split point uncertainty was employed. OCECgo-computed critical values of attenuation decline ranged from of 1.342% to 2.059% resulting in split point uncertainties from 0.10% to 4.50% of TC mass. While the employed values of attenuation decline are indeed somewhat subjective − particularly the employed factor to estimate uncertainty in the split point due to that of optical properties − these examples highlight the dependence of split point uncertainty upon the specific analysis data. For example, split point uncertainty is sensitive to the slope of the AVEC plot in the vicinity of the nominal split point. Consider dataset “A” corresponding to the example data in Figure 3 and dataset “D”; despite having similar critical attenuation declines, the relatively shallow and steep slopes of their respective AVEC plots (e.g., see Figure 3b for dataset “A”) yield the largest and smallest split point uncertainties of 4.50% and 0.10% of TC mass. Additionally, the example data show that the influence of uncertainty in the split point depends largely on its scale relative to the nominal OC and EC masses. Consider example datasets “B” and “C”, which have near-identical split point uncertainties (≈1.22% of TC mass); dataset “C” contains ≈43% OC while dataset “B” contains ≈8%; the lower relative quantity of OC in the latter results in an almost doubling of OC mass uncertainty. Critically, these results highlight the requirement to directly consider split point uncertainty in the context of the analysis’ AVEC data and overall carbon masses.
Manufacturer-reported uncertainties in carbon masses are also shown in Table 4. These estimates do not directly consider uncertainties in the calibration and split point, but rather are calculated using the fixed relations shown in Eq. (1)26, where mi represents the nominal mass of the specific carbon component.

(1) 

These fixed relations permit estimated uncertainties in OC and/or EC mass to be artificially less than that of TC mass ― this condition occurs when OC or EC mass is less than one third of TC mass, as is the case for datasets “A”, “B”, and “D”. This result is non-physical, since relative uncertainty in OC and EC masses must be bounded below by the that of TC mass, a consequence of the propagation of split point uncertainty into the calculated OC and EC masses. Cells highlighted red and yellow in the table correspond to under- and over-estimations of carbon mass uncertainty when using Eq. (1) from the manufacturer. The fixed equations under-estimate TC mass uncertainty for all four examples, a consequence of computed TC masses being sufficiently large. In most cases, the fixed equations also under-estimate EC and OC mass uncertainty, except where OC (dataset “B”) and EC (dataset “D”) were sufficiently small to cause over-estimation via Eq. (1). This asymptotic increase in uncertainty via Eq. (1) agrees with the present software since uncertainty in OC and EC masses due to that of the split point are dependent upon their absolute magnitude; however, uncertainties via the fixed equations fail to track with those of the present software, which directly considers and propagates split point uncertainty in the context of specific analysis data.
Use of a MC framework in the presented software tool is necessary to accurately propagate component uncertainties through the non-linear algorithms of thermal-optical OC/EC analyses. It is important to note however, through their inherently random nature, that MC methods are not deterministic and tend to yield inconsistent results if the employed number of MC draws/repetitions (see protocol steps 4.3.3 and 6.5.2) is insufficient ― akin to a statistically undersized sample. Therefore, there is an inherent consistency vs. computation time trade-off to be considered when processing data using OCECgo. It is thus recommended to users to perform preliminary processing and troubleshooting of the data using a small number of MC draws (e.g., 104). Once computations yield satisfactory results, the user should then increase the number of MC draws (to 106-108) to yield a result that is less-affected by the discrete and random nature of the MC method. Beyond the necessary “operational definition” of thermal-optical OC/EC analyses, there are other limitations in the processing of these data that must also be recognized when using OCECgo to compute and report OC/EC data. Firstly, NDIR-based instruments (such as that listed in the Table of Materials) suffer from drift in the NDIR signal that must be corrected. In the present protocol (see step 6.3.2 and the OCECgo documentation), a novel approach to correct drift in the NDIR detector can be optionally employed by the user. While, in the authors’ experience this yields improved results over the manufacturer’s standard linear NDIR correction, it must be noted that uncertainty in this NDIR correction is challenging if not impossible to quantify and therefore remains an unaccounted component of uncertainty in the computation of carbon masses. In a similar vein, it is also challenging to quantify uncertainty in the necessary assumption that PC and EC share optical properties. If selected (see protocol step 6.4.1), the critical attenuation decline technique attempts to conservatively bound the effect of this assumption through a subjective expansion factor. Importantly however, this is necessarily only an estimate, and the user is suggested to assess the effect of this expansion factor (i.e., the critical attenuation decline) on their specific data. OCECgo is provided as an open-source tool so that it may easily be extended by the authors and other interested collaborators to not only interface with other instruments but to also include other useful, field-specific, functionalities. Overall, the developed open-source software tool coupled with the detailed calibration procedure is intended to help achieve more accurate measurements of OC, EC, and TC mass in aerosol samples while streamlining the robust calculation of measurement uncertainties.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) FlareNet Strategic Network (Grant # 479641), NSERC Discovery Research Grants (Grant #06632 and 522658), and Natural Resources Canada (Project Manager, Michael Layer). The authors are grateful to those who shared raw data files for use as representative examples in this work.
Materials
| Name | Company | Catalog Number | Comments |
| 10% oxygen gas in helium | Local gas supplier | - - - | Primary or certified standard preferred |
| 5% methane gas in helium | Local gas supplier | - - - | Primary or certified standard preferred |
| Distilled, de-ionized water | Harleco | 6442-85 | ASTM D1193-91 Type II or Type I (preferred) |
| Filter punch tool | Sunset Laboratories Inc. | - - - | Included with carbon analyzer |
| Filter removal tool | Sunset Laboratories Inc. | - - - | Included with carbon analyzer |
| Glass jar (4 oz.) | ULINE | S-17982P-BL | Or suitable equivalent; borosillicate glass preferred |
| Helium gas | Local gas supplier | - - - | Ultra-high purity (> 99.999%) or better preferred |
| High-accuracy thermal gas mass flow meter | Bronkhorst | EL-FLOW Prestige | For accurate measurement of sample volume (see Protocol step 5) |
| High-purity sucrose | Sigma Aldrich | S9378 | Purity ≥ 99%m/m or higher preferred |
| Lint-free tissues | Kimtech | 34155 | Or suitable equivalent |
| MatLab Runtime (R2016a or newer) | MathWorks Inc. | mathworks.com | Search "runtime compiler" and install appropriate version for the operating system |
| Non-powdered, disposable, plastic gloves | VWR | 89428-752 | Or suitable, properly-sized equivalent |
| OCECgo software | Carleton University, Energy and Emissions Research Lab. | GitHub Repository | Source and build distributions of the software are available on GitHub |
| Oxygen trap | Supelco | 22449 | Or suitable GC-quality equivalent |
| Pipette | Eppendorf | 3120000020 | Model: Research® Plus 0.5 - 10 μL - Or any single-channel, adjustable volume, manual pipette |
| Pipette tips | Eppendorf | 022492012 | Model: epT.I.P.S.® Standard, 0.1 - 20 μL |
| Precision scale / balance | AND | FX-3000IWP | Precision balance with capacity > 1 kg |
| Quartz filters | Pall | 7202 | Model: Tissuquartz 2500 QAT-UP - 47 mm |
| Semi-continuous thermal-optical organic/elemental carbon analyzer | Sunset Laboratories Inc. | - - - | Model 4 semi-continuous analyzer |
References
- Cohen, A. J., et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study. The Lancet. 389 (10082), 1907-1918 (2015).
- Grahame, T. J., Klemm, R., Schlesinger, R. B. Public health and components of particulate matter: The changing assessment of black carbon. Journal of the Air & Waste Management Association. 64 (6), 620-660 (2014).
- Kim, S. -. Y., et al. The short-term association of selected components of fine particulate matter and mortality in the Denver Aerosol Sources and Health (DASH) study. Environmental Health. 14 (1), (2015).
- Basagaña, X., et al. Short-term effects of particulate matter constituents on daily hospitalizations and mortality in five South-European cities: Results from the MED-PARTICLES project. Environment International. 75, 151-158 (2015).
- Jacobson, M. Z. Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature. 409 (6821), 695-697 (2001).
- Sato, M., et al. Global atmospheric black carbon inferred from AERONET. Proceedings of the National Academy of Sciences. 100 (11), 6319-6324 (2003).
- Ramanathan, V., Carmichael, G. Global and regional climate changes due to black carbon. Nature Geoscience. 1 (4), 221-227 (2008).
- Bond, T. C., et al. Bounding the role of black carbon in the climate system: A scientific assessment. Journal of Geophysical Research: Atmospheres. 118 (11), 5380-5552 (2013).
- Jacobson, M. Z. Short-term effects of controlling fossil-fuel soot, biofuel soot and gases, and methane on climate, Arctic ice, and air pollution health. Journal of Geophysical Research. 115, 1-24 (2010).
- Doherty, S. J., Grenfell, T. C., Forsström, S., Hegg, D. L., Brandt, R. E., Warren, S. G. Observed vertical redistribution of black carbon and other insoluble light-absorbing particles in melting snow. Journal of Geophysical Research: Atmospheres. 118 (11), 5553-5569 (2013).
- Sterle, K. M., McConnell, J. R., Dozier, J., Edwards, R., Flanner, M. G. Retention and radiative forcing of black carbon in eastern Sierra Nevada snow. The Cryosphere. 7 (1), 365-374 (2013).
- Goelles, T., Bøggild, C. E., Greve, R. Ice sheet mass loss caused by dust and black carbon accumulation. The Cryosphere. 9 (5), 1845-1856 (2015).
- Kopp, R. E., Mauzerall, D. L. Assessing the climatic benefits of black carbon mitigation. Proceedings of the National Academy of Sciences of the United States of America. 107 (26), 11703-11708 (2010).
- Cavalli, F., Viana, M., Yttri, K. E., Genberg, J., Putaud, J. -. P. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol. Atmospheric Measurement Techniques. 3 (1), 79-89 (2010).
- Chen, L. -. W. A., et al. Multi-wavelength optical measurement to enhance thermal/optical analysis for carbonaceous aerosol. Atmospheric Measurement Techniques. 8 (1), 451-461 (2015).
- Chow, J. C., et al. Optical Calibration and Equivalence of a Multiwavelength Thermal/Optical Carbon Analyzer. Aerosol and Air Quality Research. 15 (4), 1145-1159 (2015).
- Chow, J. C., et al. Separation of brown carbon from black carbon for IMPROVE and Chemical Speciation Network PM 2.5 samples. Journal of the Air & Waste Management Association. 68 (5), 494-510 (2018).
- Sunset Laboratory Inc. . Organic Carbon and Elemental Carbon Field Instrument: Model 4 User’s Manual. , (2019).
- Turpin, B. J., Cary, R. A., Huntzicker, J. J. An In Situ, Time-Resolved Analyzer for Aerosol Organic and Elemental Carbon. Aerosol Science and Technology. 12 (1), 161-171 (1990).
- Petzold, A., et al. Recommendations for reporting “black carbon” measurements. Atmospheric Chemistry and Physics. 13 (16), 8365-8379 (2013).
- Nicolosi, E. M. G., Quincey, P., Font, A., Fuller, G. W. Light attenuation versus evolved carbon (AVEC) - A new way to look at elemental and organic carbon analysis. Atmospheric Environment. , 145-153 (2018).
- Chow, J. C., Watson, J. G., Crow, D., Lowenthal, D. H., Merrifield, T. Comparison of IMPROVE and NIOSH Carbon Measurements. Aerosol Science and Technology. 34 (1), 23-34 (2001).
- Chow, J. C., Watson, J. G., Chen, L. -. W. A., Arnott, W. P., Moosmüller, H., Fung, K. Equivalence of Elemental Carbon by Thermal/Optical Reflectance and Transmittance with Different Temperature Protocols. Environmental Science & Technology. 38 (16), 4414-4422 (2004).
- Subramanian, R., Khlystov, A., Robinson, A. Effect of peak inert-mode temperature on elemental carbon measured using thermal-optical analysis. Aerosol Science and Technology. 40 (10), 763-780 (2006).
- Wang, Y., Chung, A., Paulson, S. E. The effect of metal salts on quantification of elemental and organic carbon in diesel exhaust particles using thermal-optical evolved gas analysis. Atmospheric Chemistry and Physics. 10 (23), 11447-11457 (2010).
- Peterson, M. R. Standard Operating Procedure for the Determination of Organic, Elemental, and Total Carbon in Particulate Matter Using a Thermal/Optical-Transmittance Carbon Analyzer. Research Triangle. , (2009).
- Conrad, B. M., Johnson, M. R. Mass Absorption Cross-Section of Flare-Generated Black Carbon: Variability, Predictive Model, and Implications. Carbon. 149, 760-771 (2019).
- Akaike, H. A new look at the statistical model identification. IEEE Transactions on Automatic Control. 19 (6), 716-723 (1974).
- Saffaripour, M., et al. Raman spectroscopy and TEM characterization of solid particulate matter emitted from soot generators and aircraft turbine engines. Aerosol Science and Technology. 51 (4), 518-531 (2017).
- Lu, S., Irino, T., Igarashi, Y. Biomass burning history in East Asia during the last 4 million years recorded in elemental carbon variability at IODP site. U1423. Progress in Earth and Planetary Science. 5 (1), (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved